

Abstract
Cholesteryl esters are generated at multiple sites in the body by sterol O-acyltransferase (SOAT) 1 or SOAT2 in various cell types and lecithin cholesterol acyltransferase in plasma. Esterified cholesterol and triacylglycerol contained in lipoproteins cleared from the circulation via receptor-mediated or bulk-phase endocytosis are hydrolyzed by lysosomal acid lipase within the late endosomal/lysosomal (E/L) compartment. Then, through the successive actions of Niemann-Pick C (NPC) 2 and NPC 1, unesterified cholesterol (UC) is exported from the E/L compartment to the cytosol. Mutations in either NPC1 or NPC2 lead to continuing entrapment of UC in all organs, resulting in multisystem disease, which includes hepatic dysfunction and in some cases liver failure. These studies investigated primarily whether elimination of SOAT2 in NPC1-deficient mice impacted hepatic UC sequestration, inflammation, and transaminase activities. Measurements were made in 7-wk-old mice fed a low-cholesterol chow diet or one enriched with cholesterol starting 2 wk before study. In the chow-fed mice, NPC1:SOAT2 double knockouts, compared with their littermates lacking only NPC1, had 20% less liver mass, 28% lower hepatic UC concentrations, and plasma alanine aminotransferase and aspartate aminotransferase activities that were decreased by 48% and 36%, respectively. mRNA expression levels for several markers of inflammation were all significantly lower in the NPC1 mutants lacking SOAT2. The existence of a new class of potent and selective SOAT2 inhibitors provides an opportunity for exploring if suppression of this enzyme could potentially become an adjunctive therapy for liver disease in NPC1 deficiency.
NEW & NOTEWORTHY In Niemann-Pick type C1 (NPC1) disease, the entrapment of unesterified cholesterol (UC) in the endosomal/lysosomal compartment of all cells causes multiorgan disease, including neurodegeneration, pulmonary dysfunction, and liver failure. Some of this sequestered UC entered cells initially in the esterified form. When sterol O-acyltransferase 2, a cholesterol esterifying enzyme present in enterocytes and hepatocytes, is eliminated in NPC1-deficient mice, there is a reduction in their hepatomegaly, hepatic UC content, and cellular injury.
Keywords: esterified cholesterol, hepatomegaly, lysosomal storage disease, transaminase activity, unesterified cholesterol
INTRODUCTION
Cellular cholesterol homeostasis is regulated through the interplay of a constellation of genes, including LIPA, which encodes lysosomal acid lipase (LAL), as well as Niemann-Pick C (NPC) 2 and NPC1. Together, these proteins control cholesterol trafficking through the late endosomal/lysosomal (E/L) compartment of every cell (28). LAL hydrolyzes esterified cholesterol (EC) and triacylglycerol (TAG) contained within various classes of lipoproteins cleared from the circulation through receptor-mediated and bulk-phase endocytosis (23, 34). The unesterified cholesterol (UC) then moves via NPC2 to NPC1, which facilitates its exit from the E/L compartment (56). This UC mixes with cholesterol from other sources, including that synthesized locally. Such “metabolically active” UC can be utilized in different ways, or it can be esterified, stored in a lipid droplet, and potentially incorporated into nascent very low-density lipoproteins (VLDL) produced in hepatocytes or in chylomicrons that are assembled in enterocytes (65).
Mutations in LIPA, NPC2, or NPC1 are rare and have far-reaching consequences in most organs. In the case of LIPA, mutations that result in almost a complete loss of LAL activity cause Wolman disease (WD) and death in infancy. Other mutations where some residual LAL activity remains lead to cholesteryl ester storage disease (CESD), a milder, later-onset disorder (22). In WD and CESD, there is pronounced hepatomegaly and a massive accumulation of EC in many organs, especially the liver (3, 16, 18). The long-term complications of CESD are marked dyslipidemia and liver disease leading to cirrhosis (58). An enzyme replacement therapy for pediatric and adult patients called Sebelipase alfa has been approved (7, 69). Although NPC disease is significantly more common than WD/CESD, it is nevertheless rare, with the prevalence of NPC1 disease far exceeding that caused by mutations in NPC2 (77). In NPC1 disease, the major cause of morbidity and mortality is neurodegeneration, but there are subsets of patients where death results from either liver failure or pulmonary dysfunction (77). The latter is more prevalent with NPC2 deficiency (24). In both disorders UC becomes sequestered in the E/L compartment of all cells. At least in mice lacking NPC1, this sequestration is clearly discernable in the first day after birth (82). The resulting expansion of tissue UC content varies in different organs but is particularly pronounced in the liver, spleen, and lungs (57, 78, 80, 81). In the case of the central nervous system (CNS), UC entrapment leads to Purkinje cell death and neurodegeneration (31, 82). The amount of UC in the brain as a whole decreases in advanced disease because of demyelination in several major regions (82). In patients with NPC1, the type and severity of the ensuing neurological symptoms vary considerably with age (77). Studies in NPC1-deficient mouse and cat models have shown that this process can be substantially decreased by the delivery of 2-hydroxypropyl-β-cyclodextrin (2HPβCD), a cholesterol-solubilizing agent, directly into the CNS (4, 79). Such findings have led to this treatment modality being evaluated in a clinical trial with young patients with NPC1 (46). Earlier experiments using weekly systemic administration of 2HPβCD in Npc1−/− mice starting at 7 days of age found there was normalization of cholesterol levels in nearly every organ, particularly the liver, and prolonged lifespan (57). Later studies, also using an Npc1−/− mouse model, delineated the impact of 2HPβCD administration on rates of cholesterol esterification, cholesterol synthesis, and conversion of cholesterol to bile acids by the liver (35). Evidence from in vitro studies suggests that β-cyclodextrins work from inside the late E/L compartment (62).
A range of other treatment strategies aimed mainly at countering liver disease in NPC1-deficient animal models has been investigated. One of these found that restricting dietary cholesterol intake in a feline NPC1 model did not diminish hepatic UC sequestration (68). However, in NPC1-deficient mice given ezetimibe, a potent cholesterol absorption inhibitor that blocks the absorption of dietary cholesterol and the reabsorption of biliary cholesterol, the severity of liver disease was attenuated, especially if treatment started from the first day after birth (5). More recently, there is a growing interest in the potential of histone deacetylase inhibitors for NPC disease treatment (54), with a new study showing favorable effects of one such inhibitor, vorinostat, in the livers of an NPC1 mutant mouse model (41). For NPC2 deficiency, a protein replacement therapy effectively reduced tissue cholesterol levels in the liver, spleen, and lungs but not the brain of treated mice (44). In another very different approach, several novel and potent inhibitors of LAL were developed as potential agents for reducing EC hydrolysis and therefore, UC sequestration in NPC deficiency (63). It is unclear if these agents have been tested in NPC1-deficient animal models.
Instead of suppressing LAL activity, an alternate and more physiological approach to reducing the amount of UC sequestered in tissues would be to lower the EC content of the various classes of lipoproteins that are processed in the E/L compartment, following their uptake from the plasma. In humans, rabbits, and mice the proportion of the cholesterol in low-density lipoproteins (LDLs) that is esterified significantly exceeds that which is unesterified (25, 29, 50). EC is generated by three enzymes at different sites in the body. One is lecithin cholesterol acyltransferase, present in the plasma compartment (42, 64, 66). Another is sterol O-acyltransferase (SOAT) 1, located mainly in steroidogenic tissues, kidneys, sebaceous glands, and macrophages (9, 30). Available data suggest that the small amounts of EC present in the CNS are produced locally, mostly by SOAT1 (53). The third enzyme is SOAT2, which is found in enterocytes and hepatocytes (8, 30, 48). It is responsible for generating a significant proportion of the EC found in VLDL remnants (VLDLr), LDL, and chylomicron remnants (CMr). Accordingly, SOAT2 has long been a target in dyslipidemia management (61). Pioneering research in the Rudel and Tomoda laboratories has shown that suppression or elimination of SOAT2 activity in animal models of cardiovascular disease markedly lowers the cholesterol content of atherogenic lipoproteins (43, 45).
Studies in a mouse model for CESD showed that pharmacological or genetic suppression of SOAT2 function resulted in a marked reduction in hepatic EC sequestration with significantly improved liver function (36, 38). Therefore, in the current studies we investigated the potential impact of eliminating SOAT2-driven cholesterol esterification on hepatic UC entrapment and liver function in an NPC1-mutant mouse model. The rationale for evaluating this strategy was that, by lowering the EC content both of chylomicrons reaching the liver from the small intestine, and of nascent VLDL particles entering the circulation from the liver and returning there in VLDLr and LDL particles, the amount of UC ultimately sequestered in the liver could potentially be reduced (65). The data presented here showing less hepatic UC accumulation and improved liver function in the NPC1:SOAT2 double-knockout mice suggest that treatment with a SOAT2 inhibitor, starting in early stage NPC1 deficiency, may have an adjunctive therapeutic benefit.
