

Key Clinical Message
Compound heterozygosity of a previously described pathogenic variant and a second novel nucleotide substitution (NR_023343.1:n.116A>C) affecting a highly conserved nucleotide in the noncoding RNU4ATAC gene could be identified in a patient with overlapping features of Roifman Syndrome. These data extend the spectrum of pathogenic variants in RNU4ATAC.
Keywords: clinical exome sequencing, minor intron splicing, NR_023343.1:n.116A>C, NR_023343.1:n.13C>T, RNU4ATAC, Roifman Syndrome, snRNA U4atac
1. INTRODUCTION
Roifman Syndrome (RFMN) is a rare multisystem disorder characterized by pre‐ and postnatal growth retardation, microcephaly, and distinctive facial features, such as a long philtrum and thin upper lip. Recurrent infections and antibody deficiency are reported for all affected individuals described so far. Further, developmental delay as well as mental retardation was observed in most affected individuals. Hepatosplenomegaly as well as neonatal jaundice was also present in some of the affected individuals (Table 1). To our knowledge, only 12 cases of Roifman Syndrome have been described to date. The syndrome was first reported by Roifman in 1999.1 Merico et al2 identified compound heterozygosity of pathogenic single‐nucleotide variants (SNVs) in RNU4ATAC to be causative for Roifman Syndrome in 2015. RNU4ATAC codes for the small nuclear RNA (snRNA) U4atac, which is part of the minor spliceosome complex that catalyzes the splicing of an atypical class of introns (U12‐ or “ATAC”‐type) from eukaryotic pre‐mRNA. Approximately 800 human genes contain U12‐type introns.3 Pathogenic variants in RNU4ATAC are associated with the recessive disorder microcephalic osteodysplastic primordial dwarfism, type 1 (MOPD1, OMIM #210710) and the less severe Roifman Syndrome.
Table 1.
Comparing current to established cases of Roifman Syndrome
| Sex | Patient | Roif1 | Rob7 | Vries9 | Gray11 | Bogaert5 | Dinur6 | |||
|---|---|---|---|---|---|---|---|---|---|---|
| F | 4 M | M | M | F | M | M | F | M | F | |
| Growth retardation | ||||||||||
| Intrauterine | + | 4/4 | + | + | + | + | NA | NA | + | + |
| Postnatal | + | 4/4 | + | + | + | + | + | + | + | + |
| Facial features and extremities | ||||||||||
| Microcephaly | + | 4/4 | + | NR | + | + | + | + | + | + |
| Long philtrum | − | 4/4 | + | + | + | + | + | + | + | − |
| Thin upper lip | − | 4/4 | + | + | + | + | + | + | + | − |
| Clinodactyly 5th finger | NR | 4/4 | NR | + | − | − | − | − | + | + |
| Transverse palmar crease | − | 4/4 | NR | − | + | − | − | − | NR | NR |
| Brachydactyly | + | NR | + | NR | + | + | + | − | + | + |
| Musculoskeletal | ||||||||||
| Hypotonia | + | 4/4 | + | + | + | + | − | − | − | + |
| Epiphyseal dysplasia hips | + | 4/4 | NR | + | + | + | + | − | + | + |
| Dysplasia long bones | NR | 3/4 | NR | + | − | − | NR | NR | ||
| Changes in vertebral plates | NR | 4/4 | NR | + | − | − | + | − | NR | NR |
| Short metacarpals | NR | NR | + | NR | + | + | + | − | NR | NR |
| Ophthalmologic | ||||||||||
| Retinal dystrophy | − | 2/4 | − | − | − | + | + | + | − | + |
| Hepatic | ||||||||||
| Hepatosplenomegaly | + | 4/4 | + | NR | + | + | − | − | − | − |
| Neonatal jaundice | + | NR | NR | NR | + | + | NR | NR | ||
| Eczema/eosinophilia | ||||||||||
| Eczema | + | 4/4 | − | + | − | − | − | + | NR | NR |
| Elevated eosinophils | +/− | 3/4 | − | − | − | − | − | − | ||
| Development | ||||||||||
| Gross motor delay | + | 4/4 | + | + | + | + | NR | NR | + | + |
| Speech delay | + | NR | NR | NR | NR | NR | NR | NR | + | + |
| Intellectual delay | − | 3/3 | + | + | − | ND | − | − | NR | NR |
| Immunological | ||||||||||
| Antibody deficiency | NR | 4/4 | + | + | + | + | + | + | + | + |
| Metabolic/genetic | ||||||||||
| Normal karyotype | + | 4/4 | + | + | + | + | NR | NR | NR | NR |
| Normal metabolic screen | + | 3/3 | + | + | + | + | NR | NR | NR | NR |
| Hypercholesterolemia | NR | NR | NR | NR | NR | + | NR | NR | NR | NR |
| Other | ||||||||||
| Structural cardiac anomaly | VSD | NR | NR | NR | VSD | NR | − | − | − | − |
| Conductive hearing loss | − | NR | NR | NR | + | − | NR | NR | − | − |
| Bronchiectasis | − | NR | NR | NR | NR | NR | + | + | NR | NR |
Recently, also individuals with Lowry Wood Syndrome (LWS) were described, with disease‐causing variants in RNU4ATAC.4 All three syndromes share phenotypes like microcephaly and intrauterine growth retardation but also show distinct features, like immunodeficiency, which has only been described in MOPD1 and RFMN. Retinal anomalies, in contrast, have so far only been observed in RFMN and LWS. Furthermore, huge differences in severity can be observed, since MOPD1 usually leads to death in the first year of life, while RFMN and LWS show a milder manifestation.
Different patterns of biallelic variants in RNU4ATAC lead to the different syndromes. Most Roifman Syndrome causal variants identified so far showed a specific pattern of compound heterozygosity, with one variant located at highly conserved positions in the stem II region and a second variant located in the 5′stem‐loop or Sm protein‐binding site. Further, one patient was described with homozygosity for a variant in the stem II region. Variants causing the more severe phenotype of MOPD1 both clusters in the 5′stem‐loop and a few are described at the Sm protein‐binding site and proximal portion of the 3′stem‐loop. For Lowry Wood Syndrome, no specific pattern of pathogenic variants could be identified in the three affected individuals described so far.2, 4, 5, 6
Here we describe a patient with Roifman Syndrome, caused by compound heterozygosity of a previously reported pathogenic variant and a second SNV, which has to our knowledge not yet been described.
2. METHODS
2.1. Chromosomal microarray analysis
DNA was obtained from peripheral blood of all patients. Approximately 200 ng genomic DNA were required as input material for microarray preparation. We used the Infinium® CytoSNP‐850 K (Illumina, San Diego, CA, USA), which targets around 850 000 SNPs and enables the detection of CNVs as small as 10 kb. Imaging of the BeadChip was carried out on the iScan Reader. Data were analyzed using BlueFuse Multi v4.4.
2.2. High throughput sequencing
DNA was obtained from peripheral blood of all patients. For panel enrichment, approximately 45 ng of genomic DNA were required. We used the TruSight One Illumina kit (Illumina), which targets the coding sequences of 4813 genes, following the manufacturer's instructions. Sequencing was carried out on an Illumina NextSeq 500 system (Illumina) as 150 bp paired‐end runs using v2.0 SBS chemistry. Reads were aligned to the human reference genome (GRCh37/hg19) using BWA (v 0.7.8‐r455) with standard parameters. Duplicate reads and reads that did not map unambiguously were removed. The percentage of reads overlapping targeted regions and coverage statistics of targeted regions were calculated using Shell scripts. Single‐nucleotide variants and small insertions and deletions (INDELs) were called using SAMtools (v1.3.1). We used the following parameters: a maximum read depth of 10 000 (parameter ‐d), a maximum per sample depth of 10000 for INDEL calling (parameter ‐L), adjustment of mapping quality (parameter ‐C) and recalculation of per‐Base Alignment Quality (parameter ‐E). Additionally, we required putative SNVs to fulfill the following criteria: a minimum of 20% of reads showing the variant base and the variant base is indicated by reads coming from different strands. For INDELs, we required that at least 15% of reads covering this position indicate the INDEL. Variant annotation was performed with snpEff (v 4.2) and Alamut‐Batch (v 1.4.4) based on the RefSeq database. Only variants (SNVs/small INDELs) in the coding region and the flanking intronic regions (±15 bp) were evaluated. Out of the 4813 targeted genes in the TruSight One sequencing panel, 1131 genes associated with multiple congenital anomalies and developmental delay (Table S1) were considered in the analysis. These genes were curated by another laboratory taking part in the study of the European Rare Disease Working Group of Illumina.
