

Key Clinical Message
Here, we report a novel mosaic mutation in the PORCN gene in a male Goltz syndrome patient. We also compare the phenotypes of all reported males with a confirmed molecular diagnosis. This report serves to further clarify the phenotype of Goltz syndrome and suggests that expression in males varies.
Keywords: focal dermal hypoplasia, Goltz syndrome, male, PORCN
1. INTRODUCTION
Goltz syndrome (GS), also known as Focal dermal hypoplasia (OMIM #305600), is a rare X‐linked dominant syndrome with variable meso‐ectodermal abnormalities.1 Common clinical findings concern the skin (patchy skin aplasia, subcutaneous fat herniation, papilloma, telangiectasia, sparse hair, syndactyly, dysplastic nails, linear hypo‐/hyperpigmentation often following the lines of Blaschko), skeleton (ectrodactyly, oligodactyly, transverse limb defects, long bone reduction defects), eyes (anophtalmos, microphthalmia, cataract, choroid or retinal coloboma), and face (facial asymmetry, hypoplastic alae nasi, abnormal ear morphology, and dental defects).2
The molecular basis of GS was first described in 2007, when it was reported that GS is caused by loss‐of‐function mutations in the PORCN gene.3, 4 PORCN is the human homolog of a Drosophila polarity gene “porcupine”, located on the X chromosome (Xp11.23). The PORCN gene encodes a 461‐amino acid 52‐kDa endoplasmic reticulum protein, the porcupine protein, thought to be important for modification and excretion of Wnt proteins.5, 6 Wnt proteins are critical for interactions between ectoderm and mesoderm during embryogenesis.4, 7 From what is known today, PORCN is the only gene associated with GS. A total of 171 different mutations are registered in the online PORCN mutation database (http://www.lovd.nl/PORCN. Accessed on February 6, 2018), and these are scattered throughout the entire coding sequence of the PORCN gene.8
The phenotype in GS male patients is not extensively depictured, and it has not been reported how many male patients exist in total with a confirmed molecular diagnosis. This report aims to further clarify the genotype‐phenotype correlation of GS in males, which is important since male patients might have been missed or misinterpreted.
2. MATERIALS AND METHODS
2.1. Study approval
The study was performed in accordance with the Declaration of Helsinki, and the local ethical board in Stockholm approved the study. Informed consent was obtained from the family according to local ethical guidelines, and the publication of clinical data and pictures has been approved by the parents.
2.2. Genetic analysis
Genomic DNA was extracted from blood lymphocytes and fibroblasts from skin biopsies from affected and normal skin by standard protocols. The PORCN gene was amplified by PCR and screened for mutations by genomic sequencing of both DNA strands of the entire coding region and the highly conserved exon‐intron splice junctions. We performed digital PCR (QuantStudio 3D; Applied Biosystems, Foster City, CA, USA) using the manufacturer's protocol. For array comparative genomic hybridization (CGH), a custom 4x180K array CGH platform was used (Oxford Gene Technologies, Oxfordshire, UK). This platform has a genomewide average base pair spacing of about 20 Kb. Experiments were performed according to the manufacturer's protocol.
3. RESULTS
3.1. Clinical report
A 3‐year‐old male was referred to the Department of Dermatology at Danderyd Hospital in Stockholm to explore multiple skin aberrations. The patient had been adopted from China, and his medical history was unknown. The patient had several abnormalities: His face was asymmetric with a right‐sided hypoplasia, widely spaced teeth, and low set ears (Figure 1A). There was hypopigmentation in a blaschkolinear distribution on his right arm (Figure 1B). Telangiectasia was present on both arms (Figure 1B) and on the left leg (Figure 1D) in a blaschkolinear distribution. He had atrophic skin with nodular fat herniation clustered on the right‐side trunk (Figure 1C). There were areas of patchy skin aplasia on his right leg (Figure 1D). His right foot showed partial ectrodactyly of the second toe (Figure 1D). He had clinodactyly on both hands and a complete syndactyly of the third and fourth fingers of the left hand (Figure 1E). All the nails of the left hand and the nails of the second and third finger of the right hand were ridged. In addition, he had a right‐sided dacryostenosis, short stature (−4 SD), inguinal hernia, and osteopathia striata. A diagnosis of GS was suspected clinically.