MATERIALS AND METHODS
Animals and diets.
Heterozygous Npc1nih/nih mice (BALB/c background) were generated from breeding stock originally supplied to us by Dr. Peter Pentchev at the Developmental and Metabolic Neurology Branch, National Institute of Neurological Disorders and Stroke (Bethesda, MD). The history of this model has been documented (51). Our colony of Soat2+/− (Acat2+/−) mice (mixed 129/Sv: C57BL/6 background) was established using breeding stock purchased from the Jackson Laboratories. These two models were used to generate Npc1+/−:Soat2+/− breeding stock from which we obtained offspring of the four genotypes (Npc1+/+:Soat2+/+; Npc1+/+:Soat2−/−; Npc1−/−:Soat2+/+; Npc1−/−:Soat2−/−) needed for this series of studies. All progenies were genotyped on or before 21 days of age, which is when they were weaned. They were fed ad libitum a cereal-based rodent chow diet (no. 7001, Envigo, Teklad, Madison, WI). This formulation had an inherent cholesterol content of 0.02% (wt/wt) and a crude fat content of 4.4% (wt/wt). It provided 13% of calories from fat and was fed as pellets in all experiments except one in which the meal form of 7001 was made to contain cholesterol at a final level of 1.0% (wt/wt). This regimen was fed to mice of the 4 specified genotypes for 15 days, starting when they were 35 days old. A comparable cholesterol-enriched diet was used in some of our previous projects with the NPC1 mouse model (5, 31, 61). Other investigators have used a cholesterol level of 1% (wt/wt) with added fat (19) or 2% (wt/wt) cholesterol with no other additions (2). For our earlier investigations of sterol metabolism in Soat2-knockout mice we used chow, either alone or containing 0.5% (wt/wt) cholesterol (59, 72). All mice were housed as previously described and were studied in the fed state at an average age of ~7 wk (72). Historically, most metabolic studies in mice deficient in NPC1 are done when they are no more than ~7 wk of age because their physical condition declines significantly after that age (6). For the parameters that the present studies focused on, particularly hepatic cholesterol levels, we and other investigators have not previously found consistent sex-related differences in either the Npc1- (31, 40, 81) or Soat2-knockout models (59, 72). Therefore, in the present studies, data from males and females within each genotype were pooled as noted in the figure legends. All experiments were approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center.
Concentration of UC and EC and of TAG in the liver.
The liver, entire small intestine, spleen, both lungs, and brain were excised, rinsed with sodium chloride solution (0.9% wt/vol), blotted, and weighed. An aliquot of liver pieces taken from different lobes (0.6 to 0.8 g) as well as both lungs and the whole brain were each added to ~60 ml of chloroform/methanol (2:1 vol/vol). These tissues were extracted in the presence of two internal standards ([4-14C]cholesteryl oleate and [1,2-3H(N)]cholesterol; PerkinElmer Life Sciences, Inc., Boston, MA). The extracts were filtered into a 100-ml volumetric flask and a 20-ml aliquot of this was dried under air. The lipids were then dissolved in 2 ml of hexane/tert-butyl methyl ether (100:1.5 vol/vol; solvent I) and placed on a silica column (Sep-Pak Vac RC, 500 mg, Water Corp., Milford, MA) that had been prewashed with 2 ml of solvent I. The column was first eluted with 18 ml of solvent I (EC) and then with 18 ml of tert-butyl methyl ether/glacial acetic acid (100:0.2 vol/vol; solvent II) to remove the UC and other lipids (4). The EC and UC fractions were dried under air, saponified in alcoholic KOH, and extracted with petroleum ether. Aliquots of this extract were used to determine recovery of the radiolabeled standards and also to quantitate the mass of cholesterol by gas chromatography. The UC, EC, and total cholesterol (TC) concentrations were calculated as mg/g wet weight of tissue. In the case of the liver, the sum of the UC and EC values was multiplied by the organ weight to obtain mg of TC per whole liver. A similar column method, combined with an enzymatic/colorimetric assay, was used to measure the mg of TAG per whole liver (6). In the mice fed the high-cholesterol diet, the liver was the only organ taken, and this was used specifically for the measurement of EC and UC concentrations. Liver histology was not performed in any of the experiments.
Liver transaminase activities.
The activities of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in freshly collected plasma anticoagulated with heparin were measured in a commercial laboratory. As the plasma was taken from mice in the fed state, neither the concentrations of TC and TAG nor the lipoprotein composition were determined.
Relative mRNA expression analysis.
mRNA levels were measured in liver only using a quantitative real-time PCR assay (75). Total RNA was treated with DNase 1 (RNase-free; Roche, Indianapolis, IN) and reverse-transcribed with random hexamers using SuperScript II RNase H-reverse transcriptase to generate cDNA. Primer Express Software (PerkinElmer Life Sciences) was used to design the primers, which all spanned an intron. Expression levels for 10 genes were determined. The first four of these were NPC1 and NPC2, LIPA, and SOAT2. The six genes used as indices of liver injury and inflammation were cluster of differentiation 68 (also known as macrosialin), SOAT1, ATP-binding cassette transporter G1, tumor necrosis factor α; macrophage inflammatory protein-1α [chemokine (c-c motif) ligand 3], and cluster of differentiation 11c (integrinα x). PCR assays were performed using an Applied Biosystems (Foster City, CA) Prism 7900-HT sequence detection system. The PCR mixture contained (in a final volume of 10 µl) 25 ng of reverse-transcribed RNA, 150 nM of each primer, and 5 µl of 2 × SYBR Green PCR Master Mix (Applied Biosystems). All analyses were determined by the comparative cycle number at threshold method (67) using cyclophilin as the internal control. Relative mRNA levels in individual animals were determined by expressing the amount of mRNA found relative to that obtained for Npc1+/+:Soat2+/+ mice, which in each case was arbitrarily set at 1.0.
Analysis of data.
Data are reported as the mean ± SE for the specified number of animals. GraphPad Prism 6.02 software (GraphPad, San Diego, CA) was used to perform all statistical analyses. Differences between means were tested for statistical significance (P < 0.05) using one-way analysis of variance (ANOVA). Newman-Keuls multiple-comparison test for statistical significance was used for ANOVA. Transformed data were used if unequal variance among the groups was evident by Bartlett’s test.
RESULTS
Degree of change in hepatic mRNA expression levels for LipA and Npc2 associated with NPC1 deficiency was comparable in the concurrent absence of SOAT2.
In the absence of NPC1, the hepatic mRNA expression levels for both LIPA (Fig. 1A) and NPC2 (Fig. 1B) were significantly increased by about the same degree irrespective of the Soat2 genotype. This finding is consistent with that from our previous study of gene expression levels in the cerebellum of 7-wk-old Npc1−/− mice. In that instance, the increases in mRNA for both LipA and Npc2 were of comparable magnitude to those shown here for the liver (31). The data in Fig. 1, C and D confirm the genotypes of the mice in the four experimental groups. They also demonstrate that the Npc1 mRNA expression level was unchanged with SOAT2 deficiency, and likewise that the mRNA level for Soat2 did not shift in the absence of NPC1.
Fig. 1.
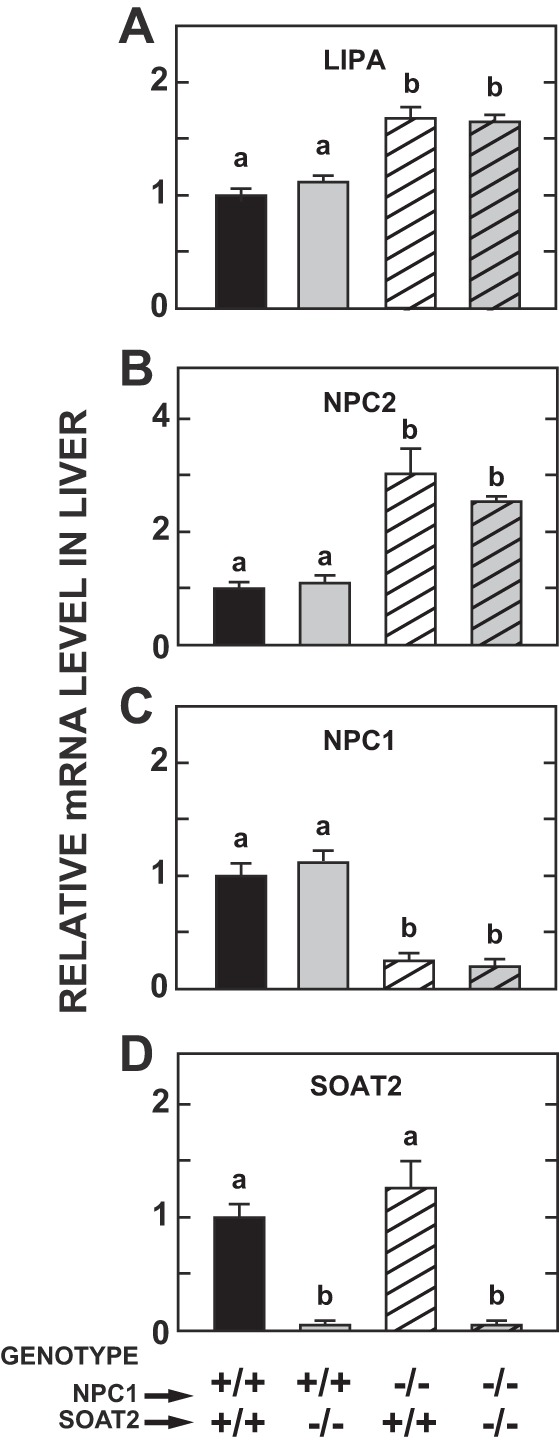
Hepatic mRNA expression levels of various genes in mice deficient in either Niemann-Pick C1 (NPC1), sterol O-acyltransferase 2 (SOAT2), or both NPC1 and SOAT2. The average age of the mice was 7 wk, and all were fed a low-cholesterol rodent chow diet. The 4 genes for which mRNA expression levels are shown are LIPA (A), NPC2 (B), NPC1 (C), and SOAT2 (D). Values are mean ± SE of data from 6 animals in each group, which contained equal or nearly equal numbers of males and females. a,bStatistically significant differences as determined by 1-way ANOVA.
Absence of SOAT2 blunted the degree of hepatomegaly in NPC1-deficient mice.
Previous studies showed that SOAT2 deficiency alone in adult chow-fed mice had no consistent impact on liver mass (36, 59). In contrast, by early adulthood, NPC1 mutant mice fed a chow diet typically exhibit an increased mass of the liver in particular, and to a lesser extent of the spleen and lungs but not of the small intestine (55, 57). These findings were replicated in the Npc1−/−:Soat2+/+ mice in the present studies (Fig. 2, A–D), although only for the liver was the difference statistically significant. In the NPC1 mutant mice, with or without SOAT2 function, the brain weight data (Fig. 2E) were reflective of the hallmark demyelination that starts occurring before 7 wk of age in this model (82). The main finding to take from Fig. 2 is that there was a clear trend toward reduced hepatomegaly in the Npc1−/−:Soat2−/− mice.
Fig. 2.
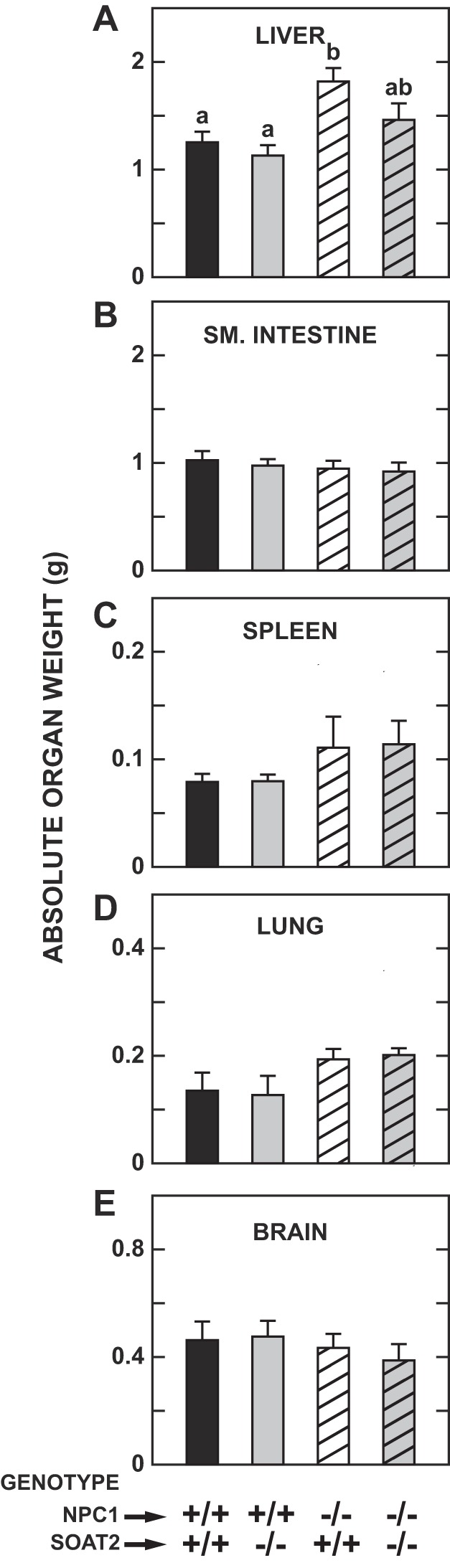
Organ weights in mice deficient in either Niemann-Pick C1 (NPC1), sterol O-acyltransferase 2 (SOAT2), or both NPC1 and SOAT2. These mice were the same as those described in Fig. 1. Their body weights (g) at study were as follows: Npc1+/+:Soat2+/+ 22.3 ± 1.2, Npc1+/+:Soat2−/− 21.1 ± 1.2, Npc1−/−:Soat2+/+ 20.4 ± 1.5, Npc1−/−:Soat2−/− 19.2 ± 1.3. Organ weights are shown for liver (A), small intestine (B), spleen (C), lung (combined) (D) and brain (E). Values are means ± SE of data from 6 animals in each group. a,b,abStatistically significant differences as determined by 1-way ANOVA.
Marked reduction in hepatic cholesterol content in the Npc1−/−:Soat2−/− mice reflected the combined effects of less liver mass and lower concentrations of UC and EC.
Hepatic UC concentrations showed the prototypical elevation seen in NPC1 deficiency, with this increment being significantly blunted in NPC1 mutants without SOAT2 (Fig. 3A). Across all four genotypes the concentration of EC in the liver was much lower than that of UC (Fig. 3B). Nevertheless, it is unequivocal that in the absence of SOAT2 there was a marked contraction in the EC level in mice with, or without, NPC1. In contrast, NPC1 deficiency alone had little impact on the hepatic EC concentration. The data for whole liver cholesterol content (Fig. 3C) show that, although the value for the double knockouts remained well above that for the wild-type controls, it was appreciably less than that in the group deficient in NPC1 only.
Fig. 3.

Hepatic concentrations of unesterified and esterified cholesterol, whole liver total cholesterol and triacylglycerol contents, and transaminase activities in mice deficient in either Niemann-Pick C1 (NPC1), sterol O-acyltransferase 2 (SOAT2), or both NPC1 and SOAT2. The livers from the same mice described in the legends to Figs. 1 and 2 were used for various analyses, including the measurement of unesterified (A) and esterified cholesterol concentrations (B). These concentrations were summed to obtain the total cholesterol concentration, which was then multiplied by the liver weight to obtain whole liver cholesterol content (C). A corresponding value for whole liver triacylglycerol content also was determined (D). Plasma activities of ALT (E) and AST (F) also were measured. For all parameters except hepatic triacylglycerol content, values are means ± SE of data from 6 animals of each genotype. In the case of liver triacylglycerol content, measurements were made in 8 mice of each genotype except for the Npc1−/−:Soat2−/− mice where there were 6. a,b,cStatistically significant differences as determined by one-way ANOVA. ALT, alanine aminotransferase; AST, aspartate aminotransferase; chol, cholesterol; conc, concentration.
Markedly lower hepatic triglyceride contents associated with NPC1 deficiency were unchanged by the concurrent absence of SOAT2.
Compared with those for cholesterol, a very different pattern was seen for liver TAG levels (Fig. 3D). As previously described for both intact liver or isolated hepatocytes, TAG levels fall appreciably in NPC1 deficiency (6, 27, 52, 73). Although SOAT2 deficiency in the mouse leads to lower hepatic TAG levels, especially when dietary cholesterol intake is elevated (1), in the current study there was no discernible additional impact of SOAT2 deficiency on hepatic TAG content in the NPC1 mutant mice.
Elimination of SOAT2 in the NPC1-deficient mice lowered liver enzyme activities and blunted the increase in mRNA hepatic expression levels for several markers of inflammation.
Other benefits to the liver in the NPC1 mutants were seen with the concurrent absence of SOAT2 function. In parallel with the reduction in hepatic UC levels seen in the double knockouts, the elevation of plasma activities of ALT (Fig. 3E) and AST (Fig. 3F) associated with NPC1 deficiency was blunted significantly when SOAT2 was absent. There was also a corresponding fall in the degree of elevation in the expression in liver of six genes that serve as markers of inflammation and macrophage presence (Fig. 4, A–F).
Fig. 4.
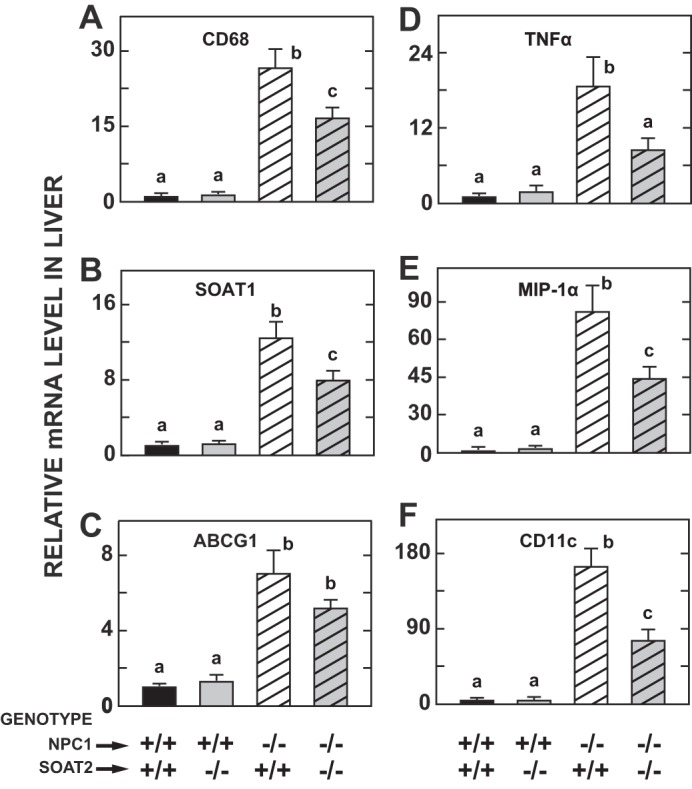
Relative levels of expression of mRNA for various markers of macrophage presence and cytokines in the livers of mice deficient in either Niemann-Pick C1 (NPC1), sterol O-acyltransferase 2 (SOAT2), or both NPC1 and SOAT2. The livers for these analyses were from the same mice used for the measurements described in Figs. 1–3. Details of the expression analysis and data presentation are given in materials and methods. The markers examined were CD68 (A), SOAT1 (B), ABCG1 (C), TNFα (D), MIP-1α (E), and CD11c (F). a,b,cStatistically significant differences as determined by 1-way ANOVA. ABCG1, ATP-binding cassette transporter G1; CD, cluster of differentiation; MIP, macrophage inflammatory protein; TNF, tumor necrosis factor.
Beneficial effects of SOAT2 deficiency in NPC1 mutant mice prevailed in the face of a marked increase in dietary cholesterol intake.
Given that intestinal cholesterol absorption appears to be unchanged in NPC1 deficiency (17), and also that substantial amounts of cholesterol can still be absorbed in the absence of SOAT2 when the dietary cholesterol intake is raised (59), it seemed possible that the benefits of SOAT2 deletion, which the NPC1 mutant mice found when they were maintained on a low cholesterol chow diet, might be attenuated or even lost if dietary cholesterol content was even marginally raised. This possibility was tested by elevating the dietary cholesterol intake of the mice of all four genotypes ~50-fold starting at 35 days of age. The cholesterol-enriched diet caused more hepatomegaly in the Npc1−/−:Soat2+/+ mice compared with their counterparts given chow only (Fig. 5A vs. Fig. 2A). However, in the double knockout mice fed cholesterol, the liver weight was not different from that seen in their counterparts given plain chow. Hepatic UC levels in the cholesterol-fed Npc1−/−:Soat2+/+ mice (Fig. 5B) were higher than in their counterparts given chow only (Fig. 3A), but in the double knockouts the UC concentrations were the same irrespective of dietary cholesterol intake. The EC concentrations increased markedly in the cholesterol-fed wild types, but they were mostly unchanged in the mice lacking just NPC1, or in their Npc1+/+:Soat2−/− and Npc1−/−:Soat2−/− littermates (Fig. 5C vs Fig. 3B). The whole liver cholesterol content data (Fig. 5D) illustrate the magnitude of the benefit that accrues from the absence of SOAT2 in this NPC1 mouse model when its intake of cholesterol is so high. This benefit was also reflected in significantly lower plasma activities of ALT (Fig. 5E) and AST (Fig. 5F).
Fig. 5.

Effect of a marked increase in dietary cholesterol intake on liver weight, concentrations of unesterified and esterified cholesterol, whole liver cholesterol content, and transaminase activities in mice deficient in either Niemann-Pick C1 (NPC1), sterol O-acyltransferase 2 (SOAT2), or both NPC1 and SOAT2. Starting at 5 wk of age, mice of these 4 genotypes were switched from the basal low-cholesterol chow diet to chow containing a high level of added cholesterol (1% wt/wt) for 2 wk. Hepatic cholesterol levels and transaminase activities were then measured. Their body weights (g) at study were as follows: Npc1+/+:Soat2+/+ 22.7 ± 0.7, Npc1+/+:Soat2−/− 22.3 ± 1.4, Npc1−/−:Soat2+/+ 19.9 ± 1.2, Npc1−/−:Soat2−/− 20.0 ± 0.9. The data are presented as follows: liver weight (A), unesterified and esterified cholesterol concentrations in liver (B and C, respectively), total cholesterol content of whole liver (D), and plasma activities of ALT and AST (E and F, respectively). Values are means ± SE of data from 4 mice in each group except for the mice deficient in both NPC1 and SOAT2 where there were 8 animals. Each group consisted of essentially equal numbers of male and female mice. a,b,cStatistically significant differences as determined by 1-way ANOVA. chol, cholesterol; conc, concentration.
Tissue UC levels in the lungs and brains of mice deficient in both NPC1 and SOAT2 were similar to those in matching mice deficient in NPC1 only.
The marked increase in UC levels in the lungs seen in the Npc1−/−:Soat2+/+ mice fed just the basal chow diet was unchanged in their Npc1−/−:Soat2−/− littermates (Table 1). Similarly, the magnitude of the fall in brain UC concentrations stemming from NPC1 deficiency was about the same irrespective of whether the mice had SOAT2.
Table 1.
Concentrations of unesterified and esterified cholesterol in the lungs and brains of mice deficient in either NPC1, SOAT2, or both NPC1 and SOAT2
| Lung, mg/g |
Brain, mg/g |
|||
|---|---|---|---|---|
| Genotype | Unesterified cholesterol | Esterified cholesterol | Unesterified cholesterol | Esterified cholesterol |
| Npc1+/+:Soat2+/+ | 5.58 ± 0.22a | 0.20 ± 0.02a | 16.64 ± 0.18a | 0.04 ± 0.01a |
| Npc1+/+:Soat2−/− | 5.48 ± 0.17a | 0.17 ± 0.02a | 15.88 ± 0.37a,b | 0.04 ± 0.01a |
| Npc1−/−:Soat2+/+ | 17.82 ± 0.45b | 0.17 ± 0.02a | 14.24 ± 0.25c | 0.05 ± 0.01a |
| Npc1−/−:Soat2−/− | 17.57 ± 0.83b | 0.14 ± 0.01a | 14.83 ± 0.05b,c | 0.05 ± 0.01a |
Values are means ± SE of data from 6 mice of each genotype. These were the same mice for which hepatic cholesterol concentration data are presented in Fig. 3. NPC1, Niemann-Pick C1; SOAT2, sterol O-acyltransferase 2.
Statistically significant differences as determined by 1-way ANOVA.
DISCUSSION
Although SOAT2 expression is localized to specific cell types in both the liver and small intestine, the focus of the present studies was primarily on the liver, which is one of the organ systems most impacted by NPC1 deficiency. Previous studies in NPC1- and NPC2-deficient mouse models showed that, compared with the level of increase in whole-organ cholesterol content seen in the liver, the increase in the small intestine was much less pronounced (56, 81). This partly reflected the fact that, unlike the mass of the liver, spleen, and lung, the small intestine weight does not increase in either NPC1- or NPC2-deficient mice, at least up to early adulthood when body weight loss and neurodegeneration become evident (56). This was again the case in the present studies (Fig. 2B). The continual sloughing of mucosal cells is likely the main reason why intestinal UC levels don’t rise as much as in the liver and several other extrahepatic organs (32).
In SOAT2-deficient mice, depending on whether their diet has a cereal or semipurified base, and also on whether it contains added cholesterol and fat, fractional cholesterol absorption is lower to a varying degree than in wild-type controls (43, 59, 72). The biggest impact SOAT2 deficiency has within the enterocyte is that it dramatically reduces the proportion of EC in nascent chylomicrons assembled there (43). Irrespective of this shift in the ratio of UC to EC within the chylomicrons, and more specifically within their remnants that are ultimately cleared by the liver (12), what matters most is that the diminished delivery of intestinally derived cholesterol into the E/L compartment of hepatocytes lacking NPC1 culminates in less UC sequestration in the liver as a whole. In essence, much the same result is achieved when Npc1−/− mice with normal levels of SOAT2 expression are given ezetimibe (5), a potent sterol absorption inhibitor known to reduce chylomicron cholesterol content (74). However, although ezetimibe treatment blunts the amount of UC becoming sequestered in livers with disrupted NPC1 function, significant entrapment still occurs because the liver also clears LDL and VLDLr from the circulation (15, 47). These particles contain SOAT2-generated EC that is used in the assembly of nascent VLDL in hepatocytes (65). EC carried in VLDLr and mature LDL particles, like that present in CMr, becomes UC through the action of LAL in the E/L compartment.
Two points regarding the relevance of the present findings to the management of liver disease in human NPC1 deficiency warrant discussion. The first has to do with the differences in shifts in lipoprotein profile typically seen in humans with NPC1 deficiency and in mouse models for this disease. Perhaps the most significant difference centers on plasma high-density lipoprotein-cholesterol levels, which fall in NPC1 patients (21) but are clearly elevated in NPC1-deficient mice (71, 80). There is also the finding that in cultured primary hepatocytes from Npc1−/− mice, the rate of secretion of VLDL was increased, and the secreted particles were enriched with EC (27). Although this observation is difficult to reconcile with the fall in circulating LDL-cholesterol levels in NPC1 patients, this does not mean that a hypothetical suppression of SOAT2 activity in NPC1 patients would be without benefit. The data from the present studies infer that a sustained pharmacological reduction in the EC content of CMr reaching the liver, and of nascent VLDL particles entering the circulation and being ultimately cleared largely by the liver in the form of VLDLr and mature LDL particles, might culminate in less hepatic UC entrapment and related injury. The results of the experiment with Npc1−/−:Soat2−/− mice fed a high-cholesterol diet (Fig. 5) attest to the decisive benefit to the liver that accrues from a complete absence of SOAT2 in the NPC1-deficient mouse. The second point is that although there are no SOAT2 inhibitors approved for clinical use, a new generation of selective and potent compounds designed to target SOAT2 has been developed. One of these, called PRD125, has clear efficacy in blunting hepatic EC sequestration in LAL-deficient mice (38), and in lowering circulating chylomicron- and LDL-cholesterol levels as well as plaque formation in established mouse models for atherosclerosis research (45). However, what must be emphasized is that if this class of compounds was ever to be evaluated for combating liver disease in NPC1 deficiency in humans, treatment would ideally need to be instituted at an early age. Clearly, in advanced NPC disease when large amounts of UC have already become entrapped in the liver, the institution of a pharmacological suppression of SOAT2, although potentially blunting the entrapment of additional UC, would not be expected to have any impact on the pool of UC already sequestered in the liver.
The findings of the current studies expand the list of strategies with potential utility in treating severe liver disease that is the cause of death in a subset of patients with NPC1 deficiency. However, based on studies in both mouse and feline NPC1 models, current data suggest that systemically administered 2HPβCD is presently the most effective agent for ameliorating liver disease, and it also diminishes UC entrapment in a number of extrahepatic organs (35, 56, 71). It is not known whether systemic administration of 2HPβCD has the same benefits in patients with NPC1 as it does in mouse and feline models, but this is currently being investigated (ClinicalTrials.gov identifier: NCT02939547). The main problem with 2HPβCD is that the very high doses needed to promote mobilization of sequestered UC are ototoxic (13, 14, 79), and there are also concerns about adverse effects in the lungs (10, 39, 79). Although one direction of research aimed at potentially ameliorating these problems is to use polymeric forms of 2HPβCD (11, 70) another approach, at least for targeting liver disease, might be to combine lower doses of 2HPβCD with other classes of agents that reduce hepatic UC sequestration by mechanisms different than those through which β-cyclodextrins are believed to act (33, 76). In this regard, it would be useful to determine what happens at a biochemical and molecular level in the livers of an NPC1-deficient mouse model treated with both systemically administered 2HPβCD and also a potent and selective inhibitor of SOAT2 such as PRD125 in their diet. Previous detailed kinetic studies in NPC1-deficient mice given 2HPβCD subcutaneously showed that the release of large amounts of sequestered UC, particularly in the liver, resulted in a rapid and pronounced compensatory inhibition of de novo cholesterol synthesis along with marked increases in tissue EC concentrations (35, 71) and biliary UC levels (37). Presumably, if SOAT2 function were blunted in the face of 2HPβCD treatment, then in the liver more of the UC released from the E/L compartment would be either converted to bile acid and/or delivered unmetabolized directly into the bile. In the case of other organs also severely impacted by NPC1 deficiency, especially the brain and lungs, the suppression of SOAT2-driven EC formation alone would be of negligible benefit largely because these tissues, unlike the liver, do not clear EC-rich CMr from the circulation (12) and also exhibit low to negligible rates of LDL clearance (47).
The final point warranting discussion centers on the interesting question of how NPC1 and SOAT2 deficiency each impact hepatic TAG content. In early studies by the Pentchev laboratory using a BALB/c mouse model with an autosomal recessive lipid storage disorder that was subsequently recognized to be NPC1 deficiency, the triglyceride concentration in the liver, kidney, and spleen was much lower in the mutants than in their wild type controls (52). Several other laboratories later reported markedly lower hepatic TAG concentrations in the livers or primary hepatocytes from Npc1−/− mice (6, 27, 73). This feature was not confirmed in one laboratory (20), but it was again evident in the current studies (Fig. 3D). The metabolic basis for lower hepatic TAG levels is not understood although one study using hepatocytes concluded that increased channeling of acetyl-CoA units into the cholesterol biosynthetic pathway limits their availability for incorporation into the fatty acids needed for TAG production (73). Although limited data show that hepatic fatty acid synthesis in Npc1−/− mice may be lower than in their Npc1+/+ controls (6), it is doubtful that this is the principal explanation for the very low TAG levels in NPC1 deficiency particularly as fatty acid flux through NPC1-deficient lysosomes is normal (49). In other studies using mice deficient in sterol 27-hydroxylase, a key enzyme in the bile acid biosynthesis, hepatic fatty acid synthesis rates and TAG concentrations are elevated in the face of an increase in cholesterol synthesis far exceeding that seen in the livers of NPC1-deficient mice (60). SOAT2 is also known to be a player in determining hepatic TAG levels. Alger et al. (1) showed that in Soat2−/− mice hepatic TAG levels increased far less than in their Soat2+/+ controls when fed a diet with moderately elevated levels of fat and cholesterol. This was attributed to higher levels of TAG mobilization in the livers of the Soat2−/− mice. In the current studies, there were actually no statistically significant differences in hepatic TAG content among mice of the four genotypes studied (Fig. 3D). However, there was a clear trend toward lower levels in the Npc1−/−:Soat2+/+ mice, and even marginally lower contents in their Npc1−/−:Soat2−/− littermates. The early studies evaluating systemic 2HPβCD treatment in NPC1-deficient mice did not report hepatic TAG levels. In addition, there appear also to be no published data showing whether hepatic TAG levels in human NPC1 deficiency fall as they do in mouse models for this disease. In the longer term, these questions will presumably be answered as part of current and future clinical studies aimed at evaluating the efficacy of systemic 2HPβCD in managing the impact of NPC1 deficiency on hepatic function. For now, however, new studies in NPC1-knockout mice are needed to determine if there is any shift in the expression levels of mRNA or protein for diacylglycerol acyltransferase 1 and 2 (DGAT1 and DGAT2), which play a critical role in the terminal phase of chylomicron assembly (26), and ultimately lipid delivery to the liver.
GRANTS
This research was supported by National Institutes of Health Grants R01-HL-009610 (to S. Turley) and DK-078592 (to J. Repa) and the Ara Parseghian Medical Research Foundation (to J. Repa).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.M.L., R.D.J., J.J.R., and S.D.T. conceived and designed research; A.M.L. and S.D.T. performed experiments; A.M.L., J.J.R., and S.D.T. analyzed data; A.M.L., R.D.J., J.J.R., and S.D.T. interpreted results of experiments; A.M.L. prepared figures; S.D.T. drafted manuscript; A.M.L., R.D.J., J.J.R., and S.D.T. edited and revised manuscript; A.M.L., R.D.J., J.J.R., and S.D.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Carolyn Crumpton, Ernest Tong, Monti Schneiderman, and Stephen Ostermann for technical assistance.
Present address of R. D. Jones: Dept. of Pathology, Northwestern University, Chicago, IL.
REFERENCES
- 1.Alger HM, Brown JM, Sawyer JK, Kelley KL, Shah R, Wilson MD, Willingham MC, Rudel LL. Inhibition of acyl-coenzyme A:cholesterol acyltransferase 2 (ACAT2) prevents dietary cholesterol-associated steatosis by enhancing hepatic triglyceride mobilization. J Biol Chem 285: 14267–14274, 2010. doi: 10.1074/jbc.M110.118422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amigo L, Mendoza H, Castro J, Quiñones V, Miquel JF, Zanlungo S. Relevance of Niemann-Pick type C1 protein expression in controlling plasma cholesterol and biliary lipid secretion in mice. Hepatology 36: 819–828, 2002. doi: 10.1053/jhep.2002.35617. [DOI] [PubMed] [Google Scholar]
- 3.Aqul A, Lopez AM, Posey KS, Taylor AM, Repa JJ, Burns DK, Turley SD. Hepatic entrapment of esterified cholesterol drives continual expansion of whole body sterol pool in lysosomal acid lipase-deficient mice. Am J Physiol Gastrointest Liver Physiol 307: G836–G847, 2014. doi: 10.1152/ajpgi.00243.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aqul A, Liu B, Ramirez CM, Pieper AA, Estill SJ, Burns DK, Liu B, Repa JJ, Turley SD, Dietschy JM. Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J Neurosci 31: 9404–9413, 2011. doi: 10.1523/JNEUROSCI.1317-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beltroy EP, Liu B, Dietschy JM, Turley SD. Lysosomal unesterified cholesterol content correlates with liver cell death in murine Niemann-Pick type C disease. J Lipid Res 48: 869–881, 2007. doi: 10.1194/jlr.M600488-JLR200. [DOI] [PubMed] [Google Scholar]
- 6.Beltroy EP, Richardson JA, Horton JD, Turley SD, Dietschy JM. Cholesterol accumulation and liver cell death in mice with Niemann-Pick type C disease. Hepatology 42: 886–893, 2005. doi: 10.1002/hep.20868. [DOI] [PubMed] [Google Scholar]
- 7.Burton BK, Balwani M, Feillet F, Barić I, Burrow TA, Camarena Grande C, Coker M, Consuelo-Sánchez A, Deegan P, Di Rocco M, Enns GM, Erbe R, Ezgu F, Ficicioglu C, Furuya KN, Kane J, Laukaitis C, Mengel E, Neilan EG, Nightingale S, Peters H, Scarpa M, Schwab KO, Smolka V, Valayannopoulos V, Wood M, Goodman Z, Yang Y, Eckert S, Rojas-Caro S, Quinn AG. A phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency. N Engl J Med 373: 1010–1020, 2015. doi: 10.1056/NEJMoa1501365. [DOI] [PubMed] [Google Scholar]
- 8.Cases S, Novak S, Zheng Y-W, Myers HM, Lear SR, Sande E, Welch CB, Lusis AJ, Spencer TA, Krause BR, Erickson SK, Farese RV Jr. ACAT-2, a second mammalian acyl-CoA:cholesterol acyltransferase. Its cloning, expression, and characterization. J Biol Chem 273: 26755–26764, 1998. doi: 10.1074/jbc.273.41.26755. [DOI] [PubMed] [Google Scholar]
- 9.Chang CCY, Sakashita N, Ornvold K, Lee O, Chang ET, Dong R, Lin S, Lee C-YG, Strom SC, Kashyap R, Fung JJ, Farese RV Jr, Patoiseau JF, Delhon A, Chang TY. Immunological quantitation and localization of ACAT-1 and ACAT-2 in human liver and small intestine. J Biol Chem 275: 28083–28092, 2000. doi: 10.1074/jbc.M003927200. [DOI] [PubMed] [Google Scholar]
- 10.Chien YH, Shieh YD, Yang CY, Lee NC, Hwu WL. Lung toxicity of hydroxypropyl-β-cyclodextrin infusion. Mol Genet Metab 109: 231–232, 2013. doi: 10.1016/j.ymgme.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 11.Collins CJ, Loren BP, Alam MS, Mondjinou Y, Skulsky JL, Chaplain CR, Haldar K, Thompson DH. Pluronic based β-cyclodextrin polyrotaxanes for treatment of Niemann-Pick Type C disease. Sci Rep 7: 46737, 2017. doi: 10.1038/srep46737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper AD. Hepatic uptake of chylomicron remnants. J Lipid Res 38: 2173–2192, 1997. [PubMed] [Google Scholar]
- 13.Crumling MA, Liu L, Thomas PV, Benson J, Kanicki A, Kabara L, Hälsey K, Dolan D, Duncan RK. Hearing loss and hair cell death in mice given the cholesterol-chelating agent hydroxypropyl-β-cyclodextrin. PLoS One 7: e53280, 2012. doi: 10.1371/journal.pone.0053280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davidson CD, Fishman YI, Puskás I, Szemán J, Sohajda T, McCauliff LA, Sikora J, Storch J, Vanier MT, Szente L, Walkley SU, Dobrenis K. Efficacy and ototoxicity of different cyclodextrins in Niemann-Pick C disease. Ann Clin Transl Neurol 3: 366–380, 2016. doi: 10.1002/acn3.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dietschy JM, Turley SD, Spady DK. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J Lipid Res 34: 1637–1659, 1993. [PubMed] [Google Scholar]
- 16.Dincsoy HP, Rolfes DB, McGraw CA, Schubert WK. Cholesterol ester storage disease and mesenteric lipodystrophy. Am J Clin Pathol 81: 263–269, 1984. doi: 10.1093/ajcp/81.2.263. [DOI] [PubMed] [Google Scholar]
- 17.Dixit SS, Sleat DE, Stock AM, Lobel P. Do mammalian NPC1 and NPC2 play a role in intestinal cholesterol absorption? Biochem J 408: 1–5, 2007. doi: 10.1042/BJ20071167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du H, Heur M, Duanmu M, Grabowski GA, Hui DY, Witte DP, Mishra J. Lysosomal acid lipase-deficient mice: depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. J Lipid Res 42: 489–500, 2001. [PubMed] [Google Scholar]
- 19.Erickson RP, Bhattacharyya A, Hunter RJ, Heidenreich RA, Cherrington NJ. Liver disease with altered bile acid transport in Niemann-Pick C mice on a high-fat, 1% cholesterol diet. Am J Physiol Gastrointest Liver Physiol 289: G300–G307, 2005. doi: 10.1152/ajpgi.00568.2004. [DOI] [PubMed] [Google Scholar]
- 20.Garver WS, Jelinek D, Oyarzo JN, Flynn J, Zuckerman M, Krishnan K, Chung BH, Heidenreich RA. Characterization of liver disease and lipid metabolism in the Niemann-Pick C1 mouse. J Cell Biochem 101: 498–516, 2007. doi: 10.1002/jcb.21200. [DOI] [PubMed] [Google Scholar]
- 21.Garver WS, Jelinek D, Meaney FJ, Flynn J, Pettit KM, Shepherd G, Heidenreich RA, Vockley CM, Castro G, Francis GA. The National Niemann-Pick Type C1 Disease Database: correlation of lipid profiles, mutations, and biochemical phenotypes. J Lipid Res 51: 406–415, 2010. doi: 10.1194/jlr.P000331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghishan FK. Inborn errors of lipid metabolism. Intracellular metabolism of cholesterol. In: Zakim and Boyer’s Hepatology A Textbook of Liver Disease (6th ed.), edited by Boyer TD, Manns MP, Sanyal AJ. Philadelphia, PA: Elvsevier, 2011, p. 1184–1189. [Google Scholar]
- 23.Goldstein JL, Dana SE, Faust JR, Beaudet AL, Brown MS. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. J Biol Chem 250: 8487–8495, 1975. [PubMed] [Google Scholar]
- 24.Griese M, Brasch F, Aldana VR, Cabrera MM, Goelnitz U, Ikonen E, Karam BJ, Liebisch G, Linder MD, Lohse P, Meyer W, Schmitz G, Pamir A, Ripper J, Rolfs A, Schams A, Lezana FJ. Respiratory disease in Niemann-Pick type C2 is caused by pulmonary alveolar proteinosis. Clin Genet 77: 119–130, 2010. doi: 10.1111/j.1399-0004.2009.01325.x. [DOI] [PubMed] [Google Scholar]
- 25.Havel RJ, Kita T, Kotite L, Kane JP, Hamilton RL, Goldstein JL, Brown MS. Concentration and composition of lipoproteins in blood plasma of the WHHL rabbit. An animal model of human familial hypercholesterolemia. Arteriosclerosis 2: 467–474, 1982. doi: 10.1161/01.ATV.2.6.467. [DOI] [PubMed] [Google Scholar]
- 26.Hung YH, Carreiro AL, Buhman KK. Dgat1 and Dgat2 regulate enterocyte triacylglycerol distribution and alter proteins associated with cytoplasmic lipid droplets in response to dietary fat. Biochim Biophys Acta 1862: 600–614, 2017. doi: 10.1016/j.bbalip.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kulinski A, Vance JE. Lipid homeostasis and lipoprotein secretion in Niemann-Pick C1-deficient hepatocytes. J Biol Chem 282: 1627–1637, 2007. doi: 10.1074/jbc.M610001200. [DOI] [PubMed] [Google Scholar]