2.3. Clinical report
A six‐year‐old female patient presents with growth retardation after a preterm delivery. She was small for gestational age. Postnatal she had a cholestatic liver disease, hyperbilirubinemia, and exocrine pancreatic insufficiency which was not detectable anymore at the age of 17 months. Further, she presents with microcephaly, a wide mouth, wide palate, orofacial hypotonia, hypoplasia of orbitae, Strabismus divergens, and astigmatism. In general, she shows unspecific facial dysmorphia with small hands and feet. Cardiac examination found a ventricular septal defect and bradycardia. Her neurologic development, especially motor, speech and language development, was delayed. She also presents with recurrent infections such as otitis and chronic bronchitis and atopic dermatitis with bilateral eczema. Recently, she was reported to be affected with spondyloepiphyseal dysplasia. (Table 1). Anthropometric values were evaluated at several time points, showing that her body height was previously and is still below the first percentile; further, her body weight was always low, varying between the first and the eighth percentile. The head circumference was not measured at all time points, but it could be observed that it was below the first percentile in the first years of life (Table 2).
Table 2.
Anthropometric values of the affected individual. Anthropometric values, like body weight, body height, and head circumference were evaluated. SD, standard deviation
| Age | Body weight | Body height | Head circumference | ||||||
|---|---|---|---|---|---|---|---|---|---|
| kg | SD (z) | Percentile | cm | SD (z) | Percentile | cm | SD (z) | Percentile | |
| 0 9/12 | 7.1 | −1.42 | P8 | 63 | −3.22 | <P1 | 40.5 | −3.86 | <P1 |
| 2 5/12 | 9.4 | −2.54 | P1 | 79 | −3.18 | <P1 | 44 | −4.6 | <P1 |
| 3 2/12 | 11 | −2.23 | P1 | 82 | −3.74 | <P1 | NR | ||
| 6 0/12 | 17 | −1.6 | P5 | 100.6 | −3.42 | <P1 | NR | ||
Using Chromosomal Microarray Analysis (CMA), no disease‐causing variant could be identified. Therefore, clinical exome sequencing was performed. The result of sequencing showed compound heterozygosity of two SNVs in the gene RNU4ATAC. The paternally inherited substitution NR_023343.1:n.13C>T in the stem II region had been described previously in two pairs of siblings and one other patient with Roifman Syndrome, while the maternally inherited substitution NR_023343.1:n.116A>C in the Sm protein‐binding site had, to our knowledge, not been described before. However, two other SNVs affecting the same nucleotide (NR_023343.1:n.116A>T and NR_023343.1:n. 116A>G) had been described in a pair of siblings affected by Roifman Syndrome and another unrelated patient.5, 6 According to phastCons and phyloP, both base substitutions are located at highly conserved positions in RNU4ATAC. They have either been identified with a very low frequency or not been identified in the healthy population. Neither variant has been detected in a homozygous state.
3. DISCUSSION
To our knowledge, twelve individuals affected with Roifman Syndrome have been described to date, of which ten cases are genetically confirmed.1, 2, 5, 6, 7, 8, 9, 10, 11 The case reported here is therefore the 13th case of Roifman Syndrome. Compared to the previously described cases, the individual described here presents with some common features of Roifman Syndrome, like intrauterine and postnatal growth retardation, microcephaly and brachydactyly, hypotonia, epiphyseal dysplasia of the hips, hepatosplenomegaly, and gross motor delay. Further, she shows some less common features, like eczema, elevated eosinophils, and a ventricular septal defect (VSD). She is missing, however, some typical facial features, like a long philtrum and thin upper lip (Table 1).
