

Abstract
High concentrations of propionate and its metabolites are found in several diseases that are often associated with the development of cardiac dysfunction, such as obesity, diabetes, propionic acidemia, and methylmalonic acidemia. In the present work, we employed a stable isotope-based metabolic flux approach to understand propionate-mediated perturbation of cardiac energy metabolism. Propionate led to accumulation of propionyl-CoA (increased by ~101-fold) and methylmalonyl-CoA (increased by 36-fold). This accumulation caused significant mitochondrial CoA trapping and inhibited fatty acid oxidation. The reduced energy contribution from fatty acid oxidation was associated with increased glucose oxidation. The enhanced anaplerosis of propionate and CoA trapping altered the pool sizes of tricarboxylic acid cycle (TCA) metabolites. In addition to being an anaplerotic substrate, the accumulation of proprionate-derived malate increased the recycling of malate to pyruvate and acetyl-CoA, which can enter the TCA for energy production. Supplementation of 3 mM l-carnitine did not relieve CoA trapping and did not reverse the propionate-mediated fuel switch. This is due to new findings that the heart appears to lack the specific enzyme catalyzing the conversion of short-chain (C3 and C4) dicarboxylyl-CoAs to dicarboxylylcarnitines. The discovery of this work warrants further investigation on the relevance of dicarboxylylcarnitines, especially C3 and C4 dicarboxylylcarnitines, in cardiac conditions such as heart failure.
Keywords: cardiac metabolism, dicarboxylylcarnitine, dicarboxylyl-CoA, propionate, stable isotope analysis
INTRODUCTION
Propionate can be derived from several sources, including the diet, metabolism by the gut microbiome, and metabolism of branched-chain amino acids (BCAAs; valine and isoleucine). The concentrations of propionate and its metabolites are relatively low in normal mammals. However, certain diseases, such as diabetes, obesity, propionic acidemia, and methylmalonic acidemia (MA), lead to dramatic increases in circulating propionate concentrations (5, 10, 25). Propionylcarnitine concentrations are doubled in diabetic individuals (29). In extreme circumstances, propionate concentrations can increase to 4–6 mM in blood in patients with propionic acidemia (41, 45), whereas a normal plasma level of propionate is around 4 µM (46). Importantly, most of the above diseases are often accociated with cardiac dysfunction (12, 21, 26, 35). Whether, and how, increased propionate in circulation directly affects the heart is unknown. Because the ability of the heart to switch rapidly between fuel sources, so-called metabolic flexibility, is critical to maintain normal cardiac function, it is possible that propionate impairs this process. The present work sought to investigate propionate metabolism in heart and its impact on cardiac metabolism in healthy rats.
Our previous work on propionate metabolism in the liver clearly demonstrated the liver CoA trapping induced by the accumulation of propionate metabolites, i.e., propionyl-CoA, methylmalonyl-CoA (MM-CoA), succinyl-CoA, and 3-hydroxylpropionyl-CoA (45). The short-chain acyl-CoAs are converted to their corresponding counterparts, i.e., acylcarnitines, for the purpose of cellular transportation of the acyl moiety or storage by carnitine acyltransferase. Carnitine acetyltransferase (CrAT) is one of the three main carnitine acyltransferases and catalyzes the interconversion between short-chain acyl-CoAs and acylcarnitines. Given the fact that CrAT is highly abundant in the heart and that CrAT has the highest activity for propionyl-CoA (3, 27, 37, 44), l-carnitine may correct propionate-mediated CoA trapping. The idea of l-carnitine supplement has been proposed to patients with propionic acidemia (20). However, the detailed effect and biochemical mechanism are not fully elucidated, particularly in the heart.
Increases in circulating BCAA concentrations have been observed in several cardiac diseases, including heart failure (2, 39, 40, 42). Recent work has also shown that circulating short-chain dicarboxylylcarnitines have similar associations (8, 16, 38, 40). Among short-chain dicarboxylylcarnitines, succinylcarnitine and methymalonylcarnitine (MM-carnitine) are counterparts of succinyl-CoA and MM-CoA, respectively. These acyl-CoA species are both derived from propionyl-CoA, a downstream metabolite of BCAA catabolism. However, whether short-chain dicarboxylylcarnitines are associated with increased propionyl-CoA metabolism remains to be investigated.
Using our previously described metabolic flux technique (22), we sought to elucidate propionate metabolism and its perturbation in the heart further. We investigated 1) the mechanism of perturbation of propionate on cardiac energy metabolism, 2) the effect of l-carnitine on propionate metabolism, and 3) the association of two C4 dicarboxylylcarnitines (succinylcarnitine and MM-carnitine) with the metabolism of propionate. The main findings of this work are that high propionate switches cardiac fuel metabolism from fatty acid to glucose because of CoA trapping induced by propionate metabolism. Administration of supraphysiological levels of l-carnitine fails to regenerate free CoA because cardiac tissue lacks a specific enzyme that catalyzes the interconvernsion between short-chain (C3 and C4) dicarboxylyl-CoA and dicarboxylylcarnitines. The results of this work shed light on the biochemical mechanism of propionate metabolic perturbation in patients with increased propionate and/or its metabolites.
MATERIALS AND METHODS
Materials.
2-Deoxy-d-glucose (2-DG) is from Chem-impex International, Inc. (Wood Dale, IL). 2-[1-13C]DG, l-[2H3]carnitine, and l-[2H9]carnitine were from Cambridge Isotope Laboratories. 2-Deoxyl-d-glucose-6-phosphate (2-DG6P) was from Santa Cruz Biotechnology (Dallas, TX). Pentanoyl-[2H9]CoA and MM-carnitine were synthesized in our laboratory as previously described (15, 48). All remaining chemicals, including l-[13C5,15N]valine, [13C6]glucose, [1-13C]palmitate, [1-13C]octanoate, and [13C3]propionate were purchased from Sigma (St. Louis, MO).
Heart perfusions.
All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Duke University. Fed male rats (~220 g, age: 7–8 wk) were anesthetized with 5% isoflurane, and the isolated hearts were perfused in the Langendorff mode at 37°C with nonrecirculating perfusate of Krebs-Ringer bicarbonate buffer containing (in mM) 119 NaCl, 4.8 KCl, 2.6 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 25 NaHCO3, 11 glucose, and 0.05 l-carnitine at a constant flow rate of 12 ml/min (22, 23). The hearts were allowed to beat spontaneously throughout the perfusion. The hearts were perfused with Krebs-Ringer bicarbonate buffer for 15 min and then switched to perfusate containing the labeled substrates for 25 min. At the end of each perfusion, hearts were freeze-clamped in liquid nitrogen and stored at −80°C until analysis. The effluent of perfusate sample (~10 ml) was collected during the final minute of perfusion and quickly frozen in liquid nitrogen.
The perfusions were conducted as the following groups: 1) hearts perfused with 11 mM 40% [13C6]glucose, 0.4 mM [1-13C]palmitate, 3% BSA (fatty acid free; Fisher Scientific), 100 µU/ml insulin, 50 µM l-carnitine, and 0.1 mM 2-DG ± (3 mM propionate ± 3 mM l-carnitine); 2) hearts perfused with 11 mM glucose, 0.4 mM palmitate, 3% BSA, 100 µU/ml insulin, 50 µM l-carnitine, and 3 mM [13C3]propionate; 3) hearts perfused with 11 mM glucose, 100 µU/ml insulin, 50 µM l-carnitine, 3% BSA, and 0.2 mM [1-13C]octanoate ± 3 mM propionate. Each of these groups contained four or five rats. Group 4 hearts were perfused with 11 mM glucose, 0.4 mM palmitate, 3% BSA, 100 µU/ml insulin, 50 µM l-[2H3]carnitine for 0, 2, 5, 10, 30, 40, and 60 min, with one rat per time point.
Perfused liver experiments.
Liver perfusion experiments were conducted at Case Western Reserve University. All animal protocols were approved by the IACUC of Case Western Reserve University. Male Sprague-Dawley rats were fed with Prolab Isopro RMH 3000 irradiated chow (St. Louis, MO). Livers from overnight-fasted rats (male, 160–180 g, 6–7 wk) were perfused for 60 min with recirculating Krebs-Ringer bicarbonate buffer containing 4% dialyzed, fatty acid-free bovine serum albumin and 4 mM glucose. To the basic perfusate we added either nothing (controls) or 5 mM [13C3]propionate (n = 5 or 6 in each group).
In vivo experiments.
To determine the metabolic source of MM-carnitine in whole-animal experiments (rats, ~200 g, n = 3), we performed intraperitoneal injection of 200 mg/kg of [13C3]propionate sodium salts. Blood samples (~200 µl) were drawn at 0, 10, 30, and 60 min after the injection of [13C3]propionate. After 60 min, rats were anesthetized with isoflurane, and the liver, kidney, heart, and brain were harvested and quickly freeze-clamped in liquid nitrogen.
LC-MS/MS for acylcarnitine profile.
Acylcarnitines in the cardiac tissue were methylated and profiled by the modified liquid chromatography with tandem mass spectrometry (LC-MS/MS) method (30). Briefly, ~20-mg powdered cardiac tissue samples were spiked with 0.2 nmol l-[2H9]carnitine as an internal standard. The derivatized samples were analyzed by two LC-MS platforms: LC-QTRAP 6500+-MS/MS (Sciex, Concord, Ontario) and LC-Q-Exactive+-MS (Thermo Fisher Scientific, Waltham, MA).
LC-MS/MS acyl-CoA analysis.
Acyl-CoAs were analyzed based on our previously established method (24). A ~150-mg heart tissue sample was spiked with internal standard [[2,2,3,3,4,4,5,5,5-2H9]pentanoyl-CoA (0.2 nmol)] and homogenized in 1.5 ml of extraction buffer (5% acetic acid in MeOH/H2O 50:50) using a tissuelyser (QIAGEN, Germantown, MD). The supernatant was run on a 1-ml ion exchange cartridge packed with 300 mg of 2–2(pyridyl)ethyl silica gel (Sigma). The cartridge-purified sample was used for all assays by LC-QTRAP 6500+-MS/MS. The detailed LC-MS/MS method was described in our previous publication (24).
LC-MS for 2-DG6P assay.
2-DG6P was quantified by LC-Q-Exactive+-MS. The binary pump was used to transport mobile phase (HPLC-MS-grade water) at a flow rate of 0.2 ml/min in isocratic elution mode. The column was a Microsorb-MV C18 column (100 × 4.6 mm, 3 µm) with C18 guard column and was kept at 40°C in the oven compartment. The auto sampler was maintained at 5°C, and the injection volume was 1 µl. The total running time was 7 min. The parameters for Q-Exactive+-MS equipped with a HESI probe: heat temperature, 425°C; sheath gas, 30, auxiliary gas, 13; sweep gas, 3; spray voltage, 3.5 kV for positive mode; capillary temperature was set at 320°C; and S-lens was 45. A full scan range was set at 60 to 900 (mass-to-charge ratio). The resolution was set at 70,000 (at mass-to-charge ratio 200). The maximum injection time was 200 ms. Automated gain control was targeted at 3 × 106 ions.
Gas chromatography-mass spectrometry for metabolite analysis.
A modified gas chromatography-mass spectrometry (GC-MS) method was used for small polar metabolites assay (28). A ~20-mg powdered tissue sample was spiked with 0.1 nmol M6 valine (M0, M1, . . . , Mn means the isotopologues that contain n heavy atoms in a molecule) as internal standard and homogenized in 400 µl methanol for 1 min followed by another 1 min homogenization after adding 400 µl H2O and 400 µl chloroform. The homogenized sample was centrifuged at 20,000 g for 15 min. The supernatant was dried completely with nitrogen gas and was derivatized with methoxylamine hydrochloride and MTBSTFA + 1% TBDMS for GC-MS analysis.
Western blot analysis of tissue.
Approximately 20 mg of powdered frozen cardiac tissue was homogenized in 150 µl chilled lysis buffer (cat. no. 9803-S; Cell Signaling Technology, Danvers, MA) using the Qiagen Retsch TissueLyser II system at a rate of 30 shakes/s for 1 min. Samples were centrifuged at 10,000 g for 10 min at 4°C. One hundred microliters of supernatant were collected, and protein concentration was quantified by bicinchoninic acid protein assay (Bio-Rad, Hercules, CA). Protein (50 µg) was added to standard Laemmli buffer, and samples were run on a 4%–15% Mini-Protean TGX stain-free gel (Bio-Rad) and then transferred to a PVDF membrane. Protein loading was validated on both the gel and PVDF membrane using the Gel Doc XR+ system (Bio-Rad). Membranes were blocked in 5% powdered milk in Tris-buffered saline with 0.1% Tween for 1 h. Membranes were then incubated with antibodies for the E1-α subunit of pyruvate dehydrogenase (PDH; cat. no. ab-110330; Abcam, Cambridge, MA) and serine-293 phosphorylated PDH (cat. no. AP-1062; Millipore Sigma, Burlington, MA) at 1:1,000 dilution overnight at 4°C. After washing and secondary antibody (cat. no. 926-32213 donkey anti-rabbit IR 800cw; LI-COR, Lincoln, NE) incubation, blots were scanned with the LI-COR Odyssey CLx system (LI-COR) and quantified with Odyssey 3.0 software using direct fluorescence measurement.
Calculations and statistics.
Measured mass isotopologues distributions expressed as mol percent were corrected for natural enrichment (9, 43). Statistical differences were assayed by one-way analysis of variance followed by a Tukey post hoc test using Prism software 5.02 (GraphPad Software) unless it was specified.
RESULTS
Propionate metabolism in heart and its effect on the tricarboxylic acid cycle and related metabolites.
The study sought to investigate 1) cardiac metabolism of propionate and its effect on metabolism of other fuels and 2) the efficacy of l-carnitine in relieving the stress of propionate overloading by transferring the propionyl-group from propionyl-CoA to carnitine, thereby permitting mitochondrial efflux. To this end, we performed three groups of heart perfusions: 1) control, 2) 3 mM propionate, and 3) 3 mM propionate + 3 mM l-carnitine. Each group had five repeated heart perfusions.
Like metabolites found in the liver, some propionate-specific metabolites were also identified in the heart. For instance, methylcitrate increased by ~100-fold in both propionate- and propionate + l-carnitine-perfused hearts. However, the concentration of methylcitrate was only ~3% of citrate (data not shown). 3-hydroxylpropionyl-CoA (0.1 ± 0.03 and 0.08 ± 0.008 nmol/g in hearts perfused with propionate and propionate + l-carnitine, respectively) and 3-hydroxylpropionylcarnitine (0.4 ± 0.05 and 0.7 ± 0.16 nmol/g in hearts perfused with propionate and propionate + l-carnitine, respectively) were also detected in the presence but not absence of propionate.
We then compared the changes of tricarboxylic acid cycle (TCA) and glycolytic intermediates among the three groups (Fig. 1A). Fumarate and malate were increased by propionate, whereas citrate and 2-ketoglutarate (2-KG) were decreased. Succinate and pyruvate tended to be higher in propionate-perfused hearts. Phosphoenolpyruvate (PEP), 3-phosphoglycerate (3-PG), and lactate were not significantly affected by propionate. l-Carnitine did not change the propionate-mediated changes in these metabolites. Closely related to TCA metabolism, amino acids were also profiled (Fig. 1A). Alanine and glutamate were significantly decreased by propionate or propionate + l-carnitine. In contrast, aspartate was doubled by propionate or propionate + l-carnitine. Propionyl-CoA is reported to form propionylglutamate (6), which could account for the decrease seen in glutamate with propionate infusion. In both propionate- and propionate + l-carnitine-perfused hearts, the relative concentration of propionylglutamate was 10-fold higher than that of acetylglutamate (Fig. 1, B and C). Propionylglutamate was identified by UHPLC-Q-Exactive-MS with accurate mass assay, relative retention time compared with acetylglutamate, and 13C-labeling from [13C3]propionate (data not shown).
Fig. 1.

Metabolites altered by propionate. Rat hearts were perfused with 40% 11 mM [13C6]glucose, [1-13C]palmitate, 0.1 mM 2-DG, 3% BSA, 100 µU insulin, and 50 µM l-carnitine (control, n = 5), control + 3 mM propionate (PA, n = 5), and PA + 3 mM l-carnitine (PA + Carn, n = 5). A: glycolytic, amino acids, and TCA metabolites changes (log of relative changes vs. control in the heat map) induced by 3 mM propionate or 3 mM propionate + 3 mM l-carnitine. The statistic significance is marked in the heat map. No significant difference was observed between PA and PA + Carn groups. B: relative concentration of propionylglutamate. C: relative concentration of acetylglutamate. D–G: stable isotope labelings of glucose-6-phosphate (G6P), 3-phosphoglycerate (3-PG), phosphoenolpyruvate (PEP), and pyruvate. H: scheme of the 13C flow from [13C6]glucose/[1-13C]palmitate and pyruvate recycling from malate via malic enzyme (ME). The error bar is the standard deviation of analysis. *P < 0.05, **P < 0.01, and ***P < 0.001. 2-DG, 2-Deoxy-d-glucose; 2-KG, 2-ketoglutarate; 3-PG, 3-phosphoglycerate; Cit, citrate; Fum, fumarate; Lac, lactate; Mal, malate; PEP, phosphoenolpyruvate; Pyr, pyruvate; Suc, succinate; TCA, tricarboxylic acid cycle; M0, M1, . . . , Mn means the isotopologues that contain n heavy atoms in a molecule.
Propionate enters the TCA as energy substrate via malic enzyme and PDH.
The use of labeled substrates, [13C6]glucose and [1-13C]palmitate, enabled us to measure fractional metabolic fluxes along their metabolic networks.
The breakdown of [13C6]glucose (M6) leads to labeled glycolic intermediates to pyruvate (M3; see Fig. 1H). M0, M1, . . . , Mn means the isotopologues that contain n heavy atoms in a molecule. The labeling of glucose-6-phosphate, 3-PG, and PEP, from [13C6]glucose, were similar and were not changed by propionate or propionate + l-carnitine (Fig. 1, D–F). However, pyruvate (Fig. 1G) had notably different labeling patterns compared with its precursors (glucose-6-phosphate, 3-PG, and PEP); there was increased M1 and M2 pyruvate labeling and decreased M3 pyruvate labeling in hearts perfused with propionate or propionate + l-carnitine. This suggests that M3 pyruvate derived from M6 glucose was diluted by pyruvate from other sources, which led to increased M1 and M2 pyruvate. The possible sources of M1 or M2 pyruvate could be from labeled malate or oxaloacetate via malic enzyme (Fig. 1H) or phosphoenolpyruvate carboxykinase (PEPCK), respectively. However, there was no detectable M1 or M2 PEP suggesting that pyruvate was derived from malate by malic enzyme and not from PEPCK. M3 alanine labeling also trended lower in propionate or propionate + l-carninitine perfused hearts (data not shown), possibly because of lower conversion of pyruvate to alanine, as reflected in a lower total pool size of alanine (Fig. 1A).
We also confirmed the recycling of malate to pyruvate by perfusing hearts with [13C3]propionate. Our previous liver perfusions with [13C3]propionate showed that there was substantial gluconeogenesis from M3 propionate (45). The labeling of metabolites from M3 propionate in both the heart and liver is compared in Fig. 2. The flow of 13C from [13C3]propionate is outlined in Fig. 2A, which shows the first round of M3 propionate entering the TCA from succinyl-CoA. Interestingly, metabolites downstream of the anaplerotic pathway of propionate (succinate and malate) showed higher labeling in the heart than in the liver (Fig. 2, H and I). This suggests that the anaplerotic flux (M3 succinyl-CoA/M3 propionyl-CoA × 100%) from propionate is higher in the heart (~57%) than in the liver (~40%). Conversely, the labeling of glycolytic metabolites (pyruvate, lactate, and PEP), alanine, and TCA intermediates from citrate to 2-KG were higher in liver than in heart (Fig. 2, B–G). In the M3 propionate perfused heart, no labeled PEP was observed (Fig. 2E). The labeling data of PEP and pyruvate in M3 propionate perfused hearts again confirmed metabolic flux via malic enzyme in the heart without detectable PEPCK activity. Based on the labeling of malate and pyruvate, we estimated the fractional flux from malate to pyruvate to be 21% (the average carbon labeling of pyruvate/the average carbon labeling of malate × 100%) in the heart. This indicates that propionate is not only an anaplerotic substrate but also a substantial energy substrate when it is in high concentration.
Fig. 2.

The labeling of metabolites from [13C3]propionate in the perfused hearts and livers. Rat hearts were perfused with 11 mM glucose, palmitate, 3% BSA, 100 µU insulin, 3 mM [13C3]propionate, and 50 µM l-carnitine (n = 6). Liver perfusion can be referred to our previous report (16). A: first round of 13C from [13C3]propionate entering TCA cycle and pyruvate (acetyl-CoA) in the heart. B–I: labeling of isotopologues of pyruvate, lactate, alanine, PEP, citrate, 2-KG, succinate, and malate in both heart and liver perfused with 3 or 5 mM [13C3]propionate. The error bar is the standard deviation of analysis. *P < 0.05, **P < 0.01, and ***P < 0.001. 2-KG, 2-ketoglutarate; MM-CoA, methylmalonyl-CoA; PEP, phosphoenolpyruvate; TCA, tricarboxylic acid cycle; M0, M1, . . . , Mn means the isotopologues that contain n heavy atoms in a molecule.
The much lower labeling of lactate (Fig. 2C) and alanine (Fig. 2D) than pyruvate in the M3 propionate-perfused heart suggested that pyruvate derivation from malate mainly occurred in mitochondria. In contrast to the heart, and as expected, liver had similar labeling because there were two metabolic fluxes from TCA intermediates to glycolic metabolites via PEPCK and malic enzyme in the liver for gluconeogenesis (45).
Propionate switches cardiac fuel preference from fatty acid to glucose.
With the labeled isotopes [13C6]glucose (40% of total 11 mM glucose) and 0.4 mM [1-13C]palmitate, we estimated the fractional flux of glucose and fatty acid oxidation to acetyl-CoA as shown in Fig. 3A. The oxidation of [1-13C]palmitate and [13C6]glucose generate M1 and M2 acetyl-CoA, which reflect the fractional fluxes from the oxidation of fatty acid and glucose, respectively. In the propionate-perfused hearts, the fractional flux from glucose was enhanced by ~twofold, and the contribution of palmitate to acetyl-CoA was reduced by 50% (Fig. 3B) compared with control. Adding l-carnitine did not reverse the propionate-mediated changes (Fig. 3B). The counterpart of acetyl-CoA, acetylcarnitine, showed similar changes (Fig. 3C).
Fig. 3.

Propionate enhanced metabolic flux from glucose to acetyl-CoA and TCA and had the opposite effect on fatty acid. A: scheme of the first round of 13C from [13C6]glucose and [1-13C]palmitate to acetyl-CoA and TCA cycle. B–M: rat hearts were perfused with 40% 11 mM [13C6]glucose, [1-13C]palmitate, 0.1 mM 2-DG, 3% BSA, 100 µU insulin, and 50 µM l-carnitine (control, n = 5), control + 3 mM propionate (PA, n = 5), and PA + 3 mM l-carnitine (PA + Carn, n = 5). B–G: labeling of isotopologues of acetyl-CoA, acetylcarnitine, citrate, 2-KG, succinate, and malate in the hearts from 3 groups (Control, PA, and PA + Carn). H–I: 2-DG6P concentration in the hearts and M3 lactate concentration in the effluent perfusates of 3 perfusion groups (control, PA, and PA + Carn). The error bar is the standard deviation of analysis. J–M: Western blot analysis of pyruvate dehydrogenase (PDH) and phosphorylated PDH (pPDH) in the perfused hearts. J and L: PDH and pPDH in the control and PA-perfused hearts. K and M: are PDH and pPDH in the control and propionate + l-carnitine (PA + Carn)-perfused hearts. The error bar is the standard deviation of analysis. *P < 0.05, **P < 0.01, and ***P < 0.001. 2-DG6P, 2-Deoxyl-d-glucose-6-phosphate; 2-KG, 2-ketoglutarate; TCA, tricarboxylic acid cycle; M0, M1, . . . , Mn means the isotopologues that contain n heavy atoms in a molecule.
The stable isotopologues of TCA intermediates showed a similar pattern of decreased palmitate oxidation induced by propionate or propionate + l-carnitine. Isotope enrichment of TCA intermediates is the result of multiple metabolic fluxes, including anaplerotic flux from M3 pyruvate, M1 and M2 acetyl-CoA, and multiple cycles of TCA flux itself (Fig. 3A). However, M2 is primarily attributed to the contribution of M2 acetyl-CoA from glucose oxidation. Therefore, the significant increase of isotope enrichment of M2 TCA intermediates suggested increased glucose oxidation by propionate (Fig. 3, D–G). The propionate-mediated changes of isotope labeling of TCA intermediates were again not significantly affected by the addition of l-carnitine.
We assessed glucose uptake, PDH phosphorylation, and lactate efflux to confirm the propionate-mediated glucose oxidation. We measured an index of glucose uptake by measuring 2-DG6P in hearts perfused with 0.1 mM 2-DG. Propionate or propionate + l-carnitine increased glucose uptake (Fig. 3H). The enhanced glucose uptake did not result in higher efflux of M3 lactate in the perfusate (Fig. 3I). The phosphorylation of PDH, as assessed by Western blot, was significantly lower in propionate + l-carnitine-perfused hearts, whereas it trended lower (but was not significant) in propionate-perfused hearts (Fig. 3, J–M).
To investigate whether the mechanism of propionate-mediated suppression of fatty acid oxidation in the perfused heart is due to inhibition of CPT-1, we perfused hearts with 0.2 mM [1-13C]octanoate ± 3 mM propionate, as octanoate does not require transport through the CPT system. The carbon flow of M1 octanoate metabolism to TCA and acetylcarnitine is shown in Fig. 4A. The labeling of M1 acetylcarnitine, which reflects mitochondrial oxidation of M1 octanoate, decreased when propionate was added in the perfusate (Fig. 4B). Propionate-mediated suppression of octanoate oxidation was also supported by the decreased M1 labeling of other metabolites in the TCA (Fig. 4, B–F). The suppressed octanoate oxidation by propionate demonstrated that propionate-mediated suppression of fatty acid oxidation was not due to the inhibition of CPT system.
Fig. 4.

Propionate suppressed mitochondrial metabolism of octanoate to TCA. Rat hearts were perfused in Langendorff mode with 11 mM glucose, 0.2 mM [1-13C]octanoate, 3% BSA, 100 µU insulin, and 50 µM l-carnitine (M1 OA, n = 4), control + 3 mM propionate (M1 OA + PA, n = 4). A: a scheme of the first round of 13C from [1-13C]octanoate to acetyl-CoA and TCA cycle. B–F: the labeling of isotopologues of acetylcarnitine, citrate, 2-KG, succinate, and malate. **P < 0.01, ***P < 0.001. 2-KG, 2-ketoglutarate; OAA, oxaloacetate; TCA, tricarboxylic acid cycle; M0, M1, . . . , Mn means the isotopologues that contain n heavy atoms in a molecule.
Propionate-induced propionyl-CoA and MM-CoA accumulation causes mitochondrial CoA trapping that is not fully relieved by l-carnitine.
Propionic acidemia is characterized by the accumulation of short-chain acyl-CoAs (propionyl-CoA) and a high ratio of acyl-CoA:CoA (34). We profiled the propionate-mediated changes of acyl-CoAs and acylcarnitines (Fig. 5). Both free CoA and l-carnitine concentrations were decreased in propionate-perfused hearts (Fig. 5A, PA groups vs. control), whereas propionyl-CoA and MM-CoA accumulated (Fig. 5A). Consistent with the lowered free CoA concentration, there was a significant decrease of most other acyl-CoAs, such as succinyl-CoA and acetyl-CoA (Fig. 5A), and other acyl-CoA species (Fig. 5B). Similarly, decreased tissue l-carnitine was also associated with a decrease of most other acylcarnitines (Fig. 5, A and C). To test whether CoA trapping could be mitigated in propionate-perfused hearts, we added excess l-carnitine (3 mM) to propionate (PA + Carn). This decreased propionyl-CoA by ~50% and increased propionylcarnitine by twofold in the propionate-perfused hearts (Fig. 5A). However, the l-carnitine supplementation did not alter the MM-CoA concentration and only partially restored free CoA concentration (Fig. 5A). MM-carnitine remained unchanged across the heart tissues from all three groups (Fig. 5A). These data strongly suggest that minimal MM-CoA was converted to MM-carnitine in the heart, even in the presence of excess l-carnitine. Interestingly, the propionate-induced suppression of succinyl-CoA was partially restored by adding l-carnitine (Fig. 5A). However, the fluctuation of succinyl-CoA induced by propionate or l-carnitine did not significantly change succinylcarnitine concentrations (Fig. 5A). The failure to increase MM-carnitine and succinylcarnitine concentrations in the presence of excess l-carnitine suggests that heart lacks a specific enzyme that can catalyze the conversion between short-chain (C3 and C4) dicarboxylyl-CoAs and dicarboxylylcarnitines. Similar findings were seen with malonyl-CoA and malonylcarnitine concentrations in the hearts perfused with and without propionate or propionate + l-carnitine (Fig. 5A).
Fig. 5.

Propionate-mediated changes of acyl-CoAs and acylcarnitines in the perfused hearts. Rat hearts were perfused with 40% 11 mM [13C6]glucose, [1-13C]palmitate, 0.1 mM 2-DG, 3% BSA, 100 µU insulin, and 50 µM l-carnitine (control, n = 5), control + 3 mM propionate (PA, n = 5), and PA + 3 mM l-carnitine (PA + Carn, n = 5). A: concentration changes of free (C0), acetyl- (C2), propionyl- (C3), malonyl- (C3-DC), succinyl-, and methylmalonyl- (MM-) acyl-CoAs (top) and acylcarnitines (bottom) in the perfused hearts from control, propionate, and PA + Carn groups. B and C: fold of acyl-CoA and acylcarnitine changes in the hearts from control, propionate, and PA + Carn groups. *P < 0.05, **P < 0.01, and ***P < 0.001. 2-DG, 2-Deoxy-d-glucose.
The low interconversion between short-chain (C3 and C4) dicarboxylyl-CoAs and their corresponding dicarboxylylcarnitines was also evidenced from tracing studies. Succinyl-CoA (M1, M2, M3, and M4), MM-CoA (M1), and malonyl-CoA (M1 and M2) were substantially labeled from the metabolism of [13C6]glucose and [1-13C]palmitate (Fig. 6, A–C). However, the labeling of succinylcarnitine, MM-carnitine, and malonylcarnitine was minor (Fig. 6, D–F).
Fig. 6.

Short-chain dicarboxylylcarnitines have little 13C labeling in the perfused hearts with [13C6]glucose and [1-13C]palmitate. Rat hearts were perfused with 40% 11 mM [13C6]glucose, [1-13C]palmitate, 0.1 mM 2-DG, 3% BSA, 100 µU insulin, and 50 µM l-carnitine (control, n = 5), control + 3 mM propionate (PA, n = 5), and PA + 3 mM l-carnitine (PA + Carn, n = 5). A–F: labeling of isotopologues of succinyl-CoA, MM-CoA, malonyl-CoA, succinylcarnitine, MM-carnitine, and malonylcarnitine. The error bar is the standard deviation of analysis. *P < 0.05, **P < 0.01, and ***P < 0.001. MM-, methylmalonyl; M0, M1, . . . , Mn means the isotopologues that contain n heavy atoms in a molecule.
To investigate whether small amounts of succinyl-CoA and MM-CoA are converted to succinylcarnitine and MM-carnitine, hearts were perfused with [13C3]propionate to highly enrich the labeling of M3 propionyl-CoA (97%), M3 MM-CoA (93%) and M3 succinyl-CoA (53%) (Fig. 7, A–C). The labeling of M3 propionylcarnitine, M3 MM-carnitine, and M3 succinylcarnitine were 93.7%, 4.7%, and 0.89%, respectively (Fig. 7, D–F). The fractional syntheses of MM-carnitine and succinylcarnitine from propionate were 4.8% and 1.7% in the heart, respectively. In the livers perfused with M3 propionate, the enrichments of M3 propionyl-CoA, M3 MM-CoA, and M3 succinyl-CoA were 91%, 93%, and 37%, respectively. Propionylcarnitine was highly labeled at M3 (93%) in the liver (Fig. 7D), which was similar to heart. MM-carnitine and succinylcarnitine in the liver tended to have higher labeling than in heart (Fig. 7, E and F). The fractional syntheses of MM-carnitine and succinylcarnitine in the liver were 8.9% and 24.5%, respectively. Overall, both tracing studies (labeled glucose/palmitate and propionate) confirmed the low enzyme activity in heart in converting C3 and C4 dicarboxylyl-CoAs to dicarboxylylcarnitines.
Fig. 7.

Short-chain dicarboxylylcarnitines are more labeled from [13C3]propionate in liver than in heart. Rat hearts were perfused with 11 mM glucose, palmitate, 3% BSA, 100 µU insulin, 50 µM l-carnitine, and 3 mM [13C3]propionate for 25 min. The experimental details of liver perfusion with 5 mM [13C3]propionate and labeling of acyl-CoA were reported in our previous work (45). A–C: labeling of propionyl-CoA, MM-CoA, and succinyl-CoA in the hearts perfused with 3 mM [13C3]propionate. D–F: labeling of isotopologues of propionylcarnitine, MM-carnitine, and succinylcarnitine in both hearts and livers perfused with [13C3]propionate. The error bar is the standard deviation of analysis. *P < 0.05, ***P < 0.001. MM-, methylmalonyl; M0, M1, . . . , Mn means the isotopologues that contain n heavy atoms in a molecule.
To investigate whether cardiac MM-carnitine may be derived from other tissues or organs, we performed in vivo experiment using an intraperitoneal injection of 200 mg/kg M3 propionate. Organs were harvested after 1 h, and the labeling of propionylcarnitine, MM-carnitine, and succinylcarnitine was assayed (Fig. 8). Brain, heart, kidney, and liver showed substantial M3 propionylcarnitine labeling at 1 h (Fig. 8A). The labeling of M3 propionylcarnitine in heart (~20%) and other organs also suggest that the circulating M3 propionate was not in supraphysiological concentration with the dose of 200 mg/kg intraperitoneal injection. MM-carnitine had minimal labeling in all organs assayed (Fig. 8B). Succinylcarnitine labeling was predominantly in the liver (Fig. 8C). These data again suggested that the liver exhibits relatively higher conversion of succinyl-CoA to succinylcarnitine as compared with the heart. We also assayed the total concentrations of carnitine, acetylcarnitine, propionylcarnitine, succinylcarnitine, and MM-carnitine in four organs. Heart tissue had higher concentrations of l-carnitine and acylcarnitines as compared with other tissues, except for propionylcarnitine. Heart tissue possessed the highest levels of succinylcarnitine and MM-carnitine among all organs assayed (Fig. 8, E–H). The source of cardiac succinylcarnitine and MM-carnitine is not clear since heart does not significantly synthesize short-chain dicarboxylylcarnitines (C3 and C4) according to this work.
Fig. 8.

Comparasion of [13C3]propionate metabolism in various organs by intraperitoneal injection. Rats (n = 3) were given an intraperitoneal bolus injection of [13C3]propionate (200 mg/kg). After one hour, rats were anesthetized using isoflurane and brain, heart, kidney, and liver were isolated and quickly clamped in liquid nitrogen. A–C: labeling of isotopologues of propionylcarnitine, MM-carnitine, and succinylcarnitine in brain, heart, kidney, and liver organs. D–H: are concentrations of l-carnitine, propionylcarnitine, MM-carnitine, succinylcarnitine, and acetylcarnitine in the brain, heart, kidney, and liver organs. The error bar is the standard deviation of analysis. *P < 0.05, **P < 0.01, and ***P < 0.001. MM-, methylmalonyl; M0, M1, . . . , Mn means the isotopologues that contain n heavy atoms in a molecule.
To investigate whether succinylcarnitine, MM-carnitine, and malonylcarnitine are newly synthesized from other unknown metabolic pathways, we perfused hearts with 50 µM l-[2H3]carnitine from 0 to 60 min, and then quantified the newly synthesized acylcarnitines by measuring M3 acylcarnitines. M3 acetylcarnitine (C2-carnitine), M3 propionylcarnitine (C3-carnitine), and M3 butyrylcarnitine (C4-carnitine) (Fig. 9) showed similar kinetics as M3 l-carnitine and other acylcarnitines. M3 l-carnitine was somewhat overestimated because of the presence of M3 l-carnitine in perfusate at the time of tissue harvest. However, M3 malonylcarnitine and M3 MM-carnitine showed undetectable label incorporation in the 60-min perfusion period and succinylcarnitine was labeled at a very low level (0.3%) (Fig. 9). These data suggest that there was negligible new succinylcarnitine, MM-carnitine, and malonylcarnitine synthesized in the perfused heart from 0 to 60 min.
Fig. 9.
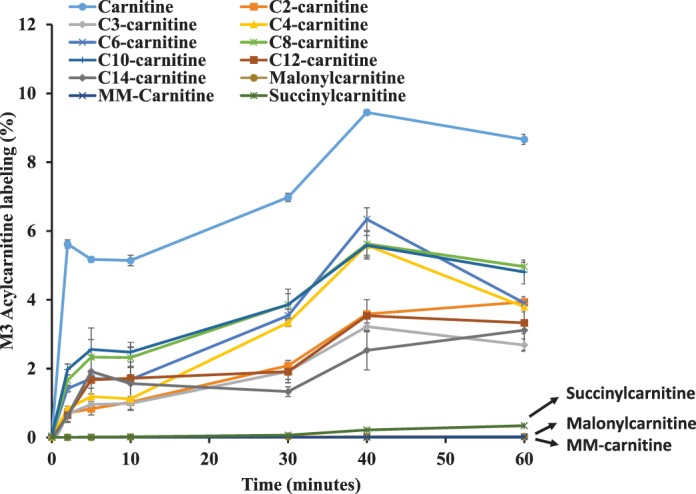
M3 acylcarnitine synthesis kinetics in the heart perfused with [2H3] l-carnitine. Rat hearts were perfused with 11 mM glucose, palmitate, 3% BSA, 100 µU insulin, and 50 µM [2H3] l-carnitine for 0, 2, 5, 10, 20, 40, and 60 min. There was one rat per time point. Labeling of M3 acylcarnitines (free carnitine, acylcarnitines with various carbon chain lengths from C2 to C14, succinylcarnitine, MM-carnitine, and malonylcarnitine) was plotted against the perfusion time. The error bar was from standard deviation of triplicate analyses. MM-, methylmalonyl; M0, M1, . . . , Mn means the isotopologues that contain n heavy atoms in a molecule.
DISCUSSION
Metabolic flexibility is essential for normal cardiac function. Although fatty acids are the principal fuel for cardiac energy production in normal conditions, the failing heart has reduced ability to utilize fatty acid and therefore relies more on glycolysis and/or ketones (1). As such, the propionate-mediated changes in fuel selection that we describe here may be relevant to cardiac disease development in individuals with increased circulating propionate concentrations. Using our established metabolic flux approach (22), this study investigated the mechanism of the cardiac fuel switch triggered by propionate. We investigated whether l-carnitine could decrease the metabolic perturbations associated with propionate overload based on the following considerations: 1) propionate causes CoA trapping by the accumulation of propionyl-CoA and MM-CoA, 2) the heart has high CrAT activity and this enzyme has the highest preference for propionyl-CoA, and 3) l-carnitine but not CoA can cross membranes and be up taken by the heart.
In this study, we show that propionate increased glucose uptake and oxidation without changing the efflux of lactate (lactate in effluent perfusate), a marker of glycolysis; propionate also suppressed fatty acid oxidation. The unchanged glycolysis could be because of: 1) the low acetyl-CoA in the propionate-perfused heart drives more pyruvate to enter the mitochondria for complete oxidation and 2) the increased aspartate derived from propionate promotes the malate shuttle and drives a redox shift to NAD+ leading to a decrease in lactate production.
The upregulated glucose oxidation is due to several factors. Suppression of fatty acid oxidation may lead to increased glucose oxidation through the glucose-fatty acid cycle (Randle cycle). Alternatively, the propionate-mediated increase of pyruvate recycling may increase PDH activity and thus increase glucose oxidation (11). The decreased acetyl-CoA/CoA ratio is also a contributor to enhance glucose oxidation.
Hypotheses to explain how propionate suppresses fatty acid oxidation include: 1) MM-CoA, which has a similar structure to malonyl-CoA, inhibits CPT-1 (33) and could competitively bind CPT-1; 2) excess propionyl-CoA leads to carnitine trapping through the increased formation of propionyl-carnitine (7, 32); and 3) excess propionyl-CoA leads to CoA trapping (18).
The inhibition of the CPT-1 by MM-CoA is unlikely based on our results. Moreover, the increased MM-CoA we observed is derived from propionyl-CoA metabolism, which occurs in mitochondria (47). Thus, the mitochondrial MM-CoA is unlikely to have access to CPT-1, which is located at the outer membrane of mitochondria. It is possible that methylcitrate could be transported out of the mitochondria and form cytosolic MM-CoA. However, we found that the methylcitrate concentration is only ~3% of the citrate concentration. Therefore, such trace amounts of cytosolic MM-CoA, which is a weak inhibitor of CPT-1, are unlikely to significantly inhibit CPT-1 and thus fatty acid oxidation (17, 31). Further evidences that the suppression of palmitate oxidation by propionate is not predominantly related to CPT are the observations that in all three perfusion groups (control, PA, and PA + Carn), levels of malonyl-CoA, which potently inhibits CPT-1, remained unchanged (Fig. 5A) and that oxidation of octanoate, which does not rely on the CPT system for transport into the mitochondria, was also significantly inhibited by propionate.
l-Carnitine is required to transport long-chain acyl-CoA species into the mitochondrial matrix for complete beta oxidation. CrAT has the highest affinity for propionyl-CoA among all other acyl-CoAs. Therefore, the accumulation of propionyl-CoA could deplete l-carnitine and potentially suppress mitochondrial oxidation of long-chain fatty acid. Indeed, we found that l-carnitine was decreased in the heart by propionate. Additionally, most other acylcarnitine species were decreased (Fig. 5, A and C), suggesting a depletion of l-carnitine. However, 3 mM l-carnitine failed to restore the propionate-suppressed palmitate oxidation as evidenced by the M1 labeling of acetyl-CoA, acetylcarnitine and other TCA intermediates in the M1 palmitate perfusions. The effect of l-carnitine extends beyond it role in transporting fatty acid into mitochondria for beta oxidation. l-Carnitine can also modulate CoA binding to PDH, which in turn regulates enzyme activity by promoting degaradtion of PDK4, a kinase that inhibits the PDH complex (36). This may partially explain why glucose oxidation remained increased in the 3 mM propionate + 3 mM l-carnitine perfused hearts (Fig. 3, B–H and J–M).
Mitochondrial CoA trapping is likely the key factor that mediates propionate-induced suppression of fatty acid oxidation. Our previous work showed that the activation of acetate to acetyl-CoA occurs only in the mitochondria (22). Propionate is similar to acetate and is activated to propionyl-CoA and is further metabolized to MM-CoA in cardiac mitochondria. The accumulation of both propionyl-CoA and MM-CoA depletes mitochondrial CoA. Additionally, propionate perfusion decreased most acyl-CoA species (Fig. 5, A and B) except malonyl-CoA (Fig. 5A). The lack of effect on malonyl-CoA concentrations is likely due to the fact that the majority of malonyl-CoA is located in the cytosol. The uneven changes of TCA metabolites in the propionate-perfused hearts indicate the low CoA concentration also lowered the concentration of acetyl-CoA and further decreased citric acid and 2-KG. As we have previously seen (33), the low acetyl-CoA and enhanced anaplerotic flux from propionate led to accumulation of metabolites in the TCA from succinate to oxaloacetate.
The minimal recovery of CoA concentrations by l-carnitine is likely due to our novel finding that the accumulated MM-CoA from propionate was minimally converted to MM-carnitine in the heart. The low activity of the conversion between dicarboxylyl-CoAs (C3 and C4) and dicarboxylylcarnitines (C3 and C4) in the heart is supported by the following: 1) malonylcarnitine, succinylcarnitine, and MM-carnitine concentrations were not affected by either propionate or propionate + l-carnitine perfusion; 2) in contrast to other acyl-CoA concentrations, the accumulation of MM-CoA in the propionate perfusions was not reduced by adding l-carnitine. Similar to MM-CoA, succinyl-CoA was not decreased by adding l-carnitine. In fact, l-carnitine increased succinyl-CoA by partially alleveviating the mitochondrial CoA trapping caused by propionate; 3) although malonyl-CoA and succinyl-CoA were significantly labeled by [13C6]glucose and [1-13C]palmitate, malonylcarnitine and succinylcarnitine were not, suggesting that de novo synthesis of these carnitine species was not occurring during the perfusions; 4) very little MM-carnitine or succinylcarnitine were labeled from 3 mM [13C3]propionate, which led to 93% M3 MM-CoA and 53% M3 succinyl-CoA in the perfused heart; 5) very little new C3 and C4 dicarboxylylcarnitines were synthesized in the hearts perfused with M3 carnitine from 0 to 60 min, whereas there was appreciable labeling of other M3 acylcarnitines; 6) our in vivo experiment showed that very little labeled MM-carnitine or succinylcarnitine were found in the heart when rats were injected with 200 mg/kg M3 propionate. All of these data strongly suggest that the heart has minimal enzyme activity to convert dicarboxylyl-CoAs (C3 and C4) to dicarboxylylcarnitines (C3 and C4). By comparison, liver had slightly higher activity in converting dicarboxylyl-CoAs (C3 and C4) and dicarboxylylcarnitines (C3 and C4), particularly succinyl-CoA to succinylcarnitine. Because the heart has much higher CrAT activity than liver, the relatively high conversion of C4 dicarboxylylcarnitines in liver is likely related to the non-CrAT carnitine acyltransferase, which is not specific to short-chain dicarboxylyl-CoAs or short-chain dicarboxylylcarnitines.
Similar situation of short-chain acyl-CoA species failing to convert to their cognate acylcarnitine species has been previously observed. Janos et al. (19) in 2014 perfused hearts with [1,2,3,4-13C4]palmitate. Although M2 malonyl-CoA was labeled at 19.9%, no malonylcarnitine labeling was observed. We had similar findings in hearts perfused with 13C-labeled substrates (glucose, palmitate, octanoate, or propionate), no malonylcarnitine was observed although malonyl-CoA was labeled. The explanation given by Janos et al. (19) was that the synthesized malonyl-CoA was located in cytosol, where it therefore had no access to CrAT or other unknown carnitine dicarboxylic acyltransferase that are located in mitochondria. This explanation would be correct if malonyl-CoA were exclusively located in cytosol. However, malonyl-CoA is found in mitochondria where it plays essential physiological roles (4). Furthermore, we showed that the low conversion between dicarboxylyl-CoA and dicarboxylylcarnitine was not only for C3 (malonyl-CoA) but also for C4 (succinyl-CoA and MM-CoA). Both succinyl-CoA and MM-CoA, derived from propionate metabolism, are located in mitochondria. Therefore, the lack of enzyme converting short-chain (C3 and C4) dicarboxylyl-CoAs to dicarboxylylcarnitines is likely the reason why we did not detect the interconversion. This agrees with previous finding that CrAT has very low activity in converting short-chain dicarboxylyl-acyl-CoA to their acylcarnitines (44). Therefore, as we observed, l-carnitine would not be effective to relieve the MM-CoA accumulation. In light of these findings, l-carnitine supplementation is unlikely to be helpful in methylmalonic acidemia patients. However, l-carnitine could be helpful for patients with propionic acidemia as suggested by Hoppel et al. (34), given the improvement in propionyl-CoA to propionylcarnitine conversion that we observed with l-carnitine supplementation.
Although we saw very little conversion of short-chain dicarboxylyl-CoA (C3 and C4) to their cognate acylcarnitine species, the heart has the highest concentrations of succinylcarnitine and MM-carnitine compared with other organs like brain, liver, and kidney (Fig. 8, F and G). The high concentration of cardiac succinylcarnitine and MM-carnitine could be due to their low metabolic activity in heart. The physiological and pathological roles of these 3 dicarboxylylcarnitines (C3 and C4) and their main source remain to be explored. Given recent reports that associate circulating dicarboxylylcarnitines with heart diseases (8, 16, 38), a better understanding of the underlying biology regulating these metabolites is needed. Our work indicates that the production of C3 and C4 dicarboxylylcarnitines in the heart is very subtle and that C3 and C4 dicarboxylylcarnitines are not direct metabolites from fatty acid oxidation. It is possible that omega oxidation of long-chain fatty acids in peroxisome or microsome could lead to Cn (n > 5) dicarboxylyl-CoAs and dicarboxylylcarnitines in the setting of mitochondrial dysfunction or when mitochondria are overloaded with fatty acid (13, 14). Moreover, the identities of dicarboxylylcarnitines in the previous reports need to be further confirmed (38), such as with high resolution mass spectrometry employed in this study or with a better liquid chromatography separation method, because dicarboxylylcarnitines and hydroxyl acylcarnitines with one or more carbons are isobaric compounds and are usually not differentiated by routine methods.
In summary, this study demonstrated that the heart lacks the specific enzymes that can catalyze the interconversion between short-chain (C3 and C4) dicarboxylyl-CoAs and their corresponding acylcarnitines. Propionate-mediated accumulation of propionyl-CoA and MM-CoA causes mitochondrial CoA trapping and alters cardiac fuel metabolism (summarized in Fig. 10), which cannot be completely alleviated by excess l-carnitine because of minimal activity of carnitine acyltransferase to MM-CoA in the heart. The corresponding metabolic changes induced by propionate in liver was also summarized in Fig. 10 based on data from this work and our previous publication (45).
Fig. 10.

Scheme of propionate-mediated perturbations in cardiac and hepatic metabolism. Cardiac and hepatic metabolic changes are summarized left and right, respectively. Propionate metabolism (red arrow) resulted in CoA trapping, which decreases fatty acid oxidation flux (blue arrow) and stimulates glucose metabolism and recycling flux from malate to pyruvate (red arrow). The interconversion between C3 and C4 dicarboxylyl-CoAs and their dicarboxylylcarnitines is negligible in heart and low in liver. The metablic results in liver (right panel) were from this work and previous publication (45). 2-KG, 2-ketoglutarate; Act-CoA, acetyl-CoA; Act-Carn, acetylcarnitine; Cit, citrate; CPT, carnitine palmitoyltransferase; Fum, fumarate; Glu, glutamate; Mal, malate; ME, malic enzyme; R-MM-CoA, (R)-methylmalonyl-CoA; S-MM-CoA: (S)-methylmalonyl-CoA; methylmalonyl-CoA; OAA, oxaloacetate; Palm-Carn, Palmitoylcarnitine; Palm-CoA, palmitoyl-CoA; PEP, phosphoenolpyruvate; PEPCK-C, cytosolic phosphoenolpyruvate carboxykinase; PEPCK-M, mitochondrial phosphoenolpyruvate carboxykinase; PDH, pyruvate dehydrogenase; Prop-Carn, propionylcarnitine; Prop-CoA, propionyl-CoA; Prop-Glu, propionylglutamate; Prop, propionate; Suc, succinate; Suc-CoA, succinyl-CoA; OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane.
GRANTS
This work was supported by Duke University startup funding to G. F. Zhang, Fundamental Research Funds for the Central Universities (grant no. 11617496) and the National Natural Science Foundation of China (grant no. 81402213) award to Y. Wang, and American Heart Association award no. 16SFRN31800000 to R. McGarrah. B. A. Christopher was supported by National Heart, Lung, and Blood Institute Grant No. T32-HL-007101.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.M., R.W.M., H.B., and G.-F.Z. conceived and designed research; Y.W., B.A.C., and K.A.W. performed experiments; Y.W., B.A.C., K.A.W., R.W.M., and H.B. analyzed data; D.M., R.W.M., H.B., and G.-F.Z. interpreted results of experiments; Y.W., R.W.M., and G.-F.Z. prepared figures; Y.W. and G.-F.Z. drafted manuscript; Y.W., B.A.C., K.A.W., D.M., R.W.M., H.B., and G.-F.Z. edited and revised manuscript; Y.W., B.A.C., K.A.W., D.M., R.W.M., H.B., and G.-F.Z. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors appreciate the suggestive discussions with Dr. Charles P. Venditti from National Human Genome Research Institute, National Institutes of Health, Bethesda, MD.
REFERENCES
- 1.Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Krüger M, Hoppel CL, Lewandowski ED, Crawford PA, Muoio DM, Kelly DP. The failing heart relies on ketone bodies as a fuel. Circulation 133: 698–705, 2016. doi: 10.1161/CIRCULATIONAHA.115.017355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhattacharya S, Granger CB, Craig D, Haynes C, Bain J, Stevens RD, Hauser ER, Newgard CB, Kraus WE, Newby LK, Shah SH. Validation of the association between a branched chain amino acid metabolite profile and extremes of coronary artery disease in patients referred for cardiac catheterization. Atherosclerosis 232: 191–196, 2014. doi: 10.1016/j.atherosclerosis.2013.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bloisi W, Colombo I, Garavaglia B, Giardini R, Finocchiaro G, Didonato S. Purification and properties of carnitine acetyltransferase from human liver. Eur J Biochem 189: 539–546, 1990. doi: 10.1111/j.1432-1033.1990.tb15520.x. [DOI] [PubMed] [Google Scholar]
- 4.Bowman CE, Rodriguez S, Selen Alpergin ES, Acoba MG, Zhao L, Hartung T, Claypool SM, Watkins PA, Wolfgang MJ. The mammalian malonyl-CoA synthetase ACSF3 is required for mitochondrial protein malonylation and metabolic efficiency. Cell Chem Biol 24: 673–684, 2017. doi: 10.1016/j.chembiol.2017.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butte NF, Liu Y, Zakeri IF, Mohney RP, Mehta N, Voruganti VS, Göring H, Cole SA, Comuzzie AG. Global metabolomic profiling targeting childhood obesity in the Hispanic population. Am J Clin Nutr 102: 256–267, 2015. doi: 10.3945/ajcn.115.111872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coude FX, Sweetman L, Nyhan WL. Inhibition by propionyl-coenzyme A of N-acetylglutamate synthetase in rat liver mitochondria. A possible explanation for hyperammonemia in propionic and methylmalonic acidemia. J Clin Invest 64: 1544–1551, 1979. doi: 10.1172/JCI109614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Degrace P, Demizieux L, Gresti J, Tsoko M, André A, Demaison L, Clouet P. Fatty acid oxidation and related gene expression in heart depleted of carnitine by mildronate treatment in the rat. Mol Cell Biochem 258: 171–182, 2004. doi: 10.1023/B:MCBI.0000012853.20116.06. [DOI] [PubMed] [Google Scholar]
- 8.Deidda M, Piras C, Dessalvi CC, Locci E, Barberini L, Torri F, Ascedu F, Atzori L, Mercuro G. Metabolomic approach to profile functional and metabolic changes in heart failure. J Transl Med 13: 297, 2015. doi: 10.1186/s12967-015-0661-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez CA, Des Rosiers C, Previs SF, David F, Brunengraber H. Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J Mass Spectrom 31: 255–262, 1996. doi:. [DOI] [PubMed] [Google Scholar]
- 10.Gao X, Zhang W, Wang Y, Pedram P, Cahill F, Zhai G, Randell E, Gulliver W, Sun G. Serum metabolic biomarkers distinguish metabolically healthy peripherally obese from unhealthy centrally obese individuals. Nutr Metab (Lond) 13: 33, 2016. doi: 10.1186/s12986-016-0095-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaimes R III, Kuzmiak-Glancy S, Brooks DM, Swift LM, Posnack NG, Kay MW. Functional response of the isolated, perfused normoxic heart to pyruvate dehydrogenase activation by dichloroacetate and pyruvate. Pflugers Arch 468: 131–142, 2016. doi: 10.1007/s00424-015-1717-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jameson E, Walter J. Cardiac arrest secondary to long QT(C) in a child with propionic acidemia. Pediatr Cardiol 29: 969–970, 2008. doi: 10.1007/s00246-007-9160-5. [DOI] [PubMed] [Google Scholar]
- 13.Janssen U, Stoffel W. Disruption of mitochondrial beta -oxidation of unsaturated fatty acids in the 3,2-trans-enoyl-CoA isomerase-deficient mouse. J Biol Chem 277: 19579–19584, 2002. doi: 10.1074/jbc.M110993200. [DOI] [PubMed] [Google Scholar]
- 14.Jin Z, Bian F, Tomcik K, Kelleher JK, Zhang GF, Brunengraber H. Compartmentation of metabolism of the C12-, C9-, and C5-n-dicarboxylates in rat liver, investigated by mass isotopomer analysis: anaplerosis from dodecanedioate. J Biol Chem 290: 18671–18677, 2015. doi: 10.1074/jbc.M115.651737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson DW. Synthesis of dicarboxylic acylcarnitines. Chem Phys Lipids 129: 161–171, 2004. doi: 10.1016/j.chemphyslip.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Kang SM, Park JC, Shin MJ, Lee H, Oh J, Ryu DH, Hwang GS, Chung JH. 1H nuclear magnetic resonance based metabolic urinary profiling of patients with ischemic heart failure. Clin Biochem 44: 293–299, 2011. doi: 10.1016/j.clinbiochem.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 17.Kashfi K, Mynatt RL, Cook GA. Hepatic carnitine palmitoyltransferase-I has two independent inhibitory binding sites for regulation of fatty acid oxidation. Biochim Biophys Acta 1212: 245–252, 1994. doi: 10.1016/0005-2760(94)90259-3. [DOI] [PubMed] [Google Scholar]
- 18.Kasumov T, Cendrowski AV, David F, Jobbins KA, Anderson VE, Brunengraber H. Mass isotopomer study of anaplerosis from propionate in the perfused rat heart. Arch Biochem Biophys 463: 110–117, 2007. doi: 10.1016/j.abb.2007.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kerner J, Minkler PE, Lesnefsky EJ, Hoppel CL. Fatty acid chain elongation in palmitate-perfused working rat heart: mitochondrial acetyl-CoA is the source of two-carbon units for chain elongation. J Biol Chem 289: 10223–10234, 2014. doi: 10.1074/jbc.M113.524314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurczynski TW, Hoppel CL, Goldblatt PJ, Gunning WT. Metabolic studies of carnitine in a child with propionic acidemia. Pediatr Res 26: 63–66, 1989. doi: 10.1203/00006450-198907000-00018. [DOI] [PubMed] [Google Scholar]
- 21.Lee TM, Addonizio LJ, Barshop BA, Chung WK. Unusual presentation of propionic acidaemia as isolated cardiomyopathy. J Inherit Metab Dis 32, Suppl 1: S97–S101, 2009. doi: 10.1007/s10545-009-1084-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Q, Deng S, Ibarra RA, Anderson VE, Brunengraber H, Zhang GF. Multiple mass isotopomer tracing of acetyl-CoA metabolism in Langendorff-perfused rat hearts: channeling of acetyl-CoA from pyruvate dehydrogenase to carnitine acetyltransferase. J Biol Chem 290: 8121–8132, 2015. doi: 10.1074/jbc.M114.631549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Q, Sadhukhan S, Berthiaume JM, Ibarra RA, Tang H, Deng S, Hamilton E, Nagy LE, Tochtrop GP, Zhang GF. 4-Hydroxy-2(E)-nonenal (HNE) catabolism and formation of HNE adducts are modulated by β oxidation of fatty acids in the isolated rat heart. Free Radic Biol Med 58: 35–44, 2013. doi: 10.1016/j.freeradbiomed.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Q, Zhang S, Berthiaume JM, Simons B, Zhang GF. Novel approach in LC-MS/MS using MRM to generate a full profile of acyl-CoAs: discovery of acyl-dephospho-CoAs. J Lipid Res 55: 592–602, 2014. doi: 10.1194/jlr.D045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manoli I, Sloan JL, Venditti CP. Isolated methylmalonic acidemia. In: GeneReviews, edited by Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, Ledbetter N. Seattle, WA: University of Washington, 1993. [Google Scholar]
- 26.Mardach R, Verity MA, Cederbaum SD. Clinical, pathological, and biochemical studies in a patient with propionic acidemia and fatal cardiomyopathy. Mol Genet Metab 85: 286–290, 2005. doi: 10.1016/j.ymgme.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 27.Marquis NR, Fritz IB. The distribution of carnitine, acetylcarnitine, and carnitine acetyltransferase in rat tissues. J Biol Chem 240: 2193–2196, 1965. [PubMed] [Google Scholar]
- 28.McDonnell E, Crown SB, Fox DB, Kitir B, Ilkayeva OR, Olsen CA, Grimsrud PA, Hirschey MD. Lipids reprogram metabolism to become a major carbon source for histone acetylation. Cell Reports 17: 1463–1472, 2016. doi: 10.1016/j.celrep.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mihalik SJ, Goodpaster BH, Kelley DE, Chace DH, Vockley J, Toledo FG, DeLany JP. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity (Silver Spring) 18: 1695–1700, 2010. doi: 10.1038/oby.2009.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Millington DS, Stevens RD. Acylcarnitines: analysis in plasma and whole blood using tandem mass spectrometry. Methods Mol Biol 708: 55–72, 2011. doi: 10.1007/978-1-61737-985-7_3. [DOI] [PubMed] [Google Scholar]
- 31.Mills SE, Foster DW, McGarry JD. Interaction of malonyl-CoA and related compounds with mitochondria from different rat tissues. Relationship between ligand binding and inhibition of carnitine palmitoyltransferase I. Biochem J 214: 83–91, 1983. doi: 10.1042/bj2140083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peschechera A, Scalibastri M, Russo F, Giarrizzo MG, Carminati P, Giannessi F, Arduini A, Ricciolini R. Carnitine depletion in rat pups from mothers given mildronate: a model of carnitine deficiency in late fetal and neonatal life. Life Sci 77: 3078–3091, 2005. doi: 10.1016/j.lfs.2005.03.029. [DOI] [PubMed] [Google Scholar]
- 33.Ragavan M, Kirpich A, Fu X, Burgess SC, McIntyre LM, Merritt ME. A comprehensive analysis of myocardial substrate preference emphasizes the need for a synchronized fluxomic/metabolomic research design. Am J Physiol Heart Circ Physiol 312: H1215–H1223, 2017. doi: 10.1152/ajpheart.00016.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roe CR, Millington DS, Maltby DA, Bohan TP, Hoppel CL. L-carnitine enhances excretion of propionyl coenzyme A as propionylcarnitine in propionic acidemia. J Clin Invest 73: 1785–1788, 1984. doi: 10.1172/JCI111387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Romano S, Valayannopoulos V, Touati G, Jais JP, Rabier D, de Keyzer Y, Bonnet D, de Lonlay P. Cardiomyopathies in propionic aciduria are reversible after liver transplantation. J Pediatr 156: 128–134, 2010. doi: 10.1016/j.jpeds.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 36.Schafer C, Young ZT, Makarewich CA, Elnwasany A, Kinter C, Kinter M, Szweda LI. Coenzyme A-mediated degradation of pyruvate dehydrogenase kinase 4 promotes cardiac metabolic flexibility after high-fat feeding in mice. J Biol Chem 293: 6915–6924, 2018. doi: 10.1074/jbc.RA117.000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seiler SE, Martin OJ, Noland RC, Slentz DH, DeBalsi KL, Ilkayeva OR, An J, Newgard CB, Koves TR, Muoio DM. Obesity and lipid stress inhibit carnitine acetyltransferase activity. J Lipid Res 55: 635–644, 2014. doi: 10.1194/jlr.M043448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shah AA, Craig DM, Sebek JK, Haynes C, Stevens RC, Muehlbauer MJ, Granger CB, Hauser ER, Newby LK, Newgard CB, Kraus WE, Hughes GC, Shah SH. Metabolic profiles predict adverse events after coronary artery bypass grafting. J Thorac Cardiovasc Surg 143: 873–878, 2012. doi: 10.1016/j.jtcvs.2011.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shah SH, Bain JR, Muehlbauer MJ, Stevens RD, Crosslin DR, Haynes C, Dungan J, Newby LK, Hauser ER, Ginsburg GS, Newgard CB, Kraus WE. Association of a peripheral blood metabolic profile with coronary artery disease and risk of subsequent cardiovascular events. Circ Cardiovasc Genet 3: 207–214, 2010. doi: 10.1161/CIRCGENETICS.109.852814. [DOI] [PubMed] [Google Scholar]
- 40.Shah SH, Sun JL, Stevens RD, Bain JR, Muehlbauer MJ, Pieper KS, Haynes C, Hauser ER, Kraus WE, Granger CB, Newgard CB, Califf RM, Newby LK. Baseline metabolomic profiles predict cardiovascular events in patients at risk for coronary artery disease. Am Heart J 163: 844–850, 2012. doi: 10.1016/j.ahj.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 41.Snyder NW, Basu SS, Worth AJ, Mesaros C, Blair IA. Metabolism of propionic acid to a novel acyl-coenzyme A thioester by mammalian cell lines and platelets. J Lipid Res 56: 142–150, 2015. doi: 10.1194/jlr.M055384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, Jeyaraj D, Youn JY, Ren S, Liu Y, Rau CD, Shah S, Ilkayeva O, Gui WJ, William NS, Wynn RM, Newgard CB, Cai H, Xiao X, Chuang DT, Schulze PC, Lynch C, Jain MK, Wang Y. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation 133: 2038–2049, 2016. doi: 10.1161/CIRCULATIONAHA.115.020226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tomcik K, Ibarra RA, Sadhukhan S, Han Y, Tochtrop GP, Zhang GF. Isotopomer enrichment assay for very short chain fatty acids and its metabolic applications. Anal Biochem 410: 110–117, 2011. doi: 10.1016/j.ab.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Violante S, Ijlst L, Ruiter J, Koster J, van Lenthe H, Duran M, de Almeida IT, Wanders RJ, Houten SM, Ventura FV. Substrate specificity of human carnitine acetyltransferase: Implications for fatty acid and branched-chain amino acid metabolism. Biochim Biophys Acta 1832: 773–779, 2013. doi: 10.1016/j.bbadis.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 45.Wilson KA, Han Y, Zhang M, Hess JP, Chapman KA, Cline GW, Tochtrop GP, Brunengraber H, Zhang GF. Inter-relations between 3-hydroxypropionate and propionate metabolism in rat liver: relevance to disorders of propionyl-CoA metabolism. Am J Physiol Endocrinol Metab 313: E413–E428, 2017. doi: 10.1152/ajpendo.00105.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolever TM, Fernandes J, Rao AV. Serum acetate:propionate ratio is related to serum cholesterol in men but not women. J Nutr 126: 2790–2797, 1996. [DOI] [PubMed] [Google Scholar]
- 47.Wongkittichote P, Ah Mew N, Chapman KA. Propionyl-CoA carboxylase - A review. Mol Genet Metab 122: 145–152, 2017. doi: 10.1016/j.ymgme.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang GF, Kombu RS, Kasumov T, Han Y, Sadhukhan S, Zhang J, Sayre LM, Ray D, Gibson KM, Anderson VA, Tochtrop GP, Brunengraber H. Catabolism of 4-hydroxyacids and 4-hydroxynonenal via 4-hydroxy-4-phosphoacyl-CoAs. J Biol Chem 284: 33521–33534, 2009. doi: 10.1074/jbc.M109.055665. [DOI] [PMC free article] [PubMed] [Google Scholar]