Figure 1.
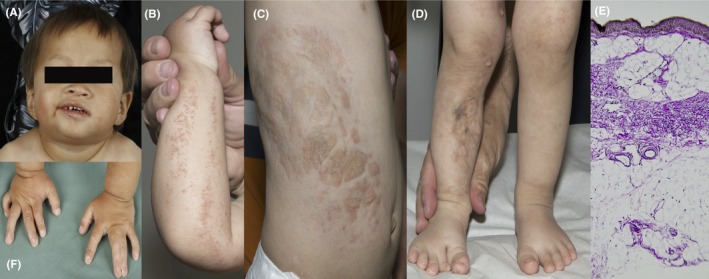
A, Right‐sided facial hypoplasia. Widely spaced teeth and low set ears. B, Hypopigmentation in a Blaschkolinear distribution flanked by telangiectasia on the right arm. C, Atrophic skin with nodular fat herniation clustered on the right‐side trunk. D, Patchy skin aplasia on the right lower leg. Partial ectrodactyly of the second toe. E, Slightly atrophic epidermis lacking adnexal structures. Collagen bundles greatly diminished and replaced by mature adipose tissue in dermis, reaching up to the epidermis in a “nevus lipomatosus superficialis‐like” manner. F, Clinodactyly on both hands and a complete syndactyly of the third and fourth fingers of the left hand. Ridged nails of all fingers of the left hand and of the second and the third finger of the right hand.
3.2. Histopathological findings
Skin biopsies were performed on the atrophic patches, and microscopy showed thinning of dermis, few collagen structures, and herniation of fat lobules in the upper dermis (Figure 1E).
3.3. Genetic findings
A previously unreported mosaic variant in exon 14, c.1274_1275del (p.Thr425Argfs*45) (NM_203475.2), was identified in samples of blood, affected skin, and normal skin by Sanger sequencing. To quantify the mosaic levels in each tissue, we performed digital PCR which showed the variant in 72% of the cells in affected skin, 49% in normal skin, and 47% in blood. The variant creates a frame shift starting at codon Thr425. The new reading frame ends in a stop codon 44 positions downstream. This PORCN variant was classified as likely pathogenic. Array CGH was normal and excluded Klinefelter syndrome.
4. DISCUSSION
This paper describes a mosaic GS male patient with a previously unreported PORCN variant and gives an overview of genetic and clinical findings in all male patients. To the best of our knowledge, this is the 24th GS male patient reported with a confirmed molecular diagnosis.
The clinical presentation of our patient is typical for GS and in accordance with the proposed diagnosis criteria.2 In addition, the detected variant leads to a stop codon, which makes the diagnosis GS very likely. However, mosaic levels were also found in unaffected tissue. We conclude that the mutation exists in both affected and unaffected tissue, but in a higher percentage in affected tissue. It has been observed in other mosaic disorders that the mutation percentage may vary and that mutations can occur even in seemingly unaffected tissue.9, 10
When reviewing the literature, we found more than 400 reported GS patients.11, 12 GS was long presumed to be lethal in males.13, 14 However, later reports have shown that approximately 10% of all GS patients are males.2, 15 The majority of surviving males can be explained by mosaicism for PORCN mutations that arise postzygotically during embryogenesis 16 or Klinefelter syndrome, since these males carry an additional X chromosome.5 There is no genotype‐phenotype correlation, which can be explained by random X‐inactivation in females or mosaicism.17, 18, 19 Of the 24 reported male patients, 19 are mosaics, one patient has Klinefelter syndrome, and four patients from two different families are non‐mosaics (Table 1). Interestingly, all four patients with non‐mosaic PORCN mutations lacked skin manifestations but had other severe symptoms compatible with GS.20, 21 In addition, diaphragm abnormalities were detected in two patients, resembling previously reported cases with pentalogy of Cantrell.18, 22
Table 1.
Overview of genetic and clinical features in all reported GS male patients with a confirmed PORCN mutation
| Patient (reference) | 1 (Madan et al)21 | 2 (Madan et al)21 | 3 (Brady et al)20 | 4 (Brady et al)20 | 5 (Alkindi et al)5 | 6 (Durack et al)7 | 7 (Rao et al)19 | 8 (Yoshihashi et al)16 | 9 (Stevenson et al) 25 | 10 (Vreeburg et al)13 | 11 (Bornholdt et al)15 | 12 (Bornholdt et al)15 | 13 (Bornholdt et al)15 | 14 (Bornholdt et al)15 | 15 (Maas et al)18 | 16 (Wang et al)4, 12 | 17 (Wang et al)4, 12 | 18 (Wang et al)4, 12 | 19 (Wang et al)4, 12 | 20 (Bostwick et al)2 | 21 (Bostwick et al)2 | 22 (Peters et al)1 | 23 (Young et al)14 | 24Our patient | Frequency |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | c.749C>T | c.470G>A | c.1093C>T 46,XXY | c.1039_1046delinsT | c.898G>T | c.129G>A | c.956dup | c.886del | c.502G>A | c.1110del | c.1186C>T | c.1315T>C | c.571C>T | c.1059_1071dup | c.370C>T | c.1064_1081del | c.1093C>G | c.956dup | c.1059_1071dup | c.853_855del | c.956dup | c.1274_1275del | – | ||
| Exon | 9 | 5 | 13 | 12 | 10 | 2 | 11 | 10 | 5 | 13 | 14 | 15 | 6 | 12 | 4 | 12 | 13 | 11 | 12 | 10 | 11 | 14 | – | ||
| Protein alteration | p.Ser250Phe | p.Gly157Asp | p.Arg365Trp | p.Leu347Trpfs*50 | p.Glu300* | p.Trp43* | p.Asn320Glufs*99 | p.Arg296Glyfs*18 | p.Gly168Arg | p.Ile371Serfs*28 | p.Arg396* | p.Trp439Arg | p.Gln191* | pThr358Profs*65 | p.Arg124* | p.Ala355_Val360del | p.Arg365Gly | p.Asn320Glufs*99 | pThr358Profs*65 | p.Thr285del | p.Asn320Glufs*99 | p.Thr425Argfs*45 | – | ||
| Mosaic | − | N/A | N/A | − | − | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | 19/24 |
| Birth weight (g) | 1900 | N/A | 3010 | 3040 | N/A | N/A | 2400 | 3290 | N/A | 1050 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 2700 | N/A | N/A | − |
| Typical skin/hair findings | − | − | − | − | +1 | +2 | +3 | +4 | +5 | +6 | +7 | +7 | +8 | +7 | +9 | +10 | +11 | +12 | +11 | +13 | +14 | +15 | +16 | + | 20/24 |
| Microphthalmia | + | + | + | + | + | − | − | − | − | − | − | − | − | − | − | − | − | − | + | + | − | − | − | − | 7/24 |
| Coloboma | + | − | + | − | + | − | + | − | + | − | + | − | − | − | − | − | − | − | + | + | − | + | + | − | 10/24 |
| Other ocular defects | +17 | − | − | +18 | +19 | − | − | + | +20 | − | − | − | +21 | − | − | − | − | − | − | − | − | +22 | + | +23 | 9/24 |
| Any ocular defect | + | + | + | + | + | − | + | + | + | − | + | − | + | − | − | − | − | − | + | + | − | + | + | + | 15/24 |
| Dental defects | N/A | N/A | − | − | +24 | +25 | +26 | − | − | − | + | − | + | − | − | + | − | − | − | + | + | − | − | + | 9/24 |
| Syndactyly | +27 | +28 | + | + | +28 | − | +29 | +30 | +31 | +32 | + | − | + | + | − | − | + | + | + | +29 | +33 | +34 | + | + | 20/24 |
| Ectrodactyly | +29 | − | − | − | +33 | − | + | − | +33 | − | − | − | − | − | − | + | + | − | + | +35 | +36 | +37 | +38 | + | 12/24 |
| Dysplastic nails | + | N/A | − | − | + | − | − | + | + | + | − | − | − | − | − | − | − | + | + | + | + | +39 | + | + | 12/24 |
| Osteopathia striata | + | N/A | − | − | − | − | − | − | + | − | − | − | − | − | − | − | − | + | − | N/A | N/A | − | N/A | + | 4/24 |
| Clavicular dysplasia | + | +40 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | N/A | N/A | − | − | − | 2/24 |
| Costovertebral defect | + | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | + | N/A | N/A | − | − | − | 2/24 |
| Diaphrapmatic hernia | − | + | + | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | N/A | N/A | − | − | − | 2/24 |
| Inguinal hernia | − | − | − | − | − | − | − | − | − | + | − | − | + | − | − | − | − | − | − | N/A | N/A | − | − | + | 3/24 |
| Cardiac anomalies | +41 | + | +42 | + | − | − | − | − | +43 | − | − | − | − | − | − | − | − | − | − | N/A | N/A | +44 | +42 | − | 7/24 |
| Pulmonary hypertension | − | − | − | − | − | − | − | − | + | − | − | − | − | − | − | − | − | − | − | − | − | − | + | − | 2/24 |
| Brain abnormality | N/A | N/A | N/A | +45 | + | − | − | − | − | +46 | − | − | +47 | − | − | − | − | − | − | N/A | N/A | +48 | − | − | 5/24 |
| Renal anomaly | +49 | +50 | − | +51 | + | − | − | +51 | − | − | − | − | − | − | − | − | − | − | − | N/A | N/A | − | − | − | 5/24 |
| Dysmorphic ears | +52 | − | − | − | + | − | − | − | +53 | +54 | − | − | − | − | − | − | − | − | − | + | − | +55 | +56 | + | 8/24 |
| Dysmorphic/asymmetric facial features | +57 | −58 | − | − | − | − | +59 | − | +60 | − | − | − | − | − | − | − | − | − | − | − | +61 | − | − | + | 5/24 |
+ =present, ‐ =absent, N/A =not available information.
1Red‐yellow atrophic cutaneous streaks with some telangiectasia in a linear pattern following Blaschko's lines on both arms and legs, the posterior neck, and the scalp, with some areas of alopecia. Clustered papillomas on the chin.
2Linear, reticulate, atrophic, and erythematous patches on the arms, thighs, and hips along the lines of Blaschko, with fat herniation in the right axilla.
3Atrophic areas of skin with yellowish nodules over depigmented macules following the lines of Blaschko. Sparse hair.
4On the right occipital scalp, an atrophic macule with small whitish spots and hair loss. Skin depressions <1 cm in diameter on the back and the right buttock. Small whitish depigmented spots, which were slightly depressed from the skin surface, distributed linearly on the trunk and arms. Streaks of brown‐pigmented macules on the dorsal aspect of the legs. Linear brown pigmentations on the dorsal aspect of the legs. Both the linear arrangement of the whitish spots and the streaks of pigmented macules followed the lines of Blaschko.
5Cutis aplasia neighboring the anterior fontanelle. Focal dermal hypoplasia following the lines of Blaschko on the lower extremities and linear lesions bilaterally on the trunk. Focal dermal hypoplasia on his right inner thigh. Small papules of the fourth toe and a small mobile mass on the posterior scalp. Sparseness of hair and eyelashes.
6Linear erythematous slightly atrophic skin lesions on the left cheek. Linear alopecia on the occipital area, following Blaschko's lines. Further atrophic slightly erythematous macules, sometimes containing telangiectasias, following Blaschko's lines on both flanks and the lateral aspects of both lower legs.
7Linear skin lesions.
8Linear skin lesions, patchy hairlessness.
9Aplasia cutis.
10Dermal hypoplasia, blaschkolinear pigmentation.
11Dermal hypoplasia.
12Dermal hypoplasia, blaschkolinear pigmentation, sparse hair.
13Hyperpigmentation, fat herniations, skin atrophy, telangiectasia.
14Hyperpigmentation, skin atrophy, telangiectasia.
15Hypoplasia, atrophy and linear hypopigmentation following the lines of Blashchko.
16Cutis aplasia, dermal hypoplasia.
17Microcornea.
18Dense intraocular tissue.
19Optic nerve atrophy and displaced lenses.
20Smaller left eye and subnormal visual evoked potentials in both eyes. Retinal flap near the ora serrata of the right eye.
21Optic atrophy.
22Bilateral nasolacrimal duct obstruction.
23Right side nasolacrimal duct obstruction.
24Widely spaced and some missing teeth.
25Very few remaining teeth, misshapen, and discoloured.
26Oligodontia.
27The left hand had syndactyly with a total of three digits. Syndactyly of the right first and second toes and the left third and fourth toes.
28Third and fourth toes bilaterally.
29Right hand.
30Cutaneous syndactyly of the right third and fourth fingers and toes and the left second and third toes.
31Syndactyly of the second and third right fingers.
32Cutaneous syndactyly of the second and third right fingers.
33Right foot.
34Both hands.
35Right hand and right foot.
36Left hand and left foot.
37Right hand and foot.
38Foot.
39Mild.
40Pseudo arthrosis of the right clavicle.
41Bicuspid aortic valve.
42Atrial septal defect.
43Small secundum atrial septal defect with spontaneous closure at one year of age, patent ductus arteriosus.
44A patent foramen ovale and an aberrant right subclavian artery were identified on echocardiogram.
45Enlarged ventricles, partial agenesis of the corpus callosum, and several intracerebral haemorrhages.
46Intraventricular right side cyst.
47Microcephaly, seizures.
48Microcephaly, myelomeningocele, Arnold‐Chiari malformation, and hydrocephalus.
49Multicystic kidney dysplasia with renal failure.
50Dysplastic kidneys, hydronephrosis, and renal failure.
51Hydronephrosis of the left kidney.
52Unfolded helices.
53Underdevelopment of the superior helices, slightly posteriorly rotated and large relative to his body.
54Asymmetric ears, the helix of the left ear showing cranial notching. The left ear slightly cup‐shaped.
55Underfolded ears with hypopigmentation of the helices.
56Simplified ears with underdevelopment of the superior helices.
57Bilateral clefts of the lip and cleft palate.
58Narrow face, midface hypoplasia, broad nasal bridge, small mandible.
59Right side hemihypotrophy.
60Slightly broad nasal tip with unusual linear erythema and dermal hypoplasia at the junction of the alae nasi, and asymmetry of the upper lip with hypoplastic tissue on the left.
61Right‐sided facial features more prominent than left‐sided.
When reviewing the literature of GS male patients with a confirmed molecular diagnosis, we noticed that skin findings, syndactyly, ocular defects, and ectrodactyly are common clinical features (Table 1). A study from 2016, including 18 patients with GS, demonstrated a higher incidence of ophthalmologic manifestations in patients with GS than previously reported (77% vs 40%).23 In our review over reported male patients, we found that 15 out of 24 (62.5%) had ocular manifestations (Table 1). However, ocular defects are not part of previously suggested clinical diagnosis criteria.2 Our suggestion is that GS should be suspected even in the absence of skin findings, if other typical signs occur, such as characteristic limb malformations or ocular defects, including coloboma or microphthalmia. Ocular manifestation should be considered as a major sign of GS, which has been suggested earlier.24
In summary, GS is a rare multisystem disorder with highly variable expressivity. This report serves to further clarify the phenotype of GS in males, which is important since there are, to our knowledge, only 24 male patients described with a confirmed molecular diagnosis. The highly variable expressivity might have caused male patients to be missed or misinterpreted until today. Our report adds to the knowledge of genotype‐phenotype correlations in male patients with PORCN mutations and highlights that PORCN mutations can be suspected in patients with characteristic limb malformations, such as ectrodactyly, or ocular manifestations, even in the absence of characteristic skin findings.
AUTHORSHIP
SF: coordinated the study, performed the genetic studies and interpreted the data. SF has reviewed the literature and written the draft of the manuscript with input from the other co‐authors. CGG: designed the study, assessed the clinical findings and has been involved in reviewing the literature, drafting the manuscript and revising it critically. KPS: contributed to the study design, assessment of clinical findings and manuscript revision. HTH: contributed to the study design, assessment of clinical findings and has been revising the manuscript critically. DC: performed the histopathological analysis and interpreted the findings. DC has contributed to the figure design and manuscript revision. IM: contributed to the study design, assessment of clinical findings and manuscript revision. TL: contributed to the study design, assessment of genetic findings, figure design and manuscript revision. AN: designed and coordinated the study. AN assessed the genetic findings and has been involved in drafting the manuscript and revising it critically. All authors have read and given final approval for the manuscript.
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGMENTS
This work was supported by grants from the Swedish Childhood Cancer Foundation, the Swedish Cancer Society, the Swedish Research Council, The Swedish Brain Foundation, Stockholm County Council and Karolinska Institutet.
Frisk S, Grandpeix‐Guyodo C, Silwerfeldt Popovic K, et al. Goltz syndrome in males: A clinical report of a male patient carrying a novel PORCN variant and a review of the literature. Clin Case Rep. 2018;6:2103–2110. 10.1002/ccr3.1783
REFERENCES
- 1. Peters T, Perrier R, Haber RM. Focal dermal hypoplasia: report of a case with myelomeningocele, Arnold‐Chiari malformation and hydrocephalus with a review of neurologic manifestations of Goltz syndrome. Pediatr Dermatol. 2014;31(2):220‐224. [DOI] [PubMed] [Google Scholar]
- 2. Bostwick B, Fang P, Patel A, Sutton VR. Phenotypic and molecular characterization of focal dermal hypoplasia in 18 individuals. Am J Med Genet C Semin Med Genet. 2016;172C(1):9‐20. [DOI] [PubMed] [Google Scholar]
- 3. Grzeschik KH, Bornholdt D, Oeffner F, et al. Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat Genet. 2007;39(7):833‐835. [DOI] [PubMed] [Google Scholar]
- 4. Wang X, Reid Sutton V, Omar Peraza‐Llanes J, et al. Mutations in X‐linked PORCN, a putative regulator of Wnt signaling, cause focal dermal hypoplasia. Nat Genet. 2007;39(7):836‐838. [DOI] [PubMed] [Google Scholar]
- 5. Alkindi S, Battin M, Aftimos S, Purvis D. Focal dermal hypoplasia due to a novel mutation in a boy with Klinefelter syndrome. Pediatr Dermatol. 2013;30(4):476‐479. [DOI] [PubMed] [Google Scholar]
- 6. Takada R, Satomi Y, Kurata T, et al. Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev Cell. 2006;11(6):791‐801. [DOI] [PubMed] [Google Scholar]
- 7. Durack A, Burrows NP, Staughton RC, Shalders K, Mellerio JE, Holden ST. Focal dermal hypoplasia: inheritance from father to daughter. Clin Exp Dermatol. 2017;42:457‐459. 10.1111/ced.13047. [DOI] [PubMed] [Google Scholar]
- 8. Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v. 2.0: the next generation in gene variant databases. Hum Mutat. 2011;32(5):557‐563. [DOI] [PubMed] [Google Scholar]
- 9. Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365(7):611‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McGoldrick P, Zhang M, van Blitterswijk M, et al. Unaffected mosaic C9orf72 case: RNA foci, dipeptide proteins, but upregulated C9orf72 expression. Neurology. 2018;90(4):e323‐e331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bree AF, Grange DK, Hicks MJ, Goltz RW. Dermatologic findings of focal dermal hypoplasia (Goltz syndrome). Am J Med Genet C Semin Med Genet. 2016;172C(1):44‐51. [DOI] [PubMed] [Google Scholar]
- 12. Wang L, Jin X, Zhao X, et al. Focal dermal hypoplasia: updates. Oral Dis. 2014;20(1):17‐24. [DOI] [PubMed] [Google Scholar]
- 13. Vreeburg M, van Geel M, van den Heuij LG, Steijlen PM, van Steensel MA. Focal dermal hypoplasia in a male patient due to mosaicism for a novel PORCN single nucleotide deletion. J Eur Acad Dermatol Venereol. 2011;25(5):592‐595. [DOI] [PubMed] [Google Scholar]
- 14. Young MP, Sawyer BL, Hartnett ME. Ophthalmologic findings in an 18‐month‐old boy with focal dermal hypoplasia. J AAPOS. 2014;18(2):205‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bornholdt D, Oeffner F, Konig A, et al. PORCN mutations in focal dermal hypoplasia: coping with lethality. Hum Mutat. 2009;30(5):E618‐E628. [DOI] [PubMed] [Google Scholar]
- 16. Yoshihashi H, Ohki H, Torii C, Ishiko A, Kosaki K. Survival of a male mosaic for PORCN mutation with mild focal dermal hypoplasia phenotype. Pediatr Dermatol. 2011;28(5):550‐554. [DOI] [PubMed] [Google Scholar]
- 17. Clements SE, Mellerio JE, Holden ST, McCauley J, McGrath JA. PORCN gene mutations and the protean nature of focal dermal hypoplasia. Br J Dermatol. 2009;160(5):1103‐1109. [DOI] [PubMed] [Google Scholar]
- 18. Maas SM, Lombardi MP, van Essen AJ, et al. Phenotype and genotype in 17 patients with Goltz‐Gorlin syndrome. J Med Genet. 2009;46(10):716‐720. [DOI] [PubMed] [Google Scholar]
- 19. Rao SS, Shenoy RD, Salian S, Girisha KM. Focal Dermal Hypoplasia with a De novo Mutation p. E300* of PORCN Gene in a Male Infant. Indian J Dermatol. 2016;61(6):700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brady PD, Van Esch H, Fieremans N, et al. Expanding the phenotypic spectrum of PORCN variants in two males with syndromic microphthalmia. Eur J Hum Genet. 2015;23(4):551‐554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Madan S, Liu W, Lu JT, et al. A non‐mosaic PORCN mutation in a male with severe congenital anomalies overlapping focal dermal hypoplasia. Mol Genet Metab Rep. 2017;12:57‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smigiel R, Jakubiak A, Lombardi MP, et al. Co‐occurrence of severe Goltz‐Gorlin syndrome and pentalogy of Cantrell ‐ Case report and review of the literature. Am J Med Genet A. 2011;155A(5):1102‐1105. [DOI] [PubMed] [Google Scholar]
- 23. Gisseman JD, Herce HH. Ophthalmologic manifestations of focal dermal hypoplasia (Goltz syndrome): A case series of 18 patients. Am J Med Genet C Semin Med Genet. 2016;172C(1):59‐63. [DOI] [PubMed] [Google Scholar]
- 24. Mary L, Scheidecker S, Kohler M, et al. Prenatal diagnosis of focal dermal hypoplasia: report of three fetuses and review of the literature. Am J Med Genet A. 2017;173(2):479‐486. [DOI] [PubMed] [Google Scholar]
- 25. Stevenson D. A., Chirpich M, Contreras Y, Hanson H, & Dent K. Goltz syndrome and PORCN mosaicism. International Journal of Dermatology. 2014;53(12):1481‐1484. doi: 10.1111/ijd.12605 [DOI] [PMC free article] [PubMed] [Google Scholar]