- 28.Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, Infante RE. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 137: 1213–1224, 2009. doi: 10.1016/j.cell.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LeBoeuf RC, Puppione DL, Schumaker VN, Lusis AJ. Genetic control of lipid transport in mice. I. Structural properties and polymorphisms of plasma lipoproteins. J Biol Chem 258: 5063–5070, 1983. [PubMed] [Google Scholar]
- 30.Lee RG, Willingham MC, Davis MA, Skinner KA, Rudel LL. Differential expression of ACAT1 and ACAT2 among cells within liver, intestine, kidney, and adrenal of nonhuman primates. J Lipid Res 41: 1991–2001, 2000. [PubMed] [Google Scholar]
- 31.Li H, Repa JJ, Valasek MA, Beltroy EP, Turley SD, German DC, Dietschy JM. Molecular, anatomical, and biochemical events associated with neurodegeneration in mice with Niemann-Pick type C disease. J Neuropathol Exp Neurol 64: 323–333, 2005. doi: 10.1093/jnen/64.4.323. [DOI] [PubMed] [Google Scholar]
- 32.Lipkin M. Proliferation and differentiation of gastrointestinal cells in normal and disease states. In: Physiology of the Gastrointestinal Tract, edited by Johnson LR. New York: Raven, 1981, p. 145–168. [Google Scholar]
- 33.Liu B. Therapeutic potential of cyclodextrins in the treatment of Niemann-Pick type C disease. Clin Lipidol 7: 289–301, 2012. doi: 10.2217/clp.12.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu B, Xie C, Richardson JA, Turley SD, Dietschy JM. Receptor-mediated and bulk-phase endocytosis cause macrophage and cholesterol accumulation in Niemann-Pick C disease. J Lipid Res 48: 1710–1723, 2007. doi: 10.1194/jlr.M700125-JLR200. [DOI] [PubMed] [Google Scholar]
- 35.Liu B, Ramirez CM, Miller AM, Repa JJ, Turley SD, Dietschy JM. Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J Lipid Res 51: 933–944, 2010. doi: 10.1194/jlr.M000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lopez AM, Chuang JC, Turley SD. Impact of loss of SOAT2 function on disease progression in the lysosomal acid lipase-deficient mouse. Steroids 130: 7–14, 2018. doi: 10.1016/j.steroids.2017.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopez AM, Terpack SJ, Posey KS, Liu B, Ramirez CM, Turley SD. Systemic administration of 2-hydroxypropyl-β-cyclodextrin to symptomatic Npc1-deficient mice slows cholesterol sequestration in the major organs and improves liver function. Clin Exp Pharmacol Physiol 41: 780–787, 2014. doi: 10.1111/1440-1681.12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lopez AM, Chuang JC, Posey KS, Ohshiro T, Tomoda H, Rudel LL, Turley SD. PRD125, a potent and selective inhibitor of sterol O-acyltransferase 2 markedly reduces hepatic cholesteryl ester accumulation and improves liver function in lysosomal acid lipase-deficient mice. J Pharmacol Exp Ther 355: 159–167, 2015. doi: 10.1124/jpet.115.227207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsuo M, Togawa M, Hirabaru K, Mochinaga S, Narita A, Adachi M, Egashira M, Irie T, Ohno K. Effects of cyclodextrin in two patients with Niemann-Pick Type C disease. Mol Genet Metab 108: 76–81, 2013. doi: 10.1016/j.ymgme.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 40.Morris MD, Bhuvaneswaran C, Shio H, Fowler S. Lysosome lipid storage disorder in NCTR-BALB/c mice. I. Description of the disease and genetics. Am J Pathol 108: 140–149, 1982. [PMC free article] [PubMed] [Google Scholar]
- 41.Munkacsi AB, Hammond N, Schneider RT, Senanayake DS, Higaki K, Lagutin K, Bloor SJ, Ory DS, Maue RA, Chen FW, Hernandez-Ono A, Dahlson N, Repa JJ, Ginsberg HN, Ioannou YA, Sturley SL. Normalization of hepatic homeostasis in the Npc1nmf164 mouse model of Niemann-Pick type C disease treated with the histone deaceyltase inhibitor Vorinostat. J Biol Chem 292: 4395–4410, 2017. doi: 10.1074/jbc.M116.770578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ng DS, Francone OL, Forte TM, Zhang J, Haghpassand M, Rubin EM. Disruption of the murine lecithin:cholesterol acyltransferase gene causes impairment of adrenal lipid delivery and up-regulation of scavenger receptor class B type I. J Biol Chem 272: 15777–15781, 1997. doi: 10.1074/jbc.272.25.15777. [DOI] [PubMed] [Google Scholar]
- 43.Nguyen TM, Sawyer JK, Kelley KL, Davis MA, Rudel LL. Cholesterol esterification by ACAT2 is essential for efficient intestinal cholesterol absorption: evidence from thoracic lymph duct cannulation. J Lipid Res 53: 95–104, 2012. doi: 10.1194/jlr.M018820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nielsen GK, Dagnaes-Hansen F, Holm IE, Meaney S, Symula D, Andersen NT, Heegaard CW. Protein replacement therapy partially corrects the cholesterol-storage phenotype in a mouse model of Niemann-Pick type C2 disease. PLoS One 6: e27287, 2011. doi: 10.1371/journal.pone.0027287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohshiro T, Ohtawa M, Nagamitsu T, Matsuda D, Yagyu H, Davis MA, Rudel LL, Ishibashi S, Tomoda H. New pyripyropene A derivatives, highly SOAT2-selective inhibitors, improve hypercholesterolemia and atherosclerosis in atherogenic mouse models. J Pharmacol Exp Ther 355: 299–307, 2015. doi: 10.1124/jpet.115.227348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ory DS, Ottinger EA, Farhat NY, King KA, Jiang X, Weissfeld L, Berry-Kravis E, Davidson CD, Bianconi S, Keener LA, Rao R, Soldatos A, Sidhu R, Walters KA, Xu X, Thurm A, Solomon B, Pavan WJ, Machielse BN, Kao M, Silber SA, McKew JC, Brewer CC, Vite CH, Walkley SU, Austin CP, Porter FD. Intrathecal 2-hydroxypropyl-β-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomized, open-label, phase 1–2 trial. Lancet 390: 1758–1768, 2017. doi: 10.1016/S0140-6736(17)31465-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osono Y, Woollett LA, Herz J, Dietschy JM. Role of the low density lipoprotein receptor in the flux of cholesterol through the plasma and across the tissues of the mouse. J Clin Invest 95: 1124–1132, 1995. doi: 10.1172/JCI117760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parini P, Davis M, Lada AT, Erickson SK, Wright TL, Gustafsson U, Sahlin S, Einarsson C, Eriksson M, Angelin B, Tomoda H, Omura S, Willingham MC, Rudel LL. ACAT2 is localized to hepatocytes and is the major cholesterol-esterifying enzyme in human liver. Circulation 110: 2017–2023, 2004. doi: 10.1161/01.CIR.0000143163.76212.0B. [DOI] [PubMed] [Google Scholar]
- 49.Passeggio J, Liscum L. Flux of fatty acids through NPC1 lysosomes. J Biol Chem 280: 10333–10339, 2005. doi: 10.1074/jbc.M413657200. [DOI] [PubMed] [Google Scholar]
- 50.Pauciullo P, Carlson LA, Eklund B, Johansson J, Olsson AG. Concentration and chemical composition of plasma lipoprotein subfractions in patients with peripheral vascular disease. Evidence for normal apolipoprotein B but low cholesteryl ester content in small VLDL. Atherosclerosis 58: 123–137, 1985. doi: 10.1016/0021-9150(85)90060-7. [DOI] [PubMed] [Google Scholar]
- 51.Pentchev PG. Niemann-Pick C research from mouse to gene. Biochim Biophys Acta 1685: 3–7, 2004. doi: 10.1016/j.bbalip.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 52.Pentchev PG, Gal AE, Booth AD, Omodeo-Sale F, Fouks J, Neumeyer BA, Quirk JM, Dawson G, Brady RO. A lysosomal storage disorder in mice characterized by a dual deficiency of sphingomyelinase and glucocerebrosidase. Biochim Biophys Acta 619: 669–679, 1980. doi: 10.1016/0005-2760(80)90116-2. [DOI] [PubMed] [Google Scholar]
- 53.Pfrieger FW, Ungerer N. Cholesterol metabolism in neurons and astrocytes. Prog Lipid Res 50: 357–371, 2011. doi: 10.1016/j.plipres.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 54.Pipalia NH, Subramanian K, Mao S, Ralph H, Hutt DM, Scott SM, Balch WE, Maxfield FR. Histone deacetylase inhibitors correct the cholesterol storage defect in most Niemann-Pick C1 mutant cells. J Lipid Res 58: 695–708, 2017. doi: 10.1194/jlr.M072140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramirez CM, Lopez AM, Le LQ, Posey KS, Weinberg AG, Turley SD. Ontogenic changes in lung cholesterol metabolism, lipid content, and histology in mice with Niemann-Pick type C disease. Biochim Biophys Acta 1841: 54–61, 2014. doi: 10.1016/j.bbalip.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramirez CM, Liu B, Aqul A, Taylor AM, Repa JJ, Turley SD, Dietschy JM. Quantitative role of LAL, NPC2, and NPC1 in lysosomal cholesterol processing defined by genetic and pharmacological manipulations. J Lipid Res 52: 688–698, 2011. doi: 10.1194/jlr.M013789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramirez CM, Liu B, Taylor AM, Repa JJ, Burns DK, Weinberg AG, Turley SD, Dietschy JM. Weekly cyclodextrin administration normalizes cholesterol metabolism in nearly every organ of the Niemann-Pick type C1 mouse and markedly prolongs life. Pediatr Res 68: 309–315, 2010. doi: 10.1203/PDR.0b013e3181ee4dd2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reiner Ž, Guardamagna O, Nair D, Soran H, Hovingh K, Bertolini S, Jones S, Ćorić M, Calandra S, Hamilton J, Eagleton T, Ros E. Lysosomal acid lipase deficiency–an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 235: 21–30, 2014. doi: 10.1016/j.atherosclerosis.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 59.Repa JJ, Buhman KK, Farese RV Jr, Dietschy JM, Turley SD. ACAT2 deficiency limits cholesterol absorption in the cholesterol-fed mouse: impact on hepatic cholesterol homeostasis. Hepatology 40: 1088–1097, 2004. doi: 10.1002/hep.20439. [DOI] [PubMed] [Google Scholar]
- 60.Repa JJ, Lund EG, Horton JD, Leitersdorf E, Russell DW, Dietschy JM, Turley SD. Disruption of the sterol 27-hydroxylase gene in mice results in hepatomegaly and hypertriglyceridemia. Reversal by cholic acid feeding. J Biol Chem 275: 39685–39692, 2000. doi: 10.1074/jbc.M007653200. [DOI] [PubMed] [Google Scholar]
- 61.Repa JJ, Li H, Frank-Cannon TC, Valasek MA, Turley SD, Tansey MG, Dietschy JM. Liver X receptor activation enhances cholesterol loss from the brain, decreases neuroinflammation, and increases survival of the NPC1 mouse. J Neurosci 27: 14470–14480, 2007. doi: 10.1523/JNEUROSCI.4823-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rosenbaum AI, Zhang G, Warren JD, Maxfield FR. Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc Natl Acad Sci USA 107: 5477–5482, 2010. doi: 10.1073/pnas.0914309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rosenbaum AI, Cosner CC, Mariani CJ, Maxfield FR, Wiest O, Helquist P. Thiadiazole carbamates: potent inhibitors of lysosomal acid lipase and potential Niemann-Pick type C disease therapeutics. J Med Chem 53: 5281–5289, 2010. doi: 10.1021/jm100499s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rousset X, Shamburek R, Vaisman B, Amar M, Remaley AT. Lecithin cholesterol acyltransferase: an anti- or pro-atherogenic factor? Curr Atheroscler Rep 13: 249–256, 2011. doi: 10.1007/s11883-011-0171-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rudel LL, Lee RG, Parini P. ACAT2 is a target for treatment of coronary heart disease associated with hypercholesterolemia. Arterioscler Thromb Vasc Biol 25: 1112–1118, 2005. doi: 10.1161/01.ATV.0000166548.65753.1e. [DOI] [PubMed] [Google Scholar]
- 66.Sakai N, Vaisman BL, Koch CA, Hoyt RF Jr, Meyn SM, Talley GD, Paiz JA, Brewer HB Jr, Santamarina-Fojo S. Targeted disruption of the mouse lecithin:cholesterol acyltransferase (LCAT) gene. Generation of a new animal model for human LCAT deficiency. J Biol Chem 272: 7506–7510, 1997. doi: 10.1074/jbc.272.11.7506. [DOI] [PubMed] [Google Scholar]
- 67.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108, 2008. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 68.Somers KL, Brown DE, Fulton R, Schultheiss PC, Hamar D, Smith MO, Allison R, Connally HE, Just C, Mitchell TW, Wenger DA, Thrall MA. Effects of dietary cholesterol restriction in a feline model of Niemann-Pick type C disease. J Inherit Metab Dis 24: 427–436, 2001. doi: 10.1023/A:1010588112003. [DOI] [PubMed] [Google Scholar]
- 69.Su K, Donaldson E, Sharma R. Novel treatment options for lysosomal acid lipase deficiency: critical appraisal of sebelipase alfa. Appl Clin Genet 9: 157–167, 2016. doi: 10.2147/TACG.S86760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tamura A, Yui N. Polyrotaxane-based systemic delivery of β-cyclodextrins for potentiating therapeutic efficacy in a mouse model of Niemann-Pick type C disease. J Control Release 269: 148–158, 2018. doi: 10.1016/j.jconrel.2017.11.016. [DOI] [PubMed] [Google Scholar]
- 71.Taylor AM, Liu B, Mari Y, Liu B, Repa JJ. Cyclodextrin mediates rapid changes in lipid balance in Npc1−/− mice without carrying cholesterol through the bloodstream. J Lipid Res 53: 2331–2342, 2012. doi: 10.1194/jlr.M028241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Turley SD, Valasek MA, Repa JJ, Dietschy JM. Multiple mechanisms limit the accumulation of unesterified cholesterol in the small intestine of mice deficient in both ACAT2 and ABCA1. Am J Physiol Gastrointest Liver Physiol 299: G1012–G1022, 2010. doi: 10.1152/ajpgi.00190.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Uronen R-L, Lundmark P, Orho-Melander M, Jauhiainen M, Larsson K, Siegbahn A, Wallentin L, Zethelius B, Melander O, Syvänen A-C, Ikonen E. Niemann-Pick C1 modulates hepatic triglyceride metabolism and its genetic variation contributes to serum triglyceride levels. Arterioscler Thromb Vasc Biol 30: 1614–1620, 2010. doi: 10.1161/ATVBAHA.110.207191. [DOI] [PubMed] [Google Scholar]
- 74.van Heek M, Compton DS, Davis HR. The cholesterol absorption inhibitor, ezetimibe, decreases diet-induced hypercholesterolemia in monkeys. Eur J Pharmacol 415: 79–84, 2001. doi: 10.1016/S0014-2999(01)00825-1. [DOI] [PubMed] [Google Scholar]
- 75.Valasek MA, Repa JJ. The power of real-time PCR. Adv Physiol Educ 29: 151–159, 2005. doi: 10.1152/advan.00019.2005. [DOI] [PubMed] [Google Scholar]
- 76.Vance JE, Karten B. Niemann-Pick C disease and mobilization of lysosomal cholesterol by cyclodextrin. J Lipid Res 55: 1609–1621, 2014. doi: 10.1194/jlr.R047837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis 5: 16, 2010. doi: 10.1186/1750-1172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vanier MT. Biochemical studies in Niemann-Pick disease. I. Major sphingolipids of liver and spleen. Biochim Biophys Acta 750: 178–184, 1983. doi: 10.1016/0005-2760(83)90218-7. [DOI] [PubMed] [Google Scholar]
- 79.Vite CH, Bagel JH, Swain GP, Prociuk M, Sikora TU, Stein VM, O’Donnell P, Ruane T, Ward S, Crooks A, Li S, Maudlin E, Stellar S, De Meulder M, Kao ML, Ory DS, Davidson CD, Vanier MT, Walkley SU. Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci Transl Med 7: 276ra26, 2015. doi: 10.1126/scitranslmed.3010101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xie C, Turley SD, Dietschy JM. Cholesterol accumulation in tissues of the Niemann-pick type C mouse is determined by the rate of lipoprotein-cholesterol uptake through the coated-pit pathway in each organ. Proc Natl Acad Sci USA 96: 11992–11997, 1999. doi: 10.1073/pnas.96.21.11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xie C, Turley SD, Pentchev PG, Dietschy JM. Cholesterol balance and metabolism in mice with loss of function of Niemann-Pick C protein. Am J Physiol 276: E336–E344, 1999. doi: 10.1152/ajpendo.1999.276.2.E336. [DOI] [PubMed] [Google Scholar]
- 82.Xie C, Burns DK, Turley SD, Dietschy JM. Cholesterol is sequestered in the brains of mice with Niemann-Pick type C disease but turnover is increased. J Neuropathol Exp Neurol 59: 1106–1117, 2000. doi: 10.1093/jnen/59.12.1106. [DOI] [PubMed] [Google Scholar]