

Abstract
Excess fat intake can increase lipid oxidation and expression of mitochondrial proteins, indicating remodeling of the mitochondrial proteome. Yet intermediates of lipid oxidation also accumulate, indicating a relative insufficiency to completely oxidize lipids. We investigated remodeling of the mitochondrial proteome to determine mechanisms of changes in lipid oxidation following high-fat feeding. C57BL/6J mice consumed a high-fat diet (HFD, 60% fat from lard) or a low-fat diet (LFD, 10% fat) for 12 wk. Mice were fasted for 4 h and then anesthetized by pentobarbital sodium overdose for tissue collection. A mitochondrial-enriched fraction was prepared from gastrocnemius muscles and underwent proteomic analysis by high-resolution mass spectrometry. Mitochondrial respiratory efficiency was measured as the ratio of ATP production to O2 consumption. Intramuscular acylcarnitines were measured by liquid chromatography-mass spectrometry. A total of 658 mitochondrial proteins were identified: 40 had higher abundance and 14 had lower abundance in mice consuming the HFD than in mice consuming the LFD. Individual proteins that changed with the HFD were primarily related to β-oxidation; there were fewer changes to the electron transfer system. Gene set enrichment analysis indicated that the HFD increased pathways of lipid metabolism and β-oxidation. Intramuscular concentrations of select acylcarnitines (C18:0) were greater in the HFD mice and reflected dietary lipid composition. Mitochondrial respiratory ATP production-to-O2 consumption ratio for lipids was not different between LFD and HFD mice. After the 60% fat diet, remodeling of the mitochondrial proteome revealed upregulation of proteins regulating lipid oxidation that was not evident for all mitochondrial pathways. The accumulation of lipid metabolites with obesity may occur without intrinsic dysfunction to mitochondrial lipid oxidation.
Keywords: lipid, mitochondria, obesity, proteomics, skeletal muscle
INTRODUCTION
Obesity is a risk factor for the development of skeletal muscle insulin resistance and is associated with excess dietary consumption of lipids. Mitochondria comprise the primary site for lipid oxidation, and alterations in mitochondrial metabolism are implicated in the development of insulin resistance (30), but the mechanisms remain unclear. A contributing factor may be a change in the abundance of proteins involved in mitochondrial lipid metabolism. Insulin-resistant ob/ob mice demonstrated a large-scale proteomic signature toward an oxidative phenotype, including a greater content of mitochondrial enzymes than lean mice (38). Even a change in the abundance of a single mitochondrial protein, such as the lipid transporter carnitine-palmitoyl transferase I, can have downstream effects on insulin resistance (6). Understanding the impact of diet-induced obesity on the mitochondrial proteome can help determine if changes in single proteins or clusters of mitochondrial proteins occur during development of insulin resistance.
An overall decline in mitochondrial protein abundance and respiratory capacity has been identified in adults with obesity and insulin resistance compared with lean adults (4, 39). Adults with type 2 diabetes were also reported to have a lower content of genes involved in oxidative phosphorylation (25). Respiration studies of permeabilized muscle fibers indicated that lower mitochondrial respiration in adults with insulin resistance was accounted for by lower markers of mitochondrial content (5). In addition, production of reactive oxygen species (ROS) by the mitochondria may contribute to insulin resistance (1). The mitochondrial proteome is susceptible to damage by ROS, which may impact respiratory efficiency. Functional impairments in the mitochondrial proteome resulting from oxidative damage may contribute to lower capacity for mitochondria to oxidize excess availability of lipids during the development of obesity.
Other evidence has demonstrated that insulin resistance can occur along with increases in mitochondrial protein abundance and respiratory capacity (13, 24). We previously reported that mitochondrial protein synthesis was higher in mice after 12 wk of high-fat diet (HFD) than low-fat diet (LFD) consumption, and the increase occurred with greater lipid oxidation when measured for the whole body or isolated mitochondria (27). Higher rates for lipid oxidation and greater mitochondrial content have also been shown in overfeeding rat models of obesity (41). Such higher flux rates through lipid oxidation are implicated in the accumulation of lipid intermediates that may impair insulin signaling (22).
A central theme for increased or decreased mitochondrial oxidation with obesity is that mitochondrial lipid oxidation capacity does not appear sufficient to oxidize excess dietary lipids. Complete lipid oxidation involves multiple pathways, including fatty acid transport, β-oxidation to yield acetyl-CoA, and production of reducing equivalents that are ultimately oxidized by the electron transfer system. A relative imbalance of proteins may allow greater fatty acid uptake relative to oxidation and contribute to accumulation of lipid intermediates (44). The mitochondrial proteome undergoes dynamic remodeling that is regulated in part by insulin (3, 32, 34) and dietary lipid availability (8). Alterations in the mitochondrial proteome may reveal underlying mechanisms of changes in lipid oxidation with obesity and insulin resistance.