The first four male individuals affected with this multisystem disorder were described as early as 1999 by Roifman, but it was Merico et al2 who described pathogenic variants in RNU4ATAC as the underlying cause for Roifman Syndrome. Whole‐genome sequencing in two affected siblings identified compound heterozygosity in RNU4ATAC as causative for Roifman Syndrome. Direct sequencing of RNU4ATAC in the other four affected individuals showed compound heterozygosity of SNVs clustering in the same structural elements of RNU4ATAC. To confirm the pathomechanism of Roifman Syndrome, 800 whole genome sequences of individuals with unrelated conditions were analyzed, in which no compound heterozygosity or homozygosity of variants in RNU4ATAC could be identified.2
The snRNA U4atac consists of six subregions, of which five are essential for U12‐dependent splicing. Elements which are crucial for base pairing with U6atac are the stem II and the stem I. Both subregions are separated by the 5′stem‐loop, which functions as a binding platform for proteins required for tri‐snRNP formation of U4atac, U6atac, and U5. The 3′stem‐loop immediately precedes a binding site for Sm proteins, which are important for snRNP assembly and import into the nucleus2, 3, 12 (Figure 1).
Figure 1.
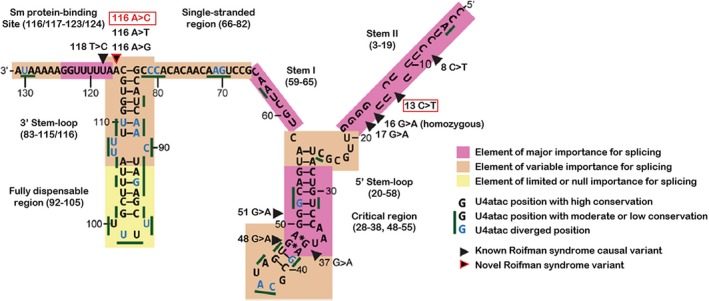
U4atac snRNA secondary structure elements and variants detected in the patient. The two variants identified in compound heterozygosity in the patient are marked with a red frame. Both variant‐positions are highly conserved. The paternally inherited variant n.13C>T is located in an element of major importance for splicing at the stem II, the maternally inherited variant n.116A>C is located in an element of variable importance for splicing at the transition of 3′stem‐loop to the Sm protein‐binding site2, 5, 12, 13
The paternally inherited variant NR_023343.1:n.13C>T is located at a highly conserved position at the stem II region of the U4atac snRNA and impairs binding to U6atac snRNA. The maternally inherited NR_023343.1:n.116A>C substitution is located at the transition of 3′stem‐loop and Sm protein‐binding site. MOPD1 causal variants have been described in the Sm protein‐binding site as well as one in the proximal portion of the 3′stem‐loop. Mutagenesis experiments demonstrated the importance of the domains of the U4atac snRNA. These experiments showed that the deletion of the distal portion (Δ92‐105) of the 3′stem‐loop does not affect minor intron splicing, while the deletion of the whole 3′stem‐loop (Δ84‐114) demonstrates a loss of in vivo activity. The loss of activity of the U4atac snRNA occurs after deletion of the whole 3′stem‐loop. Activity is retained if only the distal part of the 3′stem‐loop is deleted, pointing toward the functional importance of the proximal part of the 3′stem‐loop.12 The stem II as well as the Sm protein‐binding site have been identified as important elements in splicing, while SNVs in the proximal portion of the 3′stem‐loop had been identified as elements of variable importance.2 There are discrepant results for NR_023343.1:n.116A which, depending on the publication, either had or had not been judged as part of the Sm protein‐binding site. The core region, essential for splicing, is so far defined from NR_023343.1:n.117 to NR_023343.1:n.124.2, 3, 13 Regarding the clinical data obtained in the study presented here, NR_023343.1:n.116 also belongs to the critical region important for splicing, which has to be confirmed in further functional studies (Figure 1).
According to all the data pertaining to pathogenic variants in RNU4ATAC, it can be concluded that compound heterozygosity of NR_023343.1:n.13C>T and NR_023343.1:n.116A>C is the cause of Roifman Syndrome in this patient. A novel disease‐causing variant NR_023343.1:n.116A>C could be identified, which may help to further narrow down the element of major importance for splicing at the Sm protein‐binding site.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
AH: conducted the laboratory experiments and data analysis. JG and AL: contributed to clinical assessment of data. AL, AB‐P, and EH‐F: contributed in the supervision of the manuscript. BS: provided phenotypic data of the patient. UK: conducted the array data analysis. All authors reviewed and agreed to the content of the final manuscript.
Supporting information
ACKNOWLEDGMENTS
We would like to thank Illumina for organizing and providing reagents for the study. Acknowledgements also to the family for participating in this study.
Hallermayr A, et al. Extending the critical regions for mutations in the non‐coding gene RNU4ATAC in another patient with Roifman Syndrome. Clin Case Rep. 2018;6:2224–2228. 10.1002/ccr3.1830
REFERENCES
- 1. Roifman CM. Antibody deficiency, growth retardation, spondyloepiphyseal dysplasia and retinal dystrophy: a novel syndrome. Clin Genet. 1999;55:103‐109. [DOI] [PubMed] [Google Scholar]
- 2. Merico D, Roifman M, Braunschweig U, et al. Compound heterozygous mutations in the noncoding RNU4ATAC cause Roifman Syndrome by disrupting minor intron splicing. Nat Commun. 2015;6:8718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Turunen JJ, Niemelä EH, Verma B, Frilander MJ. The significant other: splicing by the minor spliceosome. Wiley Interdiscip Rev RNA. 2013;4(1):61‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Farach LS, Little ME, Duker AL, et al. The expanding phenotype of RNU4ATAC pathogenic variants to Lowry Wood syndrome. Am J Med Genet A. 2018;176(2):465‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bogaert DJ, Dullaers M, Kuehn HS, et al. Early‐onset primary antibody deficiency resembling common variable immunodeficiency challenges the diagnosis of Wiedeman‐Steiner and Roifman syndromes. Sci Rep. 2017;7(1):3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dinur Schejter Y, Ovadia A, Alexandrova R, et al. A homozygous mutation in the stem II domain of RNU4ATAC causes typical Roifman syndrome. NPJ Genom Med. 2017;2:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Robertson SP, Rodda C, Bankier A. Hypogonadotrophic hypogonadism in Roifman syndrome. Clin Genet. 2000;57:435‐438. [DOI] [PubMed] [Google Scholar]
- 8. Mandel K, Grunebaum E, Benson L. Noncompaction of the myocardium associated with Roifman syndrome. Cardiol Young. 2001;11(02):240‐243. [DOI] [PubMed] [Google Scholar]
- 9. de Vries PJ, McCartney DL, McCartney E, Woolf D, Wozencroft D. The cognitive and behavioural phenotype of Roifman syndrome. J Intellect Disabil Res. 2006;50(Pt 9):690‐696. [DOI] [PubMed] [Google Scholar]
- 10. Fairchild HR, Fairchild G, Tierney KM, McCartney DL, Cross JJ, de Vries PJ. Partial agenesis of the corpus callosum, hippocampal atrophy, and stable intellectual disability associated with Roifman syndrome. Am J Med Genet A. 2011;155A(10):2560‐2565. [DOI] [PubMed] [Google Scholar]
- 11. Gray P, Sillence D, Kakakios A. Is Roifman syndrome an X‐linked ciliopathy with humoral immunodeficiency? Evidence from 2 new cases. Int J Immunogenet. 2011;38(6):501‐505. [DOI] [PubMed] [Google Scholar]
- 12. Shukla GC, Cole AJ, Dietrich RC, Padgett RA. Domains of human U4atac snRNA required for U12‐dependent splicing in vivo. Nucleic Acids Res. 2002;30(21):4650‐4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nottrott S, Urlaub H, Lührmann R. Hierarchical, clustered protein interactions with U4/U6 snRNA: a biochemical role for U4/U6 proteins. EMBO J. 2002;21:5527‐5538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials