

Abstract
The mitochondrial unfolded protein response (UPRmt) is a protein quality control mechanism that strives to achieve proteostasis in the face of misfolded proteins. Because of the reliance of mitochondria on both the nuclear and mitochondrial genomes, a perturbation of the coordination of these genomes results in a mitonuclear imbalance in which holoenzymes are unable to assume mature stoichiometry and thereby activates the UPRmt. Thus, we sought to perturb this genomic coordination by using a systemic antisense oligonucleotide (in vivo morpholino) targeted to translocase of the inner membrane channel subunit 23 (Tim23), the major channel of the inner membrane. This resulted in a 40% reduction in Tim23 protein content, a 32% decrease in matrix-destined protein import, and a trend to elevate reactive oxygen species (ROS) emission under maximal respiration conditions. This import defect activated the C/EBP homologous protein (CHOP) branch of the UPRmt, as evident from increases in caseinolytic mitochondrial matrix peptidase proteolytic subunit (ClpP) and chaperonin 10 (cpn10) but not the activating transcription factor 5 (ATF5) arm. Thus, in the face of proteotoxic stress, CHOP and ATF5 could be activated independently to regain proteostasis. Our second aim was to investigate the role of proteolytically derived peptides in mediating retrograde signaling. Peptides released from the mitochondrion following basal proteolysis were isolated and incubated with import reactions. Dose- and time-dependent effect of peptides on protein import was observed. Our data suggest that mitochondrial proteolytic byproducts exert an inhibitory effect on protein import, possibly to reduce excessive protein import as a potential negative feedback mechanism. The inhibition of import into the organelle also serves a retrograde function, possibly via ROS emission, to modify nuclear gene expression and ultimately improve folding capacity.
Keywords: CHOP, in vivo morpholino, mitochondrial peptides, protein quality control
INTRODUCTION
Proteostasis refers to the maintenance of proper protein synthesis, maturation, and degradation to ensure a functional proteome (9a, 24). Proteins that are unable to assume their tertiary or quaternary structures exert toxic effects on the cell, referred to as proteotoxicity (34, 41). Within the mitochondrion, the goal of achieving protein homeostasis is complex since it is the only organelle that contains its own genome and therefore generates its own gene products. There, the maintenance of a functional stoichiometry relies on the coordination of both nuclear and mitochondrial genomes, as well as proper protein handling and maturation (5).
Despite containing their own genetic material, mitochondria rely heavily on the nuclear genome for over 99% of all mitochondrial proteins, whereas mtDNA codes for 13 proteins that are transcribed and translated within the organelle (8). Furthermore, because of the double-membrane structure of the mitochondrion, products of nuclear genes encoding mitochondrial proteins (NuGEMPs) require a sophisticated mechanism for mediating their sorting and translocation (6). Mitochondrial protein import is a complex mechanism through which proteins are recognized via their mitochondrial targeting sequence (MTS) by cytosolic chaperones and delivered to the protein import machinery (PIM) within the organelle (7, 9). Products of NuGEMPs are recognized by cytosolic chaperones that guide proteins to the translocases of the outer membrane, the TOM complex. These chaperones are also responsible for unfolding their cargo into a linear structure, or alternatively can be cotranslationally imported, to facilitate their passage through the β-barrel channel of the TOM complex, and subsequently through the translocase of the inner membrane, the TIM complex (4). Once in its final destination, such as in the matrix, the MTS is cleaved by mitochondrial processing peptidase (MPP) and the protein is refolded by mitochondrial chaperones to assume its mature conformation (22). Many of these nuclear products can then combine with mitochondrially encoded subunits to form mature holoenzymes. Previous research has shown that protein import is a dynamic process that can respond to the metabolic status of the cell, thereby promoting the balance between the nuclear and mitochondrial genomes (11, 16). However, this process is laden with potential points for protein misfolding or misassembly, which may exert proteotoxic stress on the organelle.
To combat proteotoxicity, mitochondria are equipped with a protein quality control mechanism, the unfolded protein response (UPRmt). The UPRmt is a compartment-specific response that detects misfolded proteins and either refolds them through the use of chaperones [chaperonin 10 (cpn10), heat shock protein 60 (HSP60)] or degrades them via proteases [lon peptidase (LonP), caseinolytic mitochondrial matrix peptidase proteolytic subunit (ClpP)] to eliminate the proteotoxic stress that they exert on the organelle (31). When the stress exceeds the capacity of resident quality control proteins, a retrograde signal is initiated through various mechanisms, including an increase in reactive oxygen species (ROS) to indirectly activate CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP) and C/EBPβ, which transcriptionally regulate various mitochondrial chaperones and proteases (23). Activating transcription factor (ATF)5, a recently discovered transcription factor, is also activated; however, the mechanisms involved in this activation are currently unknown in mammalian cells (14). Work in Caenorhabditis elegans (C. elegans) originally led to a proposed role for proteolytically derived peptides in mediating the nuclear translocation of ATF5. In these organisms, the byproducts of proteolysis within the mitochondrion are released into the cytosol, and this is required for the activation and nuclear translocation of the homolog of ATF5, activating transcription factor associated with stress-1 (ATFS-1), where it can promote a compensatory gene expression response (17, 18, 32). Based on this research, the UPRmt appears to be designed to selectively promote the transcription of protein quality control genes while transiently arresting other sources of proteotoxic stress (13).
However, since its discovery in 2002, our understanding of the UPRmt is primarily derived from work in lower order organisms, and our knowledge regarding its mammalian counterpart is still lacking (2, 10, 31, 36). Therefore, the first purpose of this study was to investigate the consequence of UPRmt induction in skeletal muscle by inducing a mitonuclear imbalance. This was achieved by knocking down translocase of the inner membrane channel subunit 23 (Tim23), the major channel of the TIM complex with an injectable antisense oligonucleotide. Our second purpose was to investigate the effect of peptides on the import process and their potential role as a retrograde signal. We expected that mitochondrially derived peptides would negatively influence import in a dose- and time-dependent manner.
MATERIALS AND METHODS
Animal care and in vivo morpholino treatment.
Male C57BL/6 mice (3 mo) were housed in the vivarium and given food and water ad libitum under a 12:12-h light-dark cycle throughout the entirety of the protocol. In vivo morpholinos (Gene Tools) were designed to target Tim23 and were dissolved in phosphate-buffered saline at a concentration of 5 mM as recommended by the manufacturer (Table 1). Animals (control group, n = 7; knockdown group, n = 11) were administered a dose of 12 mg/kg per day via intraperitoneal injection for 3 consecutive days, based on previous literature (12). Control animals were treated with a standard control oligo offered through Gene Tools at the same dose. Mice were euthanized 48 h following their last injection by cervical dislocation, and tissues were promptly harvested. Tibialis anterior, extensor digitorum longus, quadriceps, and triceps were collected from both hindlimbs and used immediately for mitochondrial isolations. Gastrocnemius muscles, heart, and liver were frozen in liquid nitrogen and stored at −80°C for later analysis. All animal handling and treatment was approved by the Animal Care Committee at York University.
Table 1.
In vivo morpholino sequences purchased from Gene Tools, designed according to company guidelines targeted for Tim23 knockdown and list of primer sequences used for real-time PCR analysis
| Gene | In Vivo Morpholino Sequence |
|---|---|
| Tim23 | 5′-TCT TCC GCC ACC TTC CAT GAG GTC-3′ |
| Control | 5′-CCT CTT ACC TCA GTT ACA ATT TAT A-3′ |
| Primer |
||
|---|---|---|
| Gene | Forward | Reverse |
| Ddit3 (CHOP) | 5′-CAC CAC ACC TGA AAG CAG AA-3′ | 5′-AGG TGA AAG GCA GGG ACT CA-3′ |
| ATF5 | 5′-TGG AGC GGG AGA TCC AGT A-3′ | 5′-GAC GCT GGA GAC AGA CGT ACA-3′ |
| Hspa9 (mtHsp70) | 5′-TGG CTA TTA CTG CGG GTT CT-3′ | 5′-CAT CTG CTC CAC CTC CTC T-3′ |
| Hspd1 (Hsp60) | 5′-CTG GGT GCA AGA GCC ATA TA-3′ | 5′-GAA AGG CTG CTT CTG AAC TCT-3′ |
| Hspe1 (cpn10) | 5′-GGA GTG CTG CTG CCG AAA CTG TA-3′ | 5′-TCA CAC TGA CAG GCT CAA TCT-3′ |
| Lonp1 (LonP) | 5′-CGA CTT GCA CAG CCC TAT GT-3′ | 5′-CGA ATG TTC CCG TAT GGT AGA T-3′ |
| Timm23 (Tim23) | 5′-CCCGAGGCAGATTTGAACTA-3′ | 5′-AAAGCCAGGGAGCCTAGAGTAT-3′ |
| B-actin | 5′-TGT GAC GTT ACA TCC GTA A-3′ | 5′-GCT AGG AGC CAG AGG AGT AA-3′ |
| 18S rRNA | 5′-GTAACCCGTTGAACCCCATT-3′ | 5′-CCATCCAATCGGTAGTAGCG-3′ |
All primers are designed for Mus musculus. ATF5; activating transcription factor 5; CHOP; C/EBP homologous protein; cpn10, chaperonin 10; Hsp60, heat shock protein 60; LonP, lon peptidase; mtHSP70, mitochondrial heat shock protein 70; Tim23, translocase of the inner membrane channel subunit 23.
Mitochondrial isolation.
Fresh tissues were minced and homogenized using an Ultra-Turrex Polytron (IKA, Staufen, Germany) at 9.8 Hz and subjected to differential centrifugation to separate intermyofibrillar (IMF) and subsarcolemmal mitochondria, as was done previously (29). IMF mitochondria were treated with nagarse, a proteinase from Bacillus lichenformis, to liberate the mitochondrial population. Fractions were subjected to a final centrifugation at 9,000 revolutions/min to pellet mitochondria and were resuspended in resuspension buffer containing 100 mM KCl, 10 mM MOPS, and 0.2% BSA (pH 7.4). Concentrations were determined using the Bradford method. Since the IMF mitochondria make up roughly 85% of the mitochondrial pool within skeletal muscle, we chose to study this specific subfraction (19).
Mitochondrial respiration.
Using a Clark Electrode (Strathkelvin Instruments, North Lanarkshire, Scotland), oxygen consumption was measured over time. Briefly, 50 μl of mitochondria were incubated with 250 μl of V̇o2 buffer (250 mM sucrose, 50 mM KCl, 25 mM Tris base, 10m M K2HPO4, and pH 7.4) and continuously stirred at 30°C. Mitochondrial O2 consumption was measured in the presence of 10 mM glutamate to assess basal complex I-driven respiration (state II), followed by the addition of 0.44 mM ADP for maximal (state III) respiration. Integrity of the inner membrane was assessed through the addition of NADH during state III respiration. Respiratory rates were corrected for the total amount of protein within each mitochondrial sample. Respiratory control ratios were calculated by dividing state III by state II as an indication of efficiency.
Mitochondrial ROS emission.
Mitochondria (75 μg) were incubated in a 96-well plate with V̇o2 buffer and 50 μM 2′7′-dichlorofluorescin at 37°C for 30 min. ROS emission was measured under state III and II respiratory conditions through the addition of ADP (0.44 mM) and glutamate (13.97 mM) or glutamate, respectively. Fluorescence (excitation 480 nm, emission 520 nm) was measured using a Synergy HT microplate reader (BioTek) and KC4 software (v.3.0) and was directly proportional to ROS emission.
In vitro transcription, translation, and protein import.
As previously described (38), plasmid DNA containing the ornithine transcarbamylase (OCT) cDNA and competent DH5α cells were transformed and grown on agar plates. Bacterial colonies were then amplified overnight in lysogeny broth media and treated with ampicillin. Plasmid DNA was then isolated using a HiSpeed Plasmid Maxi Kit (Qiagen, Hilden, Germany). The DNA was then linearized using Sca1 and subsequently purified using phenol and precipitated using ethanol. OCT was transcribed in vitro using SP6 polymerase for 90 min at 40°C. Translation was then carried out at 37°C by combining rabbit reticulocyte lysate, mRNA, a cocktail of amino acids minus methionine, radio-labeled 35S methionine, and water for 25 min.