The purpose of the current investigation was to examine how changes in the mitochondrial proteome can explain gains in mitochondrial lipid oxidative capacity with obesity (27). We hypothesized that proteins regulating mitochondrial lipid oxidative capacity would be upregulated with obesity and that such increases would occur across multiple pathways. We used a mouse model of high-fat feeding and then isolated mitochondria from skeletal muscle for analysis by mass spectrometry (MS). Our primary findings indicate greater abundance of proteins for fatty acid transport and β-oxidation with fewer changes in the electron transfer system. There was an accumulation of select species of intramuscular acylcarnitines, but there were no changes in phosphorylation efficiency of lipid substrates. Overall, HFD-induced obesity appeared to remodel the mitochondrial proteome toward greater fatty acid uptake and β-oxidation without intrinsic dysfunction of mitochondrial lipid oxidation.
METHODS
Mouse model.
The study was approved by the Animal Care and Use Committees at Oregon State University (approval no. 4788) and the Mayo Clinic (approval no. 385-14). Eight- to 10-wk-old male C57BL/6J mice (product no. 000664, Jackson Laboratories) were fed a HFD (60% fat) or a LFD for 12 wk (n = 8 per diet). Table 1 displays dietary lipid composition (Research Diets, New Brunswick, NJ). The percent kilocalories from total fat, carbohydrates, and protein were 10%, 70%, and 20%, respectively, for the LFD (stock no. D12450J) and 60%, 20%, and 20%, respectively, for the HFD (stock no. D12492). Diets were matched for sucrose. Cholesterol content was 279.6 and 51.6 mg/kg for the HFD and LFD, respectively. Mice were housed three to five per cage in a 12:12-h light-dark cycle at 22°C with free access to food and water. Metabolic phenotyping of these mice, including induction of obesity, development of insulin resistance as demonstrated by glucose and insulin tolerance tests, and increase in mitochondrial respiration specific to lipid substrates in the HFD mice, was previously reported (27). Tissues for the current proteomic analysis were collected from the same mice upon euthanasia. Mice were fasted for 4 h and then anesthetized by pentobarbital sodium overdose, and tissues were rapidly frozen in liquid nitrogen and stored at −80°C. Anesthetics can impair respirometry (9), but the dose of pentobarbital sodium was in a range that does not appear to impair mitochondrial respiration (40).
Table 1.
Dietary lipid composition of low- and high-fat diets
| Low-Fat Diet |
High-Fat Diet |
|||
|---|---|---|---|---|
| Absolute, g/kg diet | Relative to total fat, % | Absolute, g/kg diet | Relative to total fat, % | |
| C10 | 0 | 0.00 | 0.1 | 0.04 |
| C12 | 0 | 0.00 | 0.2 | 0.08 |
| C14 | 0.2 | 0.46 | 2.8 | 1.10 |
| C15 | 0 | 0.00 | 0.2 | 0.08 |
| C16 | 6.5 | 14.87 | 49.9 | 19.61 |
| C16:1 | 0.3 | 0.69 | 3.4 | 1.34 |
| C17 | 0.1 | 0.23 | 0.9 | 0.35 |
| C18 | 3.1 | 7.09 | 26.9 | 10.57 |
| C18:1 | 12.6 | 28.83 | 86.6 | 34.04 |
| C18:2 | 18.3 | 41.88 | 73.1 | 28.73 |
| C18:3 | 2.2 | 5.03 | 5.2 | 2.04 |
| C20 | 0 | 0.00 | 0.4 | 0.16 |
| C20:1 | 0.1 | 0.23 | 1.5 | 0.59 |
| C20:2 | 0.2 | 0.46 | 2 | 0.79 |
| C20:3 | 0 | 0.00 | 0.3 | 0.12 |
| C20:4 | 0.1 | 0.23 | 0.7 | 0.28 |
| C22:5 | 0 | 0.00 | 0.2 | 0.08 |
| Total | 43.7 | 100 | 254.5 | 100 |
| SFA | 9.9 | 22.7 | 81.5 | 32.0 |
| MUFA | 13.0 | 29.9 | 91.5 | 35.9 |
| PUFA | 20.7 | 47.4 | 81.5 | 32.0 |
Data for low-fat diet (stock no. D12450B) and high-fat diet (stock no. 12492) were provided by the manufacturer (Research Diets, New Brunswick, NJ). MUFA, monounsaturated fatty acid; PUFA, polyunsaturated fatty acid; SFA, saturated fatty acid.
Mitochondrial preparation.
Gastrocnemius muscles were processed by differential centrifugation to obtain a sample enriched for mitochondrial proteins. All buffers and tubes were kept on ice throughout the processing. Muscle samples (~90 mg) were powdered in liquid nitrogen, diluted 1:20 (wt/vol) in buffer A (in mM: 100 KCl, 50 Tris base, 5 MgCl2·6H2O, 1.8 ATP, and 1 EDTA, pH 7.2), and then homogenized using custom glass-on-glass homogenizers with 0.3-mm spacing between mortar and pestle. Samples were centrifuged for 5 min at 750 g and 4°C. The supernatant was transferred to a new tube and centrifuged for 5 min at 10,000 g and 4°C. The pellet was collected, suspended in buffer A, and then centrifuged for 5 min at 9,000 g and 4°C. The final pellet was enriched in mitochondrial proteins and suspended in lysis buffer [20 mM Tris·HCl, pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1× protease inhibitor cocktail (catalog no. P8340, Sigma)]. Protein concentrations were determined by bicinchoninic assay (Pierce BCA, Thermo Fisher Scientific, Waltham, MA), and 20 μg from each sample was separated on a 12.5% Tris-HCl Criterion gel (Bio-Rad, Hercules, CA) and stained with Bio-Safe Coomassie Stain (Bio-Rad) according to the directions. Analysis of extracts of gastrocnemius homogenates and each fraction revealed citrate synthase activity was similar between diet groups when normalized to protein concentration of muscle fractions extracted, indicating similar extraction efficiencies (data not shown).
High-resolution mass spectrometry.
Each gel lane was cut into five sections (Fig. 1). Proteins were destained, reduced, and alkylated followed by in situ digestion with 0.2 μg trypsin (Promega, Madison WI) at 37°C overnight, followed by peptide extraction with 2% trifluoroacetic acid and acetonitrile. Extractions were dried and stored at −20°C. Dried trypsin-digested samples were suspended in sample buffer (0.2% formic acid/0.1% trifluoracetic acid/0.002% Zwittergent 3–16), a portion of which was analyzed by nano-flow liquid chromatography electrospray tandem mass spectrometry (nanoLC-ESI-MS/MS) using a Thermo Ultimate 3000 RSLCnano HPLC system coupled to a Thermo Scientific Q-Exactive Mass Spectrometer (Thermo Fisher Scientific, Bremen, Germany). Chromatography was performed using solvent A (98% water/2% acetonitrile/0.2% formic acid) and solvent B (80% acetonitrile/10% isopropanol/10% water/2% formic acid), over a 2–45% B gradient for 60 min separating the peptides using an in-house column packed with Agilent Poroshell 120 EC C18 (Agilent, Santa Clara, CA). Q-Exactive mass spectrometer was set to acquire an ms1 survey scans from 350–1600 m/z at resolution 70,000 (at 200 m/z) with an AGC target of 3e6 ions. Survey scans were followed by HCD MS/MS scans on the top 15 ions at resolution 17,500 with an AGC target of 2e5 ions. Dynamic exclusion placed selected ions on an exclusion list for 45 s.
Fig. 1.

Representative gel showing molecular weight cutoffs (kDa, far-right lane) and identification of gel sections [5 sections (A–E) per sample]. Mitochondrial proteins were loaded and separated by gel electrophoresis, gels were sectioned, and proteins were digested with trypsin before mass spectrometry.
Identification of proteins and posttranslational modifications.
LC-tandem MS (LC-MS/MS) data were processed using a previously described peptide intensity-based, label-free bioinformatics method (33). MaxQuant (version 1.5.2) software was configured to detect peptide features and match the corresponding MS/MS against a Swiss-Prot mouse reference proteome (v58) supplemented with protein sequences of common sample-handling contaminants (e.g., human keratin and sheep keratin) and mouse MitoCarta 2.0 database (7). Reversed sequences were appended to the database to estimate false discovery rate (FDR) of peptide and protein identifications. MaxQuant was also instructed to use 10 ppm m/z error for both precursor and fragments, derive semitryptic peptides from the database, and identify carbamidomethylation of cysteine, oxidation of methionine, phenylalanine, and tryptophan, deamidation of asparagine and glutamine, N-terminal pyroglutamic acid, and acetylation of lