

Abstract
Necrotizing enterocolitis (NEC) is a deadly disease that occurs in 5–10% of neonates. Although NEC has been extensively studied, no single therapeutic target has been identified. Rho kinase (ROCK) is a serine/threonine kinase that affects multiple cellular processes, including tight junction (TJ) function, cellular permeability, and apoptosis. We hypothesized that ROCK inhibition would decrease cellular permeability, stabilize TJ proteins (occludin), and decrease the severity of NEC. To test this hypothesis, human colon epithelial cells (Caco-2) and human endothelial cells were studied. Cells were treated with lipopolysaccharide to simulate an in vitro model of NEC. The effect of ROCK inhibition was measured by transepithelial membrane resistance (TEER) and cellular permeability to FITC-dextran. The effects of ROCK inhibition in vivo were analyzed in the rat pup model of NEC. NEC was induced by feeding formula supplemented with Cronobacter sakazakii with or without gavaged ROCK inhibitor. Rat intestines were scored based on histological degree of injury. RNA and protein assays for occludin protein were performed for all models of NEC. Treatment with ROCK inhibitor significantly decreased cellular permeability in Caco-2 cells and increased TEER. Intestinal injury scoring revealed decreased scores in ROCK inhibitor-treated pups compared with NEC only. Both cell and rat pup models demonstrated an upregulation of occludin expression in the ROCK inhibitor-treated groups. Therefore, we conclude that ROCK inhibition protects against experimental NEC by strengthening barrier function via upregulation of occludin. These data suggest that ROCK may be a potential therapeutic target for patients with NEC.
NEW & NOTEWORTHY These studies are the first to demonstrate an upregulation of occludin tight junction protein in response to Rho kinase (ROCK) inhibition. Furthermore, we have demonstrated that ROCK inhibition in experimental models of necrotizing enterocolitis (NEC) is protective against NEC in both in vitro and in vivo models of disease.
Keywords: necrotizing enterocolitis, occludin, Rho kinase inhibitors, tight junctions
INTRODUCTION
Necrotizing enterocolitis (NEC) is a devastating gastrointestinal disorder of premature newborn infants, affecting between 5 and 10% of neonates admitted to the neonatal intensive care unit. NEC is associated with worsening neurodevelopmental outcomes and higher mortality rates in those affected. Preventative strategies, including breastmilk feeding and probiotics, have been investigated as potential therapeutic options (40); however, NEC continues to occur in medical units, and no single therapeutic has proved uniformly successful. Despite research by other investigators, the exact cause of NEC remains unclear. However, two important factors are implicated in NEC: the presence of Gram-negative bacteria and the loss of intestinal barrier function and integrity. In particular, Gram-negative bacteria, such as Cronobacter sakazakii (CS), have been linked to NEC outbreaks (25) and are known to impact intestinal barrier integrity (26, 27).
It is generally accepted that loss of normal intestinal barrier function is fundamental to the pathogenesis of inflammatory bowel disease and NEC (10, 19). The integrity of the intestinal barrier is maintained by multi-protein complexes called tight junctions (TJs). TJs develop intercellular points of contact that mediate intestinal barrier strength and the movement of water and solutes. TJs are crucial in mediating the dynamic changes of cellular permeability and cellular strength. TJs are thought to be more “leaky” in premature neonates, which may contribute to their susceptibility to NEC (40).
Both endothelial and epithelial intestinal barriers are important in the development of NEC. Highly permeable epithelial and endothelial barriers allow for increased flux of inflammatory mediators, bacterial translocation, and breakdown of intestinal architecture (33). The importance of the endothelial intestinal barrier in inflammatory bowel disease as a second line of defense against bacterial invasion and subsequent inflammatory response has been previously described (22, 38, 39). Recent mouse data have shown that the gut vascular barrier can independently inhibit bacterial invasion through its signaling mechanisms (37). The endothelium, similar to the epithelium, is maintained by TJs that mediate paracellular transport of macro- and micromolecules (29).
TJs are maintained by intracellular signaling pathways, such as those involving Rho and Rho kinase (ROCK) (43). ROCKs are serine/threonine kinases that regulate the actomyosin filaments and affect multiple cellular processes, including motility, phagocytosis, and apoptosis, through the dynamic changes in the actomyosin filaments (35). TJs include zona occludens, claudins, occludin, and junctional adhesion proteins. Within this group of TJ proteins, some are barrier forming whereas others are pore forming, allowing for the regulation of barrier permeability (20). The ROCK pathway regulates TJs, specifically occludin (23), which is a barrier-forming protein that is critical in regulating gut barrier permeability (5, 16, 18). Previous data have shown that occludin gene expression is downregulated in human NEC samples (43). The Rho pathway affects TJs in both endothelial and epithelial cell lines (42, 44). Constitutive activation of Rho GTPases has been shown to cause displacement of TJs from the cellular membrane to internal locations in epithelial cells (24). The aim of this study was to test whether inhibition of ROCK induces changes in the TJ protein occludin using in vitro and in vivo (rat pup) models of NEC (8). We hypothesized that activation of the Rho pathway would be detrimental to barrier integrity and the pharmacologic use of ROCK inhibitors would prevent experimental NEC by preserving occludin structure and function.
MATERIALS AND METHODS
Materials.
Y-27632, a ROCK inhibitor (No. Y-0503, Sigma, St. Louis, MO) was reconstituted in hydroclone water (stock concentration of 43.7 mM). Fasudil hydrochloride (FAS; No. CDS-021620, Sigma), a second ROCK inhibitor, was similarly reconstituted (stock concentration 100 μM). CS, clinical strain BAA-894 (American Type Culture Collection, Manassas, VA), was grown at 37°C in Luria broth overnight, centrifuged (3,000 revolutions/min), and washed twice in saline before being added at 1 × 108 colony-forming units/ml of rat pup formula [15 g Similac 60/40 (Ross Pediatrics) in 75 ml of Esbilac canine milk replacer (Pet-Ag)]. Lipopolysaccharide (LPS) from Escherichia coli clinical strain 0111:B4 (Sigma) was dissolved in normal saline (stock concentration of 1 mg/ml). Antibodies were obtained from the following sources: mouse anti-β-actin (1:50,000; No. A-5441, Sigma), rabbit anti-GAPDH (14C10; 1:1,000; No. 2118-S, Cell Signaling Technology, Danvers, MA), mouse anti-CD31 (1:500; No. MA-1-80069, Abcam, Cambridge, MA), mouse anti-occludin (1:1,000; No. 33-1500, Invitrogen, Carlsbad, CA), rabbit anti-ROCK1 [EP786Y] (1:500; ab45171, Abcam) for Western blot analysis, rabbit anti-occludin (1:500; No. 71-1500, Life Technologies, Carlsbad, CA) for immunofluorescence, and rabbit anti-phosphorylated myosin light chain (pMLC; 1:250; No. 3671, Cell Signaling Technology). Two human shRNA plasmids for ROCK1 knockdown were obtained from Sigma: shRNA SHCLND-NM_005406: TRCN0000194903 Vector pLKO.1 (Plasmid 1) and shRNA SHCLND-NM_005406: TRCN0000121093 Vector pLKO.1 (Plasmid 2).
Human samples.
Human samples from control patients and patients with NEC were obtained from patients undergoing resection of bowel during surgery at Ann and Robert H. Lurie Children’s Hospital. Control samples were taken from patients undergoing ostomy reversal procedures and/or intestinal resections for congenital atresia, whereas NEC samples were obtained from neonates with severe (Bell’s stage 3) NEC. The gestational age, tissue location (jejunum, ileum, etc.), and type of surgical procedure were recorded. These samples were obtained from children less than 1 yr of age on Institutional Review Board protocol (IRB No. 2013-15152). Tissue was collected in liquid nitrogen and snap frozen or preserved on optimal cutting temperature media (Sakura Finetek, Torrance, CA) at −80°C.
Animal studies.
Timed-pregnant Sprague-Dawley rats (Charles River) were induced near term at E21 with a subcutaneous injection of Pitocin 0.1–0.3 units. Rat pups were collected and separated into the experimental groups described below. The rat pups were gavaged 0.2 ml formula [15 g Similac 60/40 (Ross Pediatrics) in 75 ml of Esbilac canine milk replacer (Pet-Ag Inc.)] 3 times daily. Pups were exposed to hypoxia (5% O2-95% N2) for 5 min twice daily in a modular chamber (Billups-Rothenberg, Del Mar, CA). Rat pups in the NEC group had their formula supplemented with CS (2, 25, 27) to enhance the degree and incidence of experimental NEC. Experimental groups included controls of those fed clean rat pup formula (clean); a ROCK inhibitor (RI) + rat pup formula group with 3 mg/kg of Y-27632 (RI); a group with RI and CS bacteria in the rat pup formula (RI + NEC); and a group with CS bacteria in the rat pup formula (NEC). Four days after birth, rat pups were euthanized. The intestine and mesentery of these pups were collected for analysis. NEC was graded microscopically by a pediatric pathologist blinded to groups from grade 0 (normal) to 3 (severe) on the basis of pathological manifestations including submucosal edema, epithelial sloughing, abnormal intestinal villus architecture, and necrosis. Scores greater than or equal to 1.5 were associated with NEC. Animals were housed in the Northwestern University facilities that were fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. All procedures and protocols were approved by Northwestern University Institutional Animal Care and Use Committee and were conducted in accordance with guidelines set forth by the Guide for the Care and Use of Laboratory Animals.
Cell culture.
Human umbilical vein endothelial cells (HUVECs) were grown in Endothelial Cell Growth Kit Bovine Brain Extract (American Type Culture Collection) supplemented with 10% fetal bovine serum (FBS). The HUVECs were initially serum starved with 2% FBS. The human intestinal epithelial cell line, Caco-2, was grown in DMEM/F12 and 10% FBS. Both cell lines were plated at confluence on 24-well, 6.5 mm transwells (0.4 μm polycarbonate, Corning, Corning, NY). Each cell line was exposed to 4 conditions: control cells were not treated, RI group was given media supplemented with ROCK inhibitor Y-27632 at 5 μM or 10 μM, LPS group was treated with media supplemented with 100 µg/ml LPS, and an RI + LPS group was treated with media supplemented with RI and LPS at the concentrations described. These experiments were repeated with a different ROCK inhibitor, FAS at 10 μM, maintaining the same group conditions: control, FAS, LPS, and FAS + LPS. In experiments lasting greater than 24 h, the media with reagents were changed every 48 h. Cell cultures were maintained within the protocols of standard laboratory standards and practices.
Permeability studies.
Cells were pretreated for 1 h with Y-27632 or FAS at 10 μM concentration and then challenged with 100 µg/ml LPS. Twenty-four hours after the addition of LPS, 3-kDa fluorescein isothiocynate-labeled dextran was added to the apical layer, and the plate was placed in an incubator. After 2 h, the basal layer was collected and analyzed in triplicate using a fluorescent plate reader (Molecular Devices GeminiXS). The concentration of fluorescein isothiocynate-dextran in the basal layer was compared with the concentration applied to the apical layer.
Transcellular electrical resistance measurements.
Transendothelial and transepithelial electrical resistance (TEER) was measured in HUVECs and Caco-2 cells. Once cells had established TJs, Y-27632 or FAS was added to the apical surface of the cells for a 1-h pretreatment at a concentration of 10 μM. Controls were not exposed to Y-27632 or FAS. At subsequent intervals, the TEER was measured with a voltohmmeter (EVOM2, World Precision Instruments, Sarasota, FL). TJs were disrupted with addition of 100 µg/ml of LPS to the apical and basal layer of each transwell. TEER was measured with the voltohmmeter and compared with controls.
Immunofluorescence of intestine and mesentery.
Cells were grown and were plated at confluence on 24-well, 6.5 mm transwells as previously described in Cell culture. The cell membranes were washed with cold phosphate-buffered saline (PBS), fixed with 2% paraformaldehyde, and blocked with PBS/Triton and 5% normal goat serum. The cellular membrane was incubated with primary anti-occludin antibody (Invitrogen) or anti-CD31 antibody (Abcam) at 4°C overnight. The membrane was washed with PBS/Tween and then incubated with Alexa Fluor 488 conjugated at room temperature. The membrane was mounted with Fluoroshield with 4′,6-diamidino-2-phenylindole dihydrochloride (Sigma) and examined with a fluorescent microscope (Nikon Eclipse 90i and Nikon A1R microscope). Mean fluorescent intensity was calculated with FIJI ImageJ, and differences between groups were analyzed with ANOVA (GraphPad v. 7).
Western blot analysis.
Segments of rat pup intestine were weighed and suspended in cell lysis buffer (Cell Signaling Technology) [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM glycerophosphate, 1 mM Na3VO4, 1 µg/ml leupeptin], 1 mM PMSF, protease inhibitors [1.02 mM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF), 0.0008 mM aprotinin, 0.02 mM leupeptin, 0.04 mM bestatin, 0.015 mM pepstatin A, 0.014 mM E-64], phosphatase inhibitors (sodium vanadate, sodium molybdate, sodium tartrate), and imidazole (Sigma). Samples were homogenized on ice. After centrifugation at 4°C, supernatants were removed and stored at −80°C.
Protein was isolated from cellular monolayers grown on 12-well plates, the media was removed, 1 ml of PBS was added, and cells were scraped and transferred to a microfuge tube. Samples were microfuged at 5,000 revolutions/min at 4°C for 10 min and the supernatants removed. The cell pellets were resuspended in 500 μl of cell lysis buffer (as above in Western blot analysis). The mixture was then drawn through a 27-gauge needle and gently mixed on a rotating platform for 30 min at 4°C, followed by centrifugation 4°C for 15 min at 10,000 revolutions/min. The supernatants were removed and stored at −80°C.
Tissue and cellular samples were thawed on ice. 5× Laemmli SDS sample buffer was added and then boiled for 3 min. Samples were run on a Bio-Rad Mini-PROTEAN apparatus (Bio-Rad, Hercules, CA) at 30 mA and then transferred to a nitrocellulose membrane using standard approaches. Membranes were blocked for 1 h at room temperature by incubation with PBS-Tween containing 5% nonfat dry milk. Primary and secondary antibodies were diluted in PBS-Tween containing 5% nonfat dry milk. Protein bands were detected using ECL Western blotting detection reagents (GE Healthcare Life Sciences, Piscataway, NJ) on Amersham Hyperfilm ECL (GE Healthcare Life Sciences). Densitometry was preformed using Bio-Rad Image Laboratory software and normalized with β-actin or GAPDH. Data were collected for individual tissues and cell cultures in triplicate and reported as means ± SE.
Flow cytometry.
Cells were grown on 12-well plates and treated with varying concentrations of ROCK inhibitors. Monolayers were washed with cold PBS. Adherent cells were trypsinized, then centrifuged at 300 g at 4°C for 5 min, and the supernatants were removed. Cell pellets were suspended in 1X Binding Buffer (PE Annexin V Apoptosis Detection Kit, BD Biosciences, Franklin Lakes, NJ). Annexin V and either 7-amino-actinomycin (7-ADD) (BD Biosciences) or 4′,6-diamidino-2-phenylindole dihydrochloride (BD Biosciences) were added to cells and incubated at room temperature for 15 min. Cells were then analyzed by flow cytometry using BD LSR Fortessa Analyzer.
Quantitative reverse transcription-polymerase chain reaction.
Real-time quantitative polymerase chain reaction (qPCR) identified the expression levels of the TJ occludin in both cell lines and in the rat pup tissues. Total RNA was collected using the RNeasy Mini Kit (Qiagen, Courtaboeuf, France). RNA was converted into cDNA using GeneAMp RNA PCR Core (Life Technologies). Quantitative reverse transcription (qRT)-PCR was performed with equal amounts of cDNA using Bio-Rad CFX (Bio-Rad) with iQ SYBR Green Supermix (Bio-Rad). Normalization of the cycle threshold gene was done with respect to gene GAPDH. Primers were designed with National Institutes of Health Primer BLAST. Primer sequences are as follows: GAPDH (human) – forward: 5′-ACCACAGTCCATGCCATCAC-3′, reverse: 5′-TCCACCACCCTGTTGCTGTA-3′; occludin (human) – forward: 5′-TCAGGGAATATCCACCTATCACTTCAG-3′, reverse: 5′-CATCAGCAGCAGCCATGTACTCTTCAC-3′; GAPDH (rat) - Forward: 5′-ATCACCATCTTCCAGGAGCG-3′, reverse: 5′-TTCTGAGTGGCAGTGAGGGC-3′; occludin (rat) – forward: 5′-ACAGGTGGCGAGTCCTGCGA-3′, reverse: 5′-GCAGCAGCCATGTACTCTTCGCTC-3′.
Transfection.
Caco-2 and HUVECs were seeded in 24-well plates at 105 cells/well. They were transfected with Lipofectamine 2000 (LifeTech, Elmhurst, IL) and ROCK1 shRNA Plasmid 1 SHCLND-NM_005406: TRCN0000121093, ROCK1 shRNA Plasmid 2 SHCLND-NM_005406: TRCN000019490 (LifeTech), or sham shRNA SHC002V (LifeTech) for 72 h followed by stable selection with puromycin. Knockdown was confirmed by Western blot analysis for ROCK1 and qRT-PCR with two different ROCK1 primers: ROCK1 (human) – forward: 5′-TTGTTTGAACAGGAAGGCGGA-3′, reverse: 5′-GCCCGATGGAGACTTAGCAG-3′; and ROCK-1 (h)-PR (No. sc-29473-PR, Santa Cruz Biotechnology, Dallas, TX). After confirmation of knockdown by PCR and Western blot analysis, cells were cultured with complete media and 5 μg/ml puromycin and treated with LPS for 24 h versus controls without LPS as per our protocol.
Data analysis.
Student’s t-test and/or ANOVA were used to examine statistical differences where appropriate between groups. Data are presented as means ± SE. A statistically significant difference was accepted at P < 0.05.
RESULTS
Occludin is decreased in human intestinal samples of NEC.
To establish whether intestinal levels of occludin are changed in neonates with advanced NEC, we collected bowel samples from patients with active NEC and control specimens from patients with intestinal atresia or ostomy reversal. We evaluated occludin in our control and NEC samples obtained from neonatal patients less than 1 yr of age. As shown in Fig. 1, qRT-PCR revealed that control samples exhibited significantly higher expression of occludin compared with their NEC counterparts (P < 0.05). To determine if RNA levels correlated with the protein expression, we performed Western blot analysis of occludin in both control patients and patients with NEC, which also revealed a significant downregulation of occludin TJ protein in patients with NEC (P < 0.05).
Fig. 1.

Occludin gene and protein expression is decreased in bowel samples of patients with NEC. A: occludin gene expression in human samples was assessed via quantitative PCR. Patients with NEC had significantly lower occludin expression compared with control samples (*P < 0.05 vs. control), n = 3 per group. B: occludin protein production in human samples was analyzed by Western blot. Samples obtained from patients with NEC had significantly lower occludin protein compared with control patients (*P < 0.05), n = 3 per group. Western blot shows increased band density of occludin in control group. NEC, necrotizing enterocolitis.
LPS upregulates pMLC, and ROCK inhibition decreases production of pMLC in HUVECs and Caco-2 cells.
Given that ROCK has been recognized as important in the regulation of occludin (45), we sought to investigate whether ROCK was upregulated in our models of NEC. As a widely accepted proxy of ROCK activation, we assayed phosphorylation of myosin light chain (MLC) (31). As described in materials and methods, endothelial and epithelial cell lines were exposed to LPS and RI in various combinations. pMLC was evaluated by Western blot analysis. Fig. 2, A and B demonstrate an increase in pMLC in LPS-treated cells in both the endothelial and intestinal epithelial cell lines. Therefore, LPS treatment upregulates pMLC, and ROCK inhibitor Y-27632 downregulates pMLC during experimental NEC.
Fig. 2.
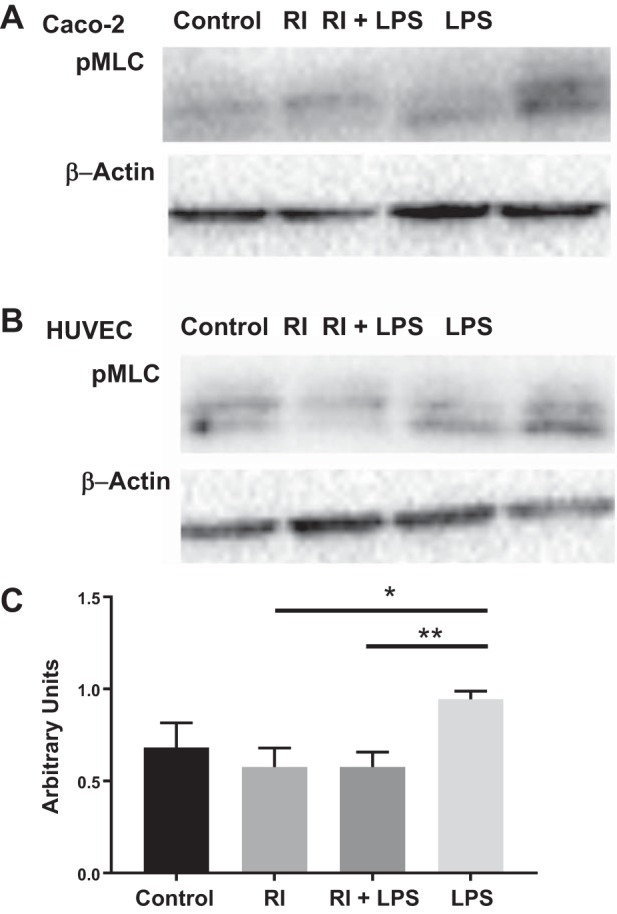
LPS increases production of phosphorylated myosin light chain (pMLC) in both HUVECs and Caco-2 cells (A and B). ROCK inhibition in the presence of LPS decreases pMLC production in both HUVECs and Caco-2 cells to values similar to that of the controls (C; **P < 0.005). Western blot of pMLC in both HUVEC and Caco-2 cells are shown with the corresponding β-actin blot. *P < 0.05. LPS, lipopolysaccharide; RI, ROCK inhibitor; ROCK, Rho kinase.
Dose response curve of ROCK inhibitor.
Given that ROCK activation (upregulation of pMLC) is significantly elevated in LPS-treated (NEC) cells, we hypothesized that inhibition may lead to protective effects against NEC. ROCK is involved in multiple cellular processes, including apoptosis, which may alter intestinal barrier function by mechanisms other than TJ modulation. To exclude apoptosis as the primary reason for changes in intestinal permeability, we examined the effects of ROCK inhibition on cellular death. A range of concentrations of ROCK inhibitors (RI and FAS at 5 μM, 10 μM, and 100 μM) were tested on both the HUVEC and Caco-2 cell lines. Apoptosis was assessed by flow cytometry for Annexin V. Experiments were completed in triplicate. In both cell lines, doses equal to or greater than 100 μm RI or 50 μM FAS significantly increased cellular death compared with controls (P < 0.05; Fig. 3). The overall effect of FAS on HUVEC apoptosis was minimal, though significant at doses greater than 50 μM FAS (Fig. 3D). Thus, we selected doses for use in our experiments at which no significant increase in cellular injury or apoptosis was identified.
Fig. 3.

Increased concentrations of ROCK inhibitor are associated with more cellular death. A: flow cytometry performed on Caco-2 cells with different concentrations of ROCK inhibitor (RI). Q3 shows percentage of cells alive; Q2 shows percentage of cells in late stage apoptosis. Controls were untreated. Decreased percentage of cells alive with increasing concentration of RI, with 100 μM RI being significantly different from control (*P < 0.05 vs. control). B: a similar trend is seen in HUVECs (*P < 0.05 vs. control). C: flow cytometry on Caco-2 cells treated with fasudil hydrochloride (FAS) demonstrate increased apoptosis after treatment with 100 μM FAS vs. controls (*P < 0.05). D: HUVECs showed increased apoptosis after treatment with 50 μM FAS (*P < 0.05). Values are means ± SE; ROCK, Rho kinase.
ROCK inhibition increases occludin in HUVECs.
To examine the effects of ROCK inhibition on the changes in TJs during endothelial LPS-mediated cellular injury, we utilized qRT-PCR to measure RNA expression of occludin in four groups of HUVECs, as previously described. As shown in Fig. 4A, we found that occludin gene expression was decreased in LPS-treated cells compared with controls (P < 0.005). Both RI and RI + LPS groups had upregulated levels of occludin. The RI + LPS group was similar to control. To determine if RNA levels correlated with the protein expression, we performed Western blot analysis of occludin. Figure 4B shows that there is increased band density in both RI groups. A trend in the control of higher occludin protein expression compared with that of LPS-treated cells was found. Both RI groups had significantly higher occludin expression compared with LPS (P < 0.05) but were not significantly different from control. An alternative ROCK inhibitor, FAS, was used to examine whether our findings were more likely because of ROCK inhibition rather than off-target pharmacological effects specific to Y-27632. Similar changes were seen in RNA and protein expression of occludin. As shown in Fig. 4C, there was increased occludin gene expression compared with controls (P < 0.05) 4 h after FAS administration. Occludin gene expression in LPS-treated cells was not downregulated at 4 h, but a significant effect was seen at 24 h (P < 0.05; Fig. 4D), as previously seen in the RI experiments. Occludin gene expression in those treated with LPS was decreased compared with the FAS + LPS treatment group at 24 h (P < 0.0005). Western blot analysis (Fig. 4E) shows increased band density in cells treated with FAS or FAS + LPS compared with LPS alone (P < 0.005). There was no difference between control and FAS in occludin protein expression. Therefore, we concluded that ROCK inhibition increases both gene and protein expression of occludin in HUVECs.
Fig. 4.

ROCK inhibition increases occludin gene and protein expression in HUVECs. A: occludin gene expression in HUVECs was assessed by quantitative PCR. LPS had significantly less occludin expression compared with controls (**P < 0.005). RI was significantly higher than controls (**P < 0.005), and RI + LPS was the same as control. RI + LPS was significantly higher than LPS (**P < 0.005). B: occludin protein expression in HUVECs was assessed via Western blot. A representative Western blot shows that control, RI, and RI + LPS groups had more occludin than LPS-treated cells. C and D: occludin gene expression in HUVECs is increased early after 4 h of FAS administration compared with controls (*P < 0.05); LPS-treated group had decreased occludin gene expression by 24 h, whereas expression in FAS + LPS treated cells remains increased vs. LPS (*P < 0.0005). E: occludin protein expression using FAS instead of RI shows more occludin in FAS (*P < 0.05) and FAS + LPS (**P < 0.005) groups compared with LPS-treated cells. *P < 0.05, **P < 0.005, ***P < 0.0005. FAS, fasudil hydrochloride; LPS, lipopolysaccharide; RI, ROCK inhibitor; ROCK, Rho kinase.
ROCK inhibition increases occludin in Caco-2 cells.
To identify the effects of ROCK inhibition during intestinal epithelial cellular injury on occludin, we measured RNA expression of occludin in our 4 experimental groups of Caco-2 cells (controls were untreated, RI were treated with 10 μM Y-27632, LPS cells were treated with 100 µg/ml of LPS, and RI + LPS were treated with both LPS and Y-27632). As shown in Fig. 5A, we found that occludin gene expression was increased in both RI groups compared with control (P < 0.05). The addition of RI to LPS-treated cells increased occludin significantly compared with LPS-treated cells (P < 0.005). The similar quantities of RNA expression found in the LPS and control samples may be because LPS-mediated downregulation of occludin does not occur until a later time point, i.e., at 120 h in Caco-2 cells (Fig. 5B). To determine if RNA levels correlated with the protein expression, we performed Western blot analysis of occludin. This analysis identified increased occludin protein in both RI treatment groups (Fig. 5C). ROCK inhibition increases both gene and protein expression of occludin in Caco-2 cells compared with both control and LPS-treated cells (P < 0.05). These findings were confirmed using a different ROCK inhibitor (10 μM FAS) instead of RI. Western blot protein analysis showed a trend toward decreased band density of occludin in LPS-treated cells compared with controls, as shown in Fig. 5D. The addition of FAS increased the levels of occludin protein expression compared with LPS (P = 0.08). This suggests that the inhibition of ROCK may strengthen the epithelial barrier via upregulation of occludin.
Fig. 5.

ROCK inhibition increases gene and protein expression of occludin in Caco-2 cells. A: occludin gene expression in Caco-2 is increased in both RI groups compared with control (*P < 0.05). With the addition of RI to LPS-treated cells, occludin gene expression increases when compared with LPS alone (**P < 0.005). B: LPS-treated Caco-2 cells had significantly decreased occludin gene expression compared with controls at 120 h (*P < 0.05). C: representative Western blot shows that both RI groups had increased occludin compared with LPS-treated cells. D: Western blot shows a trend toward decreased occludin in LPS-treated cells compared with controls, whereas the addition of FAS increases occludin protein expression (P = 0.08). FAS, fasudil hydrochloride; LPS, lipopolysaccharide; RI, ROCK inhibitor; ROCK, Rho kinase.
ROCK inhibition increases TEER and decreases permeability in Caco-2 cells.
Occludin is important in the maintenance of intestinal barrier function and integrity. To investigate the effects of ROCK inhibition on changes in cellular barrier strength, we monitored TEER and monolayer permeability in epithelial cells. Y-27632 was added to the apical layer of both cell lines at 10 μM concentration. LPS was subsequently added, and monolayer permeability was assessed after 24 h of incubation, as previously described. We identified that LPS decreased TEER in Caco-2 cells compared with control at 24 h (P < 0.0005; Fig. 6A). Furthermore, in the presence of RI, LPS treated cells were protected against the drop in TEER seen in cells treated with LPS alone by 24 h (P < 0.05). In the RI + LPS group, TEER dropped by 12% on average at 24 h. Of note, by 24 h RI increased TEER to levels similar to those of controls. To evaluate barrier function further, we assessed cellular permeability. Figure 6B shows increased cellular permeability in Caco-2 cells treated with LPS compared with controls (P < 0.05). However, ROCK inhibition decreased cellular permeability (P < 0.0005). Our previous data indicated that LPS altered occludin in Caco-2 cells by 120 h; therefore, we tested barrier permeability at 120 h. At 120 h, LPS increased permeability and RI decreased barrier permeability toward that of controls (Fig. 6B). Similar findings were identified when using FAS instead of RI for ROCK inhibition. Figure 6C shows a decrease in TEER in LPS-treated cells compared with controls (P < 0.0005), as expected from above results. The addition of FAS had a protective effect against this drop in TEER (P < 0.0005) at an earlier time point than seen in the RI + LPS group. Both FAS and FAS + LPS groups had no difference in TEER compared with controls. ROCK inhibition with FAS decreased cellular permeability (P < 0.005), as expected from the RI results. Figure 6D shows an increase in cellular permeability with LPS (P < 0.05) but a decrease in cells treated with FAS + LPS (P < 0.005). In HUVECs, ROCK inhibition had no effect on cellular permeability and TEER. Thus, ROCK inhibitors strengthen the epithelial cell barrier by reducing cellular permeability and augmentation of TEER in the intestinal epithelial Caco-2 cell line.
Fig. 6.

Permeability and TEER in Caco-2 cells. A: TEER in Caco-2 cells over 24 h in the 4 experimental groups: control, RI, RI+ LPS, and LPS. LPS significantly decreases TEER at 24 h compared with control (**P < 0.005). Addition of RI to LPS-treated cells increases TEER significantly compared with LPS-treated cells alone (*P < 0.05). B: Caco-2 cells treated with LPS have increased permeability compared with controls at 24 and 120 h (*P < 0.05). Addition of RI to LPS-treated cells decreases cellular permeability compared with LPS only treated cells both at 24 and 120 h (***P < 0.0005 and *P < 0.05, respectively). C: Caco-2 cells treated with LPS had increased TEER compared with controls at 4 h. FAS and FAS + LPS groups had no differences in TEER compared with controls (***P < 0.0005). D: similarly, there was increased permeability in LPS-treated cells that was significantly diminished in FAS-treated cells (**P < 0.005). FAS, fasudil hydrochloride; LPS, lipopolysaccharide; RI, ROCK inhibitor; ROCK, Rho kinase; TEER, transendothelial and transepithelial electrical resistance.
ROCK knockdown increases occludin in Caco-2 cells.
To exclude the possibility that occludin TJ changes occurred because of ROCK-independent off-target effects of Y-27632 and FAS, we employed genetic inhibition to knockdown ROCK in our Caco-2 cells. Prior to treatment with LPS, ROCK knockdown was confirmed using Western blot analysis of ROCK1. There was a 49% reduction in the ROCK1 protein expression in the shRNA knockdown cells compared with the sham shRNA controls, as shown in Fig. 7A. The occludin gene expression was then assayed and found to be significantly increased fourfold in the ROCK1 knockdown cells compared with wild type controls (P < 0.0005), as shown in Fig. 7B. Two different shRNA plasmids were used to create the knockdown cells, and both had similar results. To confirm these findings, protein analysis demonstrated increased occludin expression in knockdown cells treated with LPS compared with the sham controls, shown in Fig. 7C. Knockdown of ROCK appeared to enhance the production of occludin in response to LPS. To assay functionality in the ROCK1 knockdown experiment, TEER was measured for 24 h. There was a significant decrease in TEER in LPS-treated cells, as previously shown. However, ROCK1 knockdown cells with or without LPS treatment had increased TEER compared with LPS-treated cells and sham controls (P < 0.0005), as shown in Fig. 7D. Cellular permeability of ROCK1 knockdown cells showed no difference between controls and LPS-treated cells at 24 h (Fig. 7E). Therefore, we conclude that genetic ROCK inhibition increases occludin in Caco-2 cells.
Fig. 7.

Occludin is upregulated with genetic inhibition of ROCK in Caco-2 cells. A: ROCK1 protein expression in Caco-2 cells assayed with Western blot. Immunoblot density for sham shRNA cells and shRNA ROCK1 knockdown cells showed a 49% ROCK1 decrease in protein expression in knockdown cells compared with controls; representative immunoblots are shown for each group. B: occludin gene expression is increased fourfold following the knockdown of ROCK1 in Caco-2 cells compared with controls (n = 3; ***P < 0.0005). C: immunoblot density for occludin showed increased occludin expression in ROCK1 knockdown cells treated with LPS compared with untreated knockdown cells and sham controls. D: ROCK1 knockdown cells had increased TEER compared with sham controls (**P < 0.005). LPS-treated cells had significantly lower TEER compared with sham controls and with ROCK1 knockdown cells (***P < 0.0005). E: permeability to FITC-dextran was not different between ROCK1 knockdown Caco-2 cells treated with LPS vs. control. LPS, lipopolysaccharide; ROCK, Rho kinase; TEER, transendothelial and transepithelial electrical resistance.
ROCK inhibition increases occludin in the intestine of a rat pup model of NEC.
Given the improvement in barrier function in our cell line models treated with RI and the associated changes in occludin, we hypothesized that similar results would be seen in the rat pup model of NEC. Therefore, we evaluated whether rat pups with NEC demonstrated differences in intestinal occludin. Western blot analysis of rat intestine revealed that rat pups with NEC had decreased occludin in the intestine compared with the clean group and treatment group of RI + NEC (P < 0.05 and P < 0.005, respectively; Fig. 8A). To evaluate the subcellular location of occludin, we preformed immunofluorescence of the intestine. In Fig. 8B, we demonstrate that occludin (red) is preserved in the cellular membrane in the clean, RI, and RI + NEC group compared with the NEC group in both high- and low-power views. In the NEC group, the occludin (red) is internalized and no longer in the cellular membrane. These data show that ROCK inhibition stabilizes occludin protein in the epithelium of the rat intestine in an NEC model.
Fig. 8.

Occludin is upregulated with RI and localizes to the cellular membrane in the rat pup intestine. A: occludin protein expression in rat pup intestine assessed via Western blot. Rat pups inflicted with NEC had lower amounts of occludin protein in the intestine compared with RI + NEC, RI, and clean groups; n = 4 per group. B: immunofluorescence imaging of occludin (red) in the rat pup intestine. Preserved occludin staining on cellular membrane seen in clean, RI, and RI + NEC groups in both high- and low-power views. In the NEC group, there is breakdown of the villus architecture, and occludin is no longer part of the cellular membrane. **P < 0.005. Scale bar = 200 μm for ×10 images. NEC, necrotizing enterocolitis; RI, ROCK inhibitor, ROCK, Rho kinase.
ROCK inhibition protects against NEC in a rat pup model.
To determine whether enteral feeds supplemented with ROCK inhibitor Y-27632 would protect rat pups from NEC, we divided pups into four experimental groups: clean (formula), RI (formula + 3 mg/kg Y-27632), RI + NEC (formula + 3 mg/kg Y-27632 + CS), and NEC (formula + CS). Following treatment, intestinal segments were collected as previously described. In Fig. 9A, we demonstrate that the NEC group had a significantly higher average intestinal injury score compared with the clean formula group (P < 0.05). The RI + NEC group had a significantly lower average intestinal injury score from the NEC group (P < 0.05) that was comparable to injury scores found in the clean formula group. Figure 9B depicts representative hematoxylin and eosin staining of the four rat pup groups. The clean, RI, and RI + NEC groups demonstrate preserved villus architecture, whereas the NEC group had epithelial sloughing and no villus structure left. Therefore, addition of RI to the formula of the experimental NEC group protected pups against NEC.
Fig. 9.

RI decreases intestinal injury in a rat pup model of NEC. A: intestinal injury scoring of rat pup intestine from hematoxylin-eosin slides. Clean pups had significantly lower intestinal injury scores compared with the NEC group (*P < 0.05). Addition of RI to the formula decreased average intestinal injury scores in the RI + NEC group vs. NEC only (*P < 0.05); n = 17–19 pups per group. B: hematoxylin-eosin slides of rat pup intestine. Loss of villus architecture and epithelial sloughing seen in the NEC group. Preservation of epithelial barrier seen in clean, RI, and RI + NEC group. Scale bar = 200 μm. NEC, necrotizing enterocolitis; RI, ROCK inhibitor, ROCK, Rho kinase.
ROCK inhibition increases occludin in the rat mesentery in experimental NEC.
To evaluate the effects of RI on the endothelium in a rat pup model of NEC, we collected the mesentery of rat pups of each experimental group. Immunofluorescence for each of the described four treatment groups was performed. Occludin mean fluorescent intensity was significantly higher in both RI and RI + NEC compared with the clean (control) group, indicating increased occludin (P < 0.05; Fig. 10A). The mean fluorescent intensity of occludin in the clean and RI + NEC groups was significantly higher than NEC alone (P < 0.005 for both; Fig. 10B). To identify changes in gene expression, qRT-PCR for occludin was performed on samples of rat mesentery. As shown in Fig. 10C, RI and RI + NEC had significantly higher occludin RNA expression than clean control groups (P < 0.05 and P < 0.005, respectively). RI + NEC also had higher occludin expression compared with the NEC group (P < 0.005). Therefore, RI increases occludin in the mesentery in a rat model of NEC as evidenced by immunofluorescence and RNA expression.
Fig. 10.

Increased occludin gene expression and protein is found in the mesentery of intestines in a rat pup model of NEC. A: immunofluorescence imaging of mesentery in four treatment groups: clean, RI, RI + NEC and NEC groups. CD31 staining (red) was used as a marker for endothelial cells of arteries and veins of mesentery. Occludin staining (green) was brighter in RI groups indicating increased occludin, n = 3–6 per group. Scale bar = 20 μm. B: gene expression of occludin in rat pup mesentery. Clean vs. NEC (NS), clean vs. RI (**P < 0.005), clean vs. RI + NEC (**P < 0.005), RI + NEC vs. NEC (**P < 0.005); n = 4 pups per group. *P < 0.05. NEC, necrotizing enterocolitis; RI, ROCK inhibitor, ROCK, Rho kinase.
DISCUSSION
Loss of normal gut barrier function and integrity is pivotal to the pathogenesis of NEC. Preterm neonates have decreased membrane integrity, due to high intestinal permeability, that improves with increased gestational age (21). A normal increase in permeability of the neonatal intestine allows for the passage of antibodies and nutrients into the neonatal circulation; however, it also predisposes the intestine to bacterial invasion. The permeability of the intestine is regulated by the TJ of the epithelium of the brush border and the endothelium of the mesentery. TJ proteins, such as occludin, regulate the movement of leukocytes and other inflammatory mediators that contribute to barrier leakiness (29).
Previous data have shown that cellular permeability increases with increased phosphorylation of MLC, and inhibition of phosphorylation of MLC alleviates intestinal injury and stabilizes TJ (13). ROCK inhibitors decrease the phosphorylation of MLC by inactivating myosin phosphatase and relaxing the actin cytoskeleton (30). Our data show that ROCK inhibitors decrease phosphorylation of MLC in both endothelial and epithelial cells.
The Rho GTPase signaling system is important for both epithelial and endothelial permeability. RhoGTP cycles between an active and inactive form. When inflammatory mediators trigger the Rho GTPase signaling system, RhoGTP is activated. RhoGTP subsequently binds ROCK (43), which modulates multiple cellular functions through phosphorylation of serine/threonine residues affecting cellular motility, cellular permeability, and cellular death. ROCK is known to phosphorylate occludin in brain endothelial cells, leading to blood brain barrier dysfunction. Prior studies have implicated the ROCK pathway in TJ maintenance (14, 41, 42, 45).
ROCK inhibitors have been utilized to treat multiple disease processes affecting both the endothelium and epithelium. ROCK inhibitors have been approved for the treatment of cerebral vasospasm and glaucoma (17). FAS and Y-27632 are examples of ROCK inhibitors that competitively inhibit the ATP catalytic site of ROCK (3, 28). Although the ROCK inhibitors selected for this study are generally considered to be specifically ROCK inhibitors, it should be noted that pharmacological agents may have “off target” effects. For example, Y-27632 has been shown to inhibit the mitogen- and stress-activated protein kinase-1 also, which may mediate activation of CREB, an importance effector of downstream cell survival signaling previously implicated in NEC (9). To mitigate these off-target effects, we included investigation of genetic silencing of ROCK to demonstrate similar effects. Previous studies have shown the detrimental role of Rho in myocardial ischemia/reperfusion injury, demonstrating a potential need for ROCK inhibition (7). ROCK inhibitors have been tested on endotoxemic rat pups resulting in decreased intestinal cell apoptosis after drug administration (34) and decreased microvascular leak induced by LPS (32). The effect of Y-27632 was investigated in a hypoxia-reperfusion rat model of NEC (6). Although this study showed a positive impact of ROCK inhibition on rat intestinal injury and nitric oxide production, there was no analysis of the effects of ROCK inhibition on the endothelium or epithelium nor investigation into the effects of intestinal barrier integrity. We suspect that the effects of ROCK inhibition are complex and effect intestinal inflammation, permeability, and intestinal barrier breakdown during the development of NEC in neonates. Based on our data, ROCK inhibition mitigates the pathophysiologic effects of NEC and decreases intestinal injury.
Our data show that occludin is downregulated in the neonatal intestine of NEC patients (Fig. 1). Occludin plays an essential role in paracellular transport and regulation of macromolecules (1). Occludin is necessary not only for structural integrity but is also crucial as a signaling transmembrane protein (12). In both epithelial and endothelial cells, decreased occludin content is associated with increased barrier permeability (4, 46). Through its phosphorylation and dephosphorylation of serine/threonine residues, occludin changes the permeability of the cellular barrier (15, 23, 36). Therefore, an upregulation of occludin would be expected to increase intestinal barrier integrity as supported by our data. Moreover, NEC-related intestinal injury may be ameliorated.
Our data demonstrate that RI increases occludin in both Caco-2 and HUVECs. At 24 h, in Caco-2 cells we found that cellular permeability was decreased and TEER was increased with the addition of RI. We also observed that LPS-treated Caco-2 cells had a drop in cellular resistance at 24 h but no change in cellular permeability or occludin until 120 h. This suggests that LPS induces a change in TJ function by redistributing occludin before the downregulation of expression seen at 120 h. The addition of RI to LPS-treated cells helped mitigate both the decrease in TEER at 24 h and the subsequent rise in permeability at 120 h seen in Caco-2 cells. RI quickly upregulated occludin in Caco-2 cells in the first 24 h and decreased permeability of Caco-2 cells compared with untreated controls. In HUVECs, these effects were not identified, possibly because of the inherent “leaky” nature of the cell line and the low resistance generated by this cell line in culture. The differences seen between the two cell lines could also be accounted for by the inherent differences in TJs in epithelial and endothelial cell lines (47).
These observations corresponded with the changes identified in the rat pup model, which showed the upregulation of occludin with in the setting of NEC. Addition of RI to formula gavage feeds upregulated occludin levels in the intestine of NEC-treated pups. Subsequently, the RI + NEC group also had significantly decreased intestinal injury scores compared with the NEC only group. We believe that the protective effects of RI stem from its capacity to upregulate occludin at TJ sites. Therefore, we conclude that RI strengthens the intestinal barrier and protects against NEC.
TJ integrity safeguards the neonatal system from bacterial invasion. Understanding of both endothelial and epithelial models is beneficial to gain insight into the multifactorial pathogenesis of NEC. Epithelial cells are the first barrier against bacterial invasion, whereas a disrupted endothelium promotes intestinal edema and further tissue injury (11). Analysis of the rat intestinal mesentery showed increased occludin expression and immunofluorescence of occludin in blood vessels in both ROCK inhibitor groups. Our in vivo model showed increased occludin in both the mesentery and intestine, which mirrored our in vitro data in HUVECs and Caco-2 cells. Therefore, the effects of occludin are unlikely to be limited to a single cell type but may instead play a role in a complex system. It is also possible that other TJ proteins may be involved during the pathogenesis of NEC, and this will be an area of future study. Given our findings, we propose that ROCK inhibitors should be investigated further to promote intestinal health in NEC.
GRANTS
This work was supported by the Northwestern University]\-Flow Cytometry Core Facility supported by Cancer Center Support Grant NCI CA060553. Funding support was received from the American Pediatric Surgical Association (to C. J. Hunter) and National Institute of Diabetes and Digestive and Kidney Diseases K08-DK-106450 (to C. J. Hunter).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.S.G. and C.J.H. conceived and designed research; J.S.G., G.A., C.Y., and D.R.W. performed experiments; J.S.G., G.A., C.Y., D.R.W., and C.J.H. analyzed data; J.S.G., G.A., C.Y., D.R.W., and C.J.H. interpreted results of experiments; J.S.G., G.A., and C.Y. prepared figures; J.S.G. and C.J.H. drafted manuscript; G.A. and C.J.H. edited and revised manuscript; J.S.G., G.A., C.Y., D.R.W., and C.J.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge Dr. Pauline Chou, Staff Pathologist at Ann and Robert H. Lurie Children’s Hospital, Linda Zekas, APN, and the staff at the core imaging facility at Northwestern University.
REFERENCES
- 1.Al-Sadi R, Khatib K, Guo S, Ye D, Youssef M, Ma T. Occludin regulates macromolecule flux across the intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol 300: G1054–G1064, 2011. doi: 10.1152/ajpgi.00055.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almajed FS, Forsythe SJ. Cronobacter sakazakii clinical isolates overcome host barriers and evade the immune response. Microb Pathog 90: 55–63, 2016. doi: 10.1016/j.micpath.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 3.Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton 67: 545–554, 2010. doi: 10.1002/cm.20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antonetti DA, Barber AJ, Khin S, Lieth E, Tarbell JM, Gardner TW; Penn State Retina Research Group . Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content: vascular endothelial growth factor decreases occludin in retinal endothelial cells. Diabetes 47: 1953–1959, 1998. doi: 10.2337/diabetes.47.12.1953. [DOI] [PubMed] [Google Scholar]
- 5.Balda MS, Flores-Maldonado C, Cereijido M, Matter K. Multiple domains of occludin are involved in the regulation of paracellular permeability. J Cell Biochem 78: 85–96, 2000. doi: 10.1002/(SICI)1097-4644(20000701)78:1<85::AID-JCB8>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 6.Balyemez G, Sivasli E, Ceylan H, Tutar E, Ekiz S, Tarakcioglu M, Demiryurek AT, Coskun MY. Protective effects of Y-27632 on hypoxia/reoxygenation-induced intestinal injury in newborn rats. J Pediatr Surg 46: 1490–1494, 2011. doi: 10.1016/j.jpedsurg.2010.11.034. [DOI] [PubMed] [Google Scholar]
- 7.Bao W, Hu E, Tao L, Boyce R, Mirabile R, Thudium DT, Ma XL, Willette RN, Yue TL. Inhibition of Rho-kinase protects the heart against ischemia/reperfusion injury. Cardiovasc Res 61: 548–558, 2004. doi: 10.1016/j.cardiores.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Barlow B, Santulli TV. Importance of multiple episodes of hypoxia or cold stress on the development of enterocolitis in an animal model. Surgery 77: 687–690, 1975. [PubMed] [Google Scholar]
- 9.Blackwood BP, Wood DR, Yuan C, Nicolas J, De Plaen IG, Farrow KN, Chou P, Turner JR, Hunter CJ. A role for cAMP and protein kinase A in experimental necrotizing enterocolitis. Am J Pathol 187: 401–417, 2017. doi: 10.1016/j.ajpath.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab Invest 86: 191–201, 2006. doi: 10.1038/labinvest.3700373. [DOI] [PubMed] [Google Scholar]
- 11.Cromer WE, Mathis JM, Granger DN, Chaitanya GV, Alexander JS. Role of the endothelium in inflammatory bowel diseases. World J Gastroenterol 17: 578–593, 2011. doi: 10.3748/wjg.v17.i5.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cummins PM. Occludin: one protein, many forms. Mol Cell Biol 32: 242–250, 2012. doi: 10.1128/MCB.06029-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cunningham KE, Turner JR. Myosin light chain kinase: pulling the strings of epithelial tight junction function. Ann N Y Acad Sci 1258: 34–42, 2012. doi: 10.1111/j.1749-6632.2012.06526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dörfel MJ, Huber O. Modulation of tight junction structure and function by kinases and phosphatases targeting occludin. J Biomed Biotechnol 2012: 807356, 2012. doi: 10.1155/2012/807356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elias BC, Suzuki T, Seth A, Giorgianni F, Kale G, Shen L, Turner JR, Naren A, Desiderio DM, Rao R. Phosphorylation of Tyr-398 and Tyr-402 in occludin prevents its interaction with ZO-1 and destabilizes its assembly at the tight junctions. J Biol Chem 284: 1559–1569, 2009. doi: 10.1074/jbc.M804783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feldman GJ, Mullin JM, Ryan MP. Occludin: structure, function and regulation. Adv Drug Deliv Rev 57: 883–917, 2005. doi: 10.1016/j.addr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 17.Feng Y, LoGrasso PV, Defert O, Li R. Rho kinase (ROCK) inhibitors and their therapeutic potential. J Med Chem 59: 2269–2300, 2016. doi: 10.1021/acs.jmedchem.5b00683. [DOI] [PubMed] [Google Scholar]
- 18.Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 123: 1777–1788, 1993. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gephart SM, McGrath JM, Effken JA, Halpern MD. Necrotizing enterocolitis risk: state of the science. Adv Neonatal Care 12: 77–87, 2012. doi: 10.1097/ANC.0b013e31824cee94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Günzel D, Yu AS. Claudins and the modulation of tight junction permeability. Physiol Rev 93: 525–569, 2013. doi: 10.1152/physrev.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halpern MD, Denning PW. The role of intestinal epithelial barrier function in the development of NEC. Tissue Barriers 3: e1000707, 2015. doi: 10.1080/21688370.2014.1000707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heidemann J, Domschke W, Kucharzik T, Maaser C. Intestinal microvascular endothelium and innate immunity in inflammatory bowel disease: a second line of defense? Infect Immun 74: 5425–5432, 2006. doi: 10.1128/IAI.00248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirase T, Kawashima S, Wong EY, Ueyama T, Rikitake Y, Tsukita S, Yokoyama M, Staddon JM. Regulation of tight junction permeability and occludin phosphorylation by Rhoa-p160ROCK-dependent and -independent mechanisms. J Biol Chem 276: 10423–10431, 2001. doi: 10.1074/jbc.M007136200. [DOI] [PubMed] [Google Scholar]
- 24.Hopkins AM, Walsh SV, Verkade P, Boquet P, Nusrat A. Constitutive activation of Rho proteins by CNF-1 influences tight junction structure and epithelial barrier function. J Cell Sci 116: 725–742, 2003. doi: 10.1242/jcs.00300. [DOI] [PubMed] [Google Scholar]
- 25.Hunter CJ, Bean JF. Cronobacter: an emerging opportunistic pathogen associated with neonatal meningitis, sepsis and necrotizing enterocolitis. J Perinatol 33: 581–585, 2013. doi: 10.1038/jp.2013.26. [DOI] [PubMed] [Google Scholar]
- 26.Hunter CJ, Chokshi N, Ford HR. Evidence vs experience in the surgical management of necrotizing enterocolitis and focal intestinal perforation. J Perinatol 28, Suppl 1: S14–S17, 2008. doi: 10.1038/jp.2008.44. [DOI] [PubMed] [Google Scholar]
- 27.Hunter CJ, Singamsetty VK, Chokshi NK, Boyle P, Camerini V, Grishin AV, Upperman JS, Ford HR, Prasadarao NV. Enterobacter sakazakii enhances epithelial cell injury by inducing apoptosis in a rat model of necrotizing enterocolitis. J Infect Dis 198: 586–593, 2008. doi: 10.1086/590186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, Narumiya S. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol Pharmacol 57: 976–983, 2000. [PubMed] [Google Scholar]
- 29.Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 72: 463–493, 2010. doi: 10.1146/annurev-physiol-021909-135833. [DOI] [PubMed] [Google Scholar]
- 30.Kümper S, Marshall CJ. ROCK-driven actomyosin contractility induces tissue stiffness and tumor growth. Cancer Cell 19: 695–697, 2011. doi: 10.1016/j.ccr.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 31.Kureishi Y, Kobayashi S, Amano M, Kimura K, Kanaide H, Nakano T, Kaibuchi K, Ito M. Rho-associated kinase directly induces smooth muscle contraction through myosin light chain phosphorylation. J Biol Chem 272: 12257–12260, 1997. doi: 10.1074/jbc.272.19.12257. [DOI] [PubMed] [Google Scholar]
- 32.McGown CC, Brown NJ, Hellewell PG, Brookes ZL. ROCK induced inflammation of the microcirculation during endotoxemia mediated by nitric oxide synthase. Microvasc Res 81: 281–288, 2011. doi: 10.1016/j.mvr.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 33.Nowicki PT. Ischemia and necrotizing enterocolitis: where, when, and how. Semin Pediatr Surg 14: 152–158, 2005. doi: 10.1053/j.sempedsurg.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 34.Ozdemir D, Cilaker S, Tugyan K, Dagdelen MK, Derinoz O, Guneli E. The effect of Rho kinase inhibitor Y-27632 on endotoxemia-induced intestinal apoptosis in infant rats. J Mol Histol 43: 81–87, 2012. doi: 10.1007/s10735-011-9379-6. [DOI] [PubMed] [Google Scholar]
- 35.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 4: 446–456, 2003. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 36.Sakakibara A, Furuse M, Saitou M, Ando-Akatsuka Y, Tsukita S. Possible involvement of phosphorylation of occludin in tight junction formation. J Cell Biol 137: 1393–1401, 1997. doi: 10.1083/jcb.137.6.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spadoni I, Zagato E, Bertocchi A, Paolinelli R, Hot E, Di Sabatino A, Caprioli F, Bottiglieri L, Oldani A, Viale G, Penna G, Dejana E, Rescigno M. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 350: 830–834, 2015. doi: 10.1126/science.aad0135. [DOI] [PubMed] [Google Scholar]
- 38.Sun Z, Wang X, Andersson R. Role of intestinal permeability in monitoring mucosal barrier function. History, methodology, and significance of pathophysiology. Dig Surg 15: 386–397, 1998. doi: 10.1159/000018651. [DOI] [PubMed] [Google Scholar]
- 39.Thomas H. Intestinal tract: gut endothelial cells—another line of defence. Nat Rev Gastroenterol Hepatol 13: 4, 2016. doi: 10.1038/nrgastro.2015.205. [DOI] [PubMed] [Google Scholar]
- 40.Thompson AM, Bizzarro MJ. Necrotizing enterocolitis in newborns: pathogenesis, prevention and management. Drugs 68: 1227–1238, 2008. doi: 10.2165/00003495-200868090-00004. [DOI] [PubMed] [Google Scholar]
- 41.Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, Madara JL. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol 273: C1378–C1385, 1997. doi: 10.1152/ajpcell.1997.273.4.C1378. [DOI] [PubMed] [Google Scholar]
- 42.Walsh SV, Hopkins AM, Chen J, Narumiya S, Parkos CA, Nusrat A. Rho kinase regulates tight junction function and is necessary for tight junction assembly in polarized intestinal epithelia. Gastroenterology 121: 566–579, 2001. doi: 10.1053/gast.2001.27060. [DOI] [PubMed] [Google Scholar]
- 43.Weitkamp JH, Rosen MJ, Zhao Z, Koyama T, Geem D, Denning TL, Rock MT, Moore DJ, Halpern MD, Matta P, Denning PW. Small intestinal intraepithelial TCRγδ+ T lymphocytes are present in the premature intestine but selectively reduced in surgical necrotizing enterocolitis. PLoS One 9: e99042, 2014. [Erratum in PLoS One 9: e105487, 2014.] doi: 10.1371/journal.pone.0099042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol 39: 187–199, 2002. doi: 10.1016/S1537-1891(03)00008-9. [DOI] [PubMed] [Google Scholar]
- 45.Yamamoto M, Ramirez SH, Sato S, Kiyota T, Cerny RL, Kaibuchi K, Persidsky Y, Ikezu T. Phosphorylation of claudin-5 and occludin by rho kinase in brain endothelial cells. Am J Pathol 172: 521–533, 2008. doi: 10.2353/ajpath.2008.070076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ye D, Guo S, Al-Sadi R, Ma TY. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology 141: 1323–1333, 2011. doi: 10.1053/j.gastro.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol 17: 564–580, 2016. doi: 10.1038/nrm.2016.80. [DOI] [PubMed] [Google Scholar]