As previously described (38), 75 μg of freshly isolated mitochondria and 18 μl of the translation mix were incubated for 30 min at 30°C to allow protein import. Mitochondria were then centrifuged through a sucrose gradient at 16,000 g for 15 min at 4°C and resuspended in breaking buffer (0.6 M sorbitol and 20 mM HEPES) and lysis buffer (10% glycerol, 2.3% SDS, 62.5mM Tris-HCl, and 5% mercaptoethanol). Samples were denatured at 95°C for 5 min and then resolved through a 12% SDS-polyacrylamide gel for ~2 h. Gels were then boiled for 5 min in 5% TCA, briefly washed with ddH2O, incubated with 10 mM Tris for 5 min, followed by 30 min in 1 M salicylic acid. Gels were then dehydrated at 80°C for 1 h, and radiolabelled bands were captured on a Fujifilm Multipurpose Storage Phospho film and visualized using a Typhoon Scanner (GE Healthcare, Little Chalfont, UK).
Protein release assay and peptide isolation.
IMF mitochondria (150 μg) from untreated C57BL/6 mice were incubated for 1 h at 30°C with or without 5 μM H2O2 in the presence of FeSO4 to perturb the organelle and promote apoptosis. To assess whether or not peptides were being released from the mitochondrial permeability transition pore (mtPTP) basally and under stress, under the same conditions, 200 μM cyclosporin A was added to block the opening of the mtPTP. Following incubation, mitochondria were pelleted by centrifugation (5 min at 14,000 g at 4°C) to collect all released material. Finally, the released fraction was separated based on size using a Spin-X Concentrator (Corning, MA), collecting released products under 3 kDa in size. Concentrations were determined spectrophotometrically using a NanoDrop 2000 (Thermo Fisher).
This protocol was also used to isolate peptides for the import reactions with the exception that H2O2 and FeSO4 were not used to perturb the mitochondrion. Peptides were isolated under basal conditions in the presence of up to 600 μg of mitochondria to increase peptide yield.
Total RNA and reverse transcription.
Approximately 100 mg of pulverized gastrocnemius tissue was combined with 1 ml of TRIzol Reagent (Invitrogen) and homogenized at 6 Hz (3 × 10 s) then mixed with chloroform. Samples were centrifuged at 16,000 g for 15 min at 4°C. The aqueous phase was then transferred to a new tube and precipitated using isopropanol at −80°C for 1 h. Samples were then once again centrifuged at 16,000 g for 15 min at 4°C, and pellets were resuspended in 30 μl of molecular grade sterile water (Wisent Bioproducts). The concentration and purity of RNA were determined by spectrophotometer (NanoDrop 2000, Thermo Fisher). Superscript III reverse transcriptase (Invitrogen) was used according to manufacturer directions to convert 1.5 μg of RNA into cDNA.
Real-time PCR.
Primers (Table 1) were designed using sequences from GenBank, Primer 3 (v.0.4.0) software (Massachusetts Institute of Technology, Cambridge, MA) and OligoAnalyzer 3.1 (Integrated DNA Technologies) for genes of interest. mRNA expression was determined by combining SYBR Green Supermix (Quanta Biosciences), forward and reverse primers (20 uM), stH2O, and cDNA (10 ng) in a 96-well plate. PCR amplification was carried out in a StepOnePlus Real-Time PCR System (Applied Biosystems), including a final melt-stage to check for nonspecific amplification. Results were corrected using two housekeeping genes: β-actin and 18S ribosomal RNA.
Protein isolation.
Frozen gastrocnemius muscle was pounded into a fine powder, of which 15–20 mg were diluted in 10× Sakamoto Buffer with the addition of protease and phosphatase inhibitors (Sigma-Aldrich). Samples were then rotated at 4°C for 1 h and then sonicated (3 × 3 s at 30% power). Samples underwent centrifugation at 14,000 g at 4°C, and the supernate was collected and stored at −80°C.
Western blotting and protein carbonylation.
As previously described (28), whole muscle protein extracts were prepared and separated in polyacrylamide SDS-PAGE gels. Proteins were then transferred onto nitrocellulose membranes and blocked in 5–10% milk in Tris-buffered saline-Tween 20. Subsequently, membranes were incubated with the appropriate concentration of primary antibodies overnight at 4°C (Table 2) and then with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Membranes were then visualized with enhanced chemiluminescence and a Carestream Model imaging system, and Image J software was used for quantifications and corrected for with the appropriate loading control. Protein carbonylation was measured using a typical Western blotting procedure along with OxiSelect protein carbonyl immunoblot kit (STA-308; Cell BioLabs).
Table 2.
List of antibodies and concentrations used for Western blotting
| Concentration |
||||
|---|---|---|---|---|
| Protein | Manufacturer | Catalog No./Reference | Primary | Secondary |
| Tim23 | BD Transduction | 611222 | 1:1,000 | 1:2,000 (M) |
| COX IV | Abcam | ab140643 | 1:4,000 | 1:4,000 (M) |
| Tfam | In house | (21) | 1:3,000 | 1:5,000 (R) |
| mtHsp70 | Enzo Life Sciences | ADI-SPA-810 | 1:1,000 | 1:2,000 (M) |
| Hsp60 | Enzo Life Sciences | ADI-SPA-806 | 1:1,000 | 1:2,000 (M) |
| cpn10 | Enzo Life Sciences | ADI-SPA-110 | 1:4,000 | 1:4,000 (R) |
| ClpP | Abcam | ab124822 | 1:1,000 | 1:1,000 (R) |
| LonP | Cell Signaling | 28020S | 1:1,000 | 1:2,000 (R) |
| LC3-II | Cell Signaling | 4108S | 1:500 | 1:1,000(R) |
| ATF4 | Santa Cruz | Sc-200 | 1:250 (5% BSA) | 1:3,000 (R) |
| ATF5 | Abcam | Ab60126 | 1:750 (5% BSA) | 1:3,000 (R) |
| BiP | Cell Signaling | 1:750 | 1:2,000 (R) | |
| CHOP | Santa Cruz | sc-7351 | 1:500 | 1:2,500 (M) |
| VDAC | Abcam | ab14734 | 1:5,000 | 1:5,000 (M) |
| Aciculin | In house | (5) | 1:3,000 | 1:3,000 (M) |
| Anti-mouse | Cell Signaling | 7076S | ||
| Anti-rabbit | Cell Signaling | 7074S | ||
All antibodies are prepared in 5% milk unless otherwise specified. ATF4/5; activating transcription factor 4/5; BiP, binding immunoglobulin protein; CHOP; C/EBP homologous protein; ClpP, caseinolytic mitochondrial matrix peptidase proteolytic subunit; COX IV, cytochrome-c oxidase subunit IV; cpn10, chaperonin 10; Hsp60, heat shock protein 60; LC3-II, microtubule-associated protein 1A/1B-light chain 3; LonP, lon peptidase; M, mouse; mtHSP70, mitochondrial heat shock protein 70; R, rabbit. Tfam, mitochondrial transcription factor A; Tim23, translocase of the inner membrane channel subunit 23; VDAC, voltage-dependent anion channel.
Statistical analysis.
Unpaired t-tests were used to detect differences between Tim23 knockdown and control mice using GraphPad Prism 6.0. When comparing the effect of peptides on the import reaction or in the proteolysis experiments, paired t-tests or two-way ANOVA were used when appropriate. The critical value was set at P ≤ 0.05. All error bars represent the SE.
RESULTS
Consequence of Tim23 knockdown.
Following in vivo morpholino treatment, Tim23 protein content was significantly reduced in the IMF fraction by an average of 40% (Fig. 1, A and B). This corresponded to a 32% reduction in OCT import into the mitochondrial matrix in vitro where precursor OCT represents newly synthesized protein product and mature OCT has its protein product and mature OCT has its mitochondrial targeting sequence (MTS) cleaved off upon entering the matrix, resulting in a smaller protein product (Fig. 1C). As expected, the degree of Tim23 knockdown was strongly correlated to the level of impairment of import into the matrix (R2 = 0.53, P = 0.01, Fig. 1D). Nuclear-encoded mitochondrial proteins were measured to assess whether or not this reduction in protein import affected the delivery of proteins to the organelle in vivo. Cytochrome-c oxidase subunit IV (COX IV), a subunit of the electron transport chain (ETC) located within the inner membrane, was not changed in the IMF fraction (Fig. 1A). However, mitochondrial transcription factor A (Tfam) and citrate synthase were significantly reduced by 25% and 30%, respectively, (Fig. 1B) suggesting that partial loss of Tim23 selectively impairs protein import into the matrix both in vivo and in vitro.
Fig. 1.

Consequence of Tim23 knockdown. A: representative Western blots for Tim23, Tfam, citrate synthase, COX IV, and the loading control, VDAC. B: graphical representation for Tim23, Tfam, and citrate synthase protein content following in vivo morpholino treatment. C: graphical representation of OCT import into the mitochondrial matrix with a corresponding blot. D: correlation between Tim23 protein content and OCT import into the mitochondrial matrix (R2 = 0.533). (n = 5–11; *P < 0.05; main effect of Tim23 KD, †P < 0.001). Dashed line in citrate synthase blot indicates images taken from two separate blots. COX IV, cytochrome-c oxidase subunit IV; Cit Syn, citrate synthase; CTL, control; KD, knockdown; OCT, ornithine transcarbamylase; Tfam, mitochondrial transcription factor A; Tim23, translocase of the inner membrane channel subunit 23; TL, OCT translation product alone; VDAC, voltage-dependent anion channel.
Mitochondrial function in the presence of an import defect.
Since Tim23 is the major channel of the TIM complex and is an integral part of the protein import process, we speculated that mitochondrial function would be impaired. To ascertain whether there was any mitochondrial dysfunction, mitochondrial respiration and ROS emission were assessed. Mitochondrial oxygen consumption driven by complex I, both in the absence and presence of ADP (state II and state III, respectively), were unchanged in the IMF fraction (Fig. 2A) of Tim23 knockdown animals. Supporting these data, there were no statistical differences in the respiratory exchange ratios between control and knockdown animals (Fig. 2B). Respiration rates were also not altered in the presence of NADH, used to address the integrity of the mitochondrial membranes (data not shown). ROS emission was expressed per µg of mitochondrial protein, as well as a function of respiration rate. Our data reveal that Tim23 knockdown increased absolute rates of ROS emission under both state II and III conditions (Fig. 2C). When expressed per unit of oxygen consumption, this elevation (Fig. 2D, P = 0.059) was retained during state III but not state II respiration. These data suggest that maximal mitochondrial function may be more susceptible to oxidative stress in the face of an import defect and that ROS may have a role in retrograde signaling in the absence of Tim23. Despite this, protein carbonylation at the whole muscle level revealed no differences (Fig. 2E), suggesting that the elevations in ROS were not sufficient to alter the oxidation of muscle proteins in general.
Fig. 2.

Mitochondrial function in the presence of an import defect. Mitochondrial respiration during both state II (complex I driven) and state III (maximal) respiratory states (A) and the respiratory control ratios (B). ROS production under the same respiratory conditions, expressed per μg of mitochondria (C) and corrected for oxygen consumption (D). *P < 0.05, main effect of KD on absolute ROS emission. Representative blot for protein carbonylation in whole muscle lysates and the corresponding Ponceau stain for loading (E). (n = 4–6.) AFU, arbitrary fluorescence units; CTL, control intermyofibrillar mitochondria; KD, knockdown intermyofibrillar mitochondria; ROS, reactive oxygen species.
Activation of the UPRmt following Tim23 knockdown.
To assess whether partial loss of Tim23 resulted in a perturbation of protein homeostasis, markers of the UPRmt were measured. To our surprise, ATF5, a novel proteolytic-dependent transcription factor, was unchanged (Fig. 3A). However, CHOP was elevated by 2.7-fold in the knockdown animals (Fig. 3B). Mitochondrial chaperones, mitochondrial HSP70 (mtHSP70) and HSP60, were unchanged in Tim23 knockdown animals (Fig. 3A), but chaperonin 10 (cpn10) was elevated by 40% (Fig. 3B). Similarly, of the resident proteases, LonP showed no change (Fig. 3A); however, ClpP was increased by 1.8-fold (Fig. 3B). Taken together, it appears that a CHOP-dependent branch of the UPRmt may have been activated following inhibition of protein import into the matrix.
Fig. 3.

Activation of the UPRmt following Tim23 knockdown. A: representative Western blots for mtHSP70, HSP60, cpn10, ClpP; LonP, CHOP, ATF5, and the loading controls, VDAC and aciculin. B: graphical representation of CHOP (P = 0.05), Cpn10, and ClpP protein content expressed as a fold over control values. C and D: representative blots for UPRER markers BiP and ATF6 (C) and ISR marker ATF4 (D) in whole muscle extracts. E: representative blots for autophagy/mitophagy markers LC3-II in isolated intermyofibrillar mitochondria and p62 and parkin in whole muscle lysates. (CTL, n = 7; KD, n = 11.) Dashed line in ClpP and p62 blots indicates images taken from two separate blots. ATF, activating transcription factor; BiP, binding immunoglobulin protein; CHOP, C/EBP homologous protein; ClpP, caseinolytic mitochondrial matrix peptidase proteolytic subunit; cpn10, chaperonin 10; CTL, control; HSP60, heat shock protein 60; IMF, separate intermyofibrillar; ISR, integrated stress response; KD, knockdown; LC3-II, microtubule-associated protein 1A/1B-light chain 3; LonP, lon peptidase; mtHSP70, mitochondrial heat shock protein 70; Tim23, translocase of the inner membrane channel subunit 23; UPRER, unfolded protein response of the endoplasmic reticulum; UPRmt, mitochondrial unfolded protein response; VDAC, voltage-dependent anion channel.
Selective stress response activation following Tim23 knockdown.
The UPRmt lies in the midst of a network of cellular stress responses, each of which can be activated independently or in conjunction with another. To investigate whether other stress responses were activated in response to Tim23 knockdown, we measured markers of the unfolded protein response of the endoplasmic reticulum (UPRER), integrated stress response (ISR), and mitophagy. Binding immunoglobulin protein (BiP), an ER chaperone, and ATF6, a transcription factor, both commonly used as a markers of ER proteotoxic stress, was unaffected following Tim23 knockdown (Fig. 3C). The ISR aims to alleviate proteotoxic stress by inhibiting protein translation while selectively upregulating the translation of specific targets, such as ATF4. However, ATF4 was not affected by Tim23 knockdown (Fig. 3D), suggesting that the ISR was not activated. Mitophagy, the process of mitochondrial recycling within the cell, is also activated following prolonged proteotoxic stress. However, there were no changes in the accumulation of microtubule-associated protein 1A/1B-light chain 3 (LC3-I/II) on the mitochondrial fraction, indicating no differences in mitochondrial encapsulation by the phagophore (Fig. 3E). Similarly, both p62 or Parkin in whole muscle lysates were changed in response to Tim23 knockdown (Fig. 3E). Taken together, this demonstrates that acute knockdown of Tim23 culminates in the activation of the UPRmt specifically, without the induction of other stress responses. This further supports the idea that the various arms of the UPR may be selectively activated to restore homeostasis.
We also sought to investigate whether partial loss of Tim23 resulted in changes in mRNA expression. We had hypothesized that various protein quality control proteins would be upregulated in similar fashion to the protein data. However, both ATF5 and CHOP transcripts remained unchanged following Tim23 knockdown, and similarly, no differences were observed in their downstream targets, namely cpn10, LonP, HSP60, and mtHSP70 (Fig. 4).
Fig. 4.
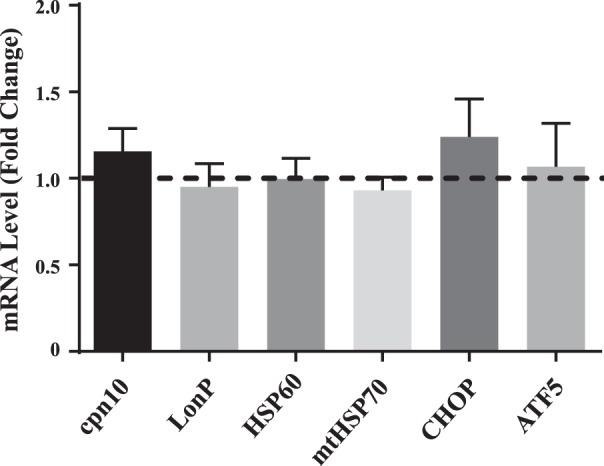
Changes in gene expression following Tim3 knockdown. mRNA expression of various UPRmt markers are expressed as fold changes over control values (n = 6–8). ATF5, activating transcription factor 5; CHOP, C/EBP homologous protein; Cpn10, chaperonin 10; HSP60, heat shock protein 60; LonP, lon peptidase; mtHSP70, mitochondrial heat shock protein 70; Tim23, translocase of the inner membrane channel subunit 23; UPRmt, mitochondrial unfolded protein response.
Peptide release from mitochondria.
To assess peptide release, mitochondria were isolated and incubated at 30°C for 30 min to allow proteolysis to occur basally or in the presence of H2O2 and FeSO4 to produce a hydroxyl radical (HO−). Our results demonstrate that proteolysis does occur, and its byproducts are exported into the cytosol when mitochondria are left unperturbed. This export is enhanced in the presence of oxidative stress (Fig. 5A). We sought to further assess how these protein fragments were exiting the mitochondrion. The addition of 200 μM cyclosporin A was used under these same conditions to block the opening of the mitochondrial permeability transition pore (mtPTP), a nonselective channel on the outer membrane. The addition of cyclosporin A did not block the release of peptides from the mitochondria, thus eliminating the mtPTP as the channel through which the peptides were exported (Fig. 5A).
Fig. 5.

Role of mitochondrially derived peptides on protein import. A: mitochondrial peptide release in the presence of H2O2 and FeSO4 with or without cyclosporin A. **P < 0.001; n = 6. B: peptides incubated with import reactions at various doses; graph represents import rates as a percentage of their respective controls. P = 0.06; 2 and 4 μg, n = 6; 6 μg, n = 12). C: import reactions in the presence of 6 μg of peptides for various time points (10, 20, and 30 min) shown as a percentage of control (10 and 20 min, n = 5; 30 min, n = 12). D: mitochondrial respiration supported by complex I both with and without ADP in the presence of 6 μg of peptides. E: ROS emission under the same respiratory states in the presence of 6 μg of peptides. Inset: absolute ROS emission per μg of mitochondria. AFU, arbitrary fluorescence units; PEP, mitochondrially released peptides; ROS, reactive oxygen species; TL, translation product alone; VEH, vehicle.
Role of mitochondrially derived peptides in mediating protein import.
The mechanism through which peptides influence retrograde signaling during the activation of the UPRmt is currently unknown. Thus, we investigated whether peptides may be influencing the UPRmt by modulating protein import. To test this hypothesis, peptides were isolated following basal proteolysis from mitochondria and incubated with radiolabeled OCT and freshly isolated mitochondria. Peptides were added to the import reaction in increasing doses (2, 4, and 6 μg) with volume-matched controls. No effect of peptides on protein import was observed at 2 and 4 μg; however, a 25% (P < 0.05) decrease in import was apparent using 6 μg of peptides (Fig. 5B). We also evaluated the time-dependence of this inhibition by incubating 6 μg of peptides for various time points (10, 20, and 30 min) since import itself is a time-dependent process. The inhibitory effect was not apparent at early times but became evident after 30 min of incubation (Fig. 5C).
We also investigated whether this effect of peptides was specific to protein import or if it had a role in modulating other mitochondrial functions. Thus, we incubated 6 μg of peptides with freshly isolated IMF mitochondria and assessed respiration and ROS emission. There was no effect of the presence of peptides on either respiration (Fig. 5D) or ROS emission (Fig. 5E) either represented per μg of mitochondria (inset) or corrected for respiratory rate, suggesting that peptides specifically modulate protein import, as previously shown in C. elegans (18, 32).
DISCUSSION
The maintenance of proteostasis is integral for the health of the cell and lifespan of the organism (26, 30, 40, 41). Proteins that are misfolded or misassembled exert negative effects referred to as proteotoxicity, and this is combatted by compartment-specific protein quality control mechanisms known as the unfolded protein response (UPR) (33, 35). Mitochondrial biogenesis requires the contribution of protein products from both the nuclear and mitochondrial genomes. Thus, a mitochondrion must coordinate the expression of the nuclear genome while also supporting the transcription and translation of its own gene products. Mitochondria are equipped with a protein import machinery (PIM) that facilitates the translocation of nuclear-encoded proteins (37), as well as a mitochondrion-specific UPR (UPRmt) that strives to maintain proteostasis (31, 43).
In this study, we sought to better understand the UPRmt by inducing an imbalance between the two genomes. To achieve this, we knocked down the expression of Tim23, the major channel of the translocase of the inner membrane (TIM complex), using a systemically injectable antisense oligonucleotide, in vivo morpholino. This treatment successfully reduced Tim23 protein in skeletal muscle IMF mitochondria to 60% of normal levels. We were not seeking a complete depletion of Tim23 levels because homozygous deletion of Tim23 is embryonic lethal and thus critical for development (1). The consequence of this depletion was a functional impairment of protein import into the matrix but not to other subcompartments, such as the inner membrane. This specificity is undoubtedly due to the complex nature of the PIM in which Tim23 mediates matrix-destined import but is not essential for translocation to other subcompartments (22). For example, Tim22 is thought to operate in conjunction with, and independently from, Tim23 to facilitate the passage of proteins directly into the inner membrane (27). In our experiments, this could also explain how mitochondrial respiration was maintained basally despite this matrix import defect, since the electron transport chain (ETC) is found within the inner membrane. These data are in accordance with previous literature from the liver of Tim23 heterozygous and wild-type mice in which no differences in mitochondrial respiration were found (29).
We had hypothesized that there would be an elevation in ROS emission in these animals because of the induction of structural imbalances in the stoichiometry of Krebs cycle and electron transport chain subunits in the presence of an import defect. Correspondingly, we did observe elevations in absolute ROS in the presence of an import defect, and a tendency for elevated ROS emission during ADP-stimulated maximal respiration in mitochondria. Thus, it appears that compositional changes brought upon by the import defect may only become evident during maximal respiration. However, this had no apparent effect on the oxidative status of the muscle, measured globally. ROS are thought to mediate retrograde signaling to the nucleus through JNK, a mitogen-activated protein kinase. Work by Horibe et al. (31) demonstrated that JNK activation was required for CHOP activation during UPRmt; however, ROS were never measured in that study. Thus, the elevated ROS levels observed during state III respiration may, in part, explain the 2.7-fold increase in CHOP seen following Tim23 knockdown.
We also hypothesized that Tim23 knockdown in vivo would result in UPRmt activation. This was based on the research by Fiorese et al. (14) who knocked down Tim23 in C. elegans to induce the UPRmt. This robustly activated ATFS-1, the homolog of ATF5. Interestingly, they also demonstrated that Tim23 knockdown also activates ATF5 when expressed in C. elegans (14). However, in our study, no detectable changes in mRNAs encoding ATF5, CHOP, or their downstream targets were observed in the tissues collected 48 h following the last injection. Alterations in mRNA levels are transient events, and it is possible that these changes were occurring at an earlier time point. Thus, we sought to examine the protein level of various UPRmt markers via immunoblotting. This analysis revealed an increase in CHOP, the stress-induced transcription factor associated with UPR activation. ClpP and cpn10, a resident protease and chaperone, respectively, were also both upregulated following Tim23 knockdown. Both ClpP and cpn10 are targets of CHOP, suggesting that the CHOP branch of the UPRmt was activated following Tim23 knockdown (20). However, ATF5, a proteolytic-dependent transcription factor, was not elevated following an acute knockdown of Tim23 in vivo and neither were its downstream targets LonP, HSP60, and mtHSP70. These results provide preliminary evidence that the various branches of the UPRmt can potentially act independently, and thus future works should address whether both transcription factors are needed for the maintenance of mitochondrial proteostasis. This appears to be different from stimuli that induce ER stress, where CHOP transcriptionally activates ATF5 to promote apoptosis (39). It is also appreciated that the UPRmt lies in the midst of cellular stress responses that may act in conjunction or independently from one another. Our data demonstrate the selective activation of the stress responses to regain homeostasis since the UPR of the endoplasmic reticulum, ISR, and mitophagy did not appear to be activated. The dynamic nature of mitophagy makes it difficult to make conclusions since measurements of flux were not made. Taken together, our results suggest that Tim23 knockdown in vivo produces an import defect that results in modest mitochondrial dysfunction. This is likely involved in inducing proteotoxic stress, leading to the activation of the CHOP branch of the UPRmt, independent of ATF5. Therefore, even though Tim23 knockdown in C. elegans activates ATFS-1, our findings demonstrate that the activation of the UPRmt in mammalian models may differ from its counterpart in C. elegans.
Much of what we know about ATF5 is through work done on ATFS-1. It is thought that, similar to ATFS-1, ATF5 is imported into the mitochondrion under basal conditions and degraded. However, in the face of proteotoxicity, proteolytically derived peptides are exported into the cytosol, thereby redirecting ATFS-1 into the nucleus where it acts as a transcription factor (17). However, it remains unclear how these peptides may influence the translocation of ATFS-1 or of other transcription factors in the face of proteotoxicity. Thus, we sought to investigate potential mechanisms through which retrograde signaling may be activated, and we chose to focus on the role of mitochondrial proteolysis.
Freshly isolated mitochondria left unperturbed will continue to proteolytically degrade resident proteins and release the byproducts of this catabolism into the cytosol (4). Therefore, we incubated mitochondria to allow this basal proteolysis to occur, and then we collected all products released from the organelle that were under 3 kDa. This includes all peptides that are 6–30 amino acids in length and therefore ~1 kDa in size (17). Our data support previous literature that shows that peptides are released from mitochondria basally (4), and we demonstrated that excessive induction of oxidative stress promotes proteolysis and the subsequent release of peptides from the organelle. Next, we asked whether the mitochondrial permeability transition pore (mtPTP) could facilitate this export since it is a channel that is opened during oxidative stress. However, treatment of mitochondria with cyclosporin A, a known inhibitor of pore formation, had no impact on peptide release (15). In C. elegans, the mitochondrial channel HAF-1 is necessary for the release of peptides from the organelle (18). Recently, Yano (42) provided evidence for a mammalian homolog, ATP binding cassette subfamily B member 10 (ABCB10), and demonstrated its role in mediating UPRmt activation in HepG2 cells; however, this channel does not seem to regulate peptide release. Therefore, it remains unclear which channel mediates the export of these proteolytically derived peptides into the cytosol.
How could released peptides mediate retrograde signaling? Even in C. elegans this remains unknown, but a few possible mechanisms have been proposed. The first possibility is that peptides are being “sensed” by ATFS-1 or its mammalian homologue ATF5 (18, 25, 32). The second is that peptide efflux rate or volume is being monitored to influence ATF5 translocation (25). We propose that peptides are influencing the import machinery to redirect the translocation of proteins from the mitochondrion to the nucleus. Work in yeast, and more recently in mammals, has demonstrated the dynamic nature of protein import and its ability to be modulated in response to its metabolic environment. This positions the PIM as a sensor of mitochondrial health status and a means of communicating this throughout the cell (16). An example of this is during the process of mitophagy, where phosphatase and tensin homolog-induced putative kinase 1 (PINK1) is regularly imported into the matrix but under stress is arrested on the outer membrane, thereby initiating mitophagy. Thus, we asked whether proteolytic byproducts could modulate protein import as a potential retrograde signal to communicate proteotoxic stress. We incubated peptides at various doses and found that import capacity, but not respiration or ROS emission, was reduced in the presence of peptides in a dose- and time-dependent manner. These findings fit with the idea that low concentrations of peptides, which are continuously exported from mitochondria due to basal proteolysis, would remain innocuous. However, higher concentrations of peptides representing stress-induced elevations in protein breakdown would exert signaling consequences. For example, peptide-dependent import inhibition could serve to redirect ATF5 or other proteins to the nucleus to induce compensatory gene expression while at the same time reducing new sources of proteotoxic stress directed toward the organelle’s PIM. It remains to be addressed whether peptides could in fact influence ATF5 directly, or through some intermediary signaling step.
In summary, our results demonstrate that Tim23 knockdown in vivo disrupts protein import, resulting in a mitonuclear imbalance that stimulates ROS emission and selectively activates the CHOP branch of the UPRmt, independent of ATF5. In addition, our data highlight a potential mechanism in which protein import is modulated by the presence of mitochondrially derived peptides in the cytosol to communicate proteotoxic stress within the mitochondrion to the nucleus. Better understanding of the UPRmt and its retrograde signals would provide much needed insight into the mitochondrial stress response which has been implicated in a variety of cancers, neurodegenerative diseases, and aging.
GRANTS
This work was supported by funding from the Natural Sciences and Engineering Research Council of Canada (to D. A. Hood). D. A. Hood is also the holder of a Canada Research Chair in Cell Physiology. A. N. Oliveira was a recipient of a Canadian Institutes of Health Research-Canada Graduate Scholarship.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.A.H. conceived and designed research; A.N.O. performed experiments; A.N.O. analyzed data; A.N.O. interpreted results of experiments; A.N.O. prepared figures; A.N.O. drafted manuscript; A.N.O. and D.A.H. edited and revised manuscript; D.A.H. approved final version of manuscript.
REFERENCES
- 1.Ahting U, Floss T, Uez N, Schneider-Lohmar I, Becker L, Kling E, Iuso A, Bender A, Hrabé M, Angelis D, Gailus-Durner V, Fuchs H, Meitinger T, Wurst W, Prokisch H, Klopstock T. Neurological phenotype and reduced lifespan in heterozygous Tim23 knockout mice, the first mouse model of defective mitochondrial import. Biochim Biophys Acta 1787: 371–376, 2009. doi: 10.1016/j.bbabio.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Aldridge JE, Horibe T, Hoogenraad NJ. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One 2: e874, 2007. doi: 10.1371/journal.pone.0000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Augustin S, Nolden M, Müller S, Hardt O, Arnold I, Langer T. Characterization of peptides released from mitochondria: evidence for constant proteolysis and peptide efflux. J Biol Chem 280: 2691–2699, 2005. doi: 10.1074/jbc.M410609200. [DOI] [PubMed] [Google Scholar]
- 5.Belkin AM, Klimanskaya IV, Lukashev ME, Lilley K, Critchley DR, Koteliansky VE. A novel phosphoglucomutase-related protein is concentrated in adherens junctions of muscle and nonmuscle cells. J Cell Sci 107: 159–173, 1994. [DOI] [PubMed] [Google Scholar]
- 6.Bohnert M, Pfanner N, van der Laan M. A dynamic machinery for import of mitochondrial precursor proteins. FEBS Lett 581: 2802–2810, 2007. doi: 10.1016/j.febslet.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 7.Bolender N, Sickmann A, Wagner R, Meisinger C, Pfanner N. Multiple pathways for sorting mitochondrial precursor proteins. EMBO Rep 9: 42–49, 2008. doi: 10.1038/sj.embor.7401126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44, D1: D1251–D1257, 2016. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell 138: 628–644, 2009. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9a.D’Amico D, Sorrentino V, Auwerx J. Cytosolic proteostasis networks of the mitochondrial stress response. Trends Biochem Sci 42: 712–725, 2017. doi: 10.1016/j.tibs.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Deepa SS, Bhaskaran S, Ranjit R, Qaisar R, Nair BC, Liu Y, Walsh ME, Fok WC, Van Remmen H. Down-regulation of the mitochondrial matrix peptidase ClpP in muscle cells causes mitochondrial dysfunction and decreases cell proliferation. Free Radic Biol Med 91: 281–292, 2016. doi: 10.1016/j.freeradbiomed.2015.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erlich AT, Tryon LD, Crilly MJ, Memme JM, Moosavi ZSM, Oliveira AN, Beyfuss K, Hood DA. Function of specialized regulatory proteins and signaling pathways in exercise-induced muscle mitochondrial biogenesis. Integr Med Res 5: 187–197, 2016. doi: 10.1016/j.imr.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferguson DP, Dangott LJ, Lightfoot JT. Lessons learned from vivo-morpholinos: How to avoid vivo-morpholino toxicity. Biotechniques 56: 251–256, 2014. doi: 10.2144/000114167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fiorese CJ, Haynes CM. Integrating the UPR into the mitochondrial maintenance network. Crit Rev Biochem Mol Biol 52: 304–313, 2017. doi: 10.1080/10409238.2017.1291577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr Biol 26: 2037–2043, 2016. doi: 10.1016/j.cub.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gutiérrez-Aguilar M, Baines CP. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochim Biophys Acta 1850: 2041–2047, 2015. doi: 10.1016/j.bbagen.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harbauer AB, Zahedi RP, Sickmann A, Pfanner N, Meisinger C. The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab 19: 357–372, 2014. doi: 10.1016/j.cmet.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 17.Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell 13: 467–480, 2007. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 18.Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell 37: 529–540, 2010. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoppeler H. Exercise-induced ultrastructural changes in skeletal muscle. Int J Sports Med 7: 187–204, 1986. doi: 10.1055/s-2008-1025758. [DOI] [PubMed] [Google Scholar]
- 20.Horibe T, Hoogenraad NJ. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS One 2: e835, 2007. doi: 10.1371/journal.pone.0000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inagaki H, Hayashi T, Matsushima Y, Lin KH, Maeda S, Ichihara S, Kitagawa Y, Saito T. Isolation of rat mitochondrial transcription factor A (r-Tfam) cDNA. DNA Seq 11: 131–135, 2000. doi: 10.3109/10425170009033980. [DOI] [PubMed] [Google Scholar]
- 22.Jensen RE, Dunn CD. Protein import into and across the mitochondrial inner membrane: role of the TIM23 and TIM22 translocons. Biochim Biophys Acta 1592: 25–34, 2002. doi: 10.1016/S0167-4889(02)00261-6. [DOI] [PubMed] [Google Scholar]
- 23.Jovaisaite V, Auwerx J. The mitochondrial unfolded protein response—synchronizing genomes. Curr Opin Cell Biol 33: 74–81, 2015. doi: 10.1016/j.ceb.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaushik S, Cuervo AM. Proteostasis and aging. Nat Med 21: 1406–1415, 2015. doi: 10.1038/nm.4001. [DOI] [PubMed] [Google Scholar]
- 25.Kirstein-Miles J, Morimoto RI. Peptides signal mitochondrial stress. Cell Metab 11: 177–178, 2010. doi: 10.1016/j.cmet.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 26.Koga H, Kaushik S, Cuervo AM. Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res Rev 10: 205–215, 2011. doi: 10.1016/j.arr.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kovermann P, Truscott KN, Guiard B, Rehling P, Sepuri NB, Müller H, Jensen RE, Wagner R, Pfanner N. Tim22, the essential core of the mitochondrial protein insertion complex, forms a voltage-activated and signal-gated channel. Mol Cell 9: 363–373, 2002. doi: 10.1016/S1097-2765(02)00446-X. [DOI] [PubMed] [Google Scholar]
- 28.Memme JM, Oliveira AN, Hood DA. Chronology of UPR activation in skeletal muscle adaptations to chronic contractile activity. Am J Physiol Cell Physiol 310: C1024–C1036, 2016. doi: 10.1152/ajpcell.00009.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Menzies KJ, Singh K, Saleem A, Hood DA. Sirtuin 1-mediated effects of exercise and resveratrol on mitochondrial biogenesis. J Biol Chem 288: 6968–6979, 2013. doi: 10.1074/jbc.M112.431155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merkwirth C, Jovaisaite V, Durieux J, Matilainen O, Jordan SD, Quiros PM, Steffen KK, Williams EG, Mouchiroud L, Tronnes SU, Murillo V, Wolff SC, Shaw RJ, Auwerx J, Dillin A. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 165: 1209–1223, 2016. doi: 10.1016/j.cell.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mottis A, Jovaisaite V, Auwerx J. The mitochondrial unfolded protein response in mammalian physiology. Mamm Genome 25: 424–433, 2014. doi: 10.1007/s00335-014-9525-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337: 587–590, 2012. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta 1833: 410–416, 2013. doi: 10.1016/j.bbamcr.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sandri M, Robbins J. Proteotoxicity: an underappreciated pathology in cardiac disease. J Mol Cell Cardiol 71: 3–10, 2014. doi: 10.1016/j.yjmcc.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem 74: 739–789, 2005. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 36.Seiferling D, Szczepanowska K, Becker C, Senft K, Hermans S, Maiti P, König T, Kukat A, Trifunovic A. Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Rep 17: 953–964, 2016. doi: 10.15252/embr.201642077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Straub SP, Stiller SB, Wiedemann N, Pfanner N. Dynamic organization of the mitochondrial protein import machinery. Biol Chem 397: 1097–1114, 2016. doi: 10.1515/hsz-2016-0145. [DOI] [PubMed] [Google Scholar]
- 38.Takahashi M, Hood DA. Protein import into subsarcolemmal and intermyofibrillar skeletal muscle mitochondria. Differential import regulation in distinct subcellular regions. J Biol Chem 271: 27285–27291, 1996. doi: 10.1074/jbc.271.44.27285. [DOI] [PubMed] [Google Scholar]
- 39.Teske BF, Fusakio ME, Zhou D, Shan J, Mcclintick JN. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol Biol Cell 24: 2477–2490, 2013. doi: 10.1091/mbc.E13-01-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tian Y, Garcia G, Bian Q, Steffen KK, Joe L, Wolff S, Meyer BJ, Dillin A. Mitochondrial stress induces chromatin reorganization to promote longevity and UPRmt. Cell 165: 1197–1208, 2016. doi: 10.1016/j.cell.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529: 326–335, 2016. doi: 10.1038/nature17041. [DOI] [PubMed] [Google Scholar]
- 42.Yano M. ABCB10 depletion reduces unfolded protein response in mitochondria. Biochem Biophys Res Commun 486: 465–469, 2017. doi: 10.1016/j.bbrc.2017.03.063. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J 21: 4411–4419, 2002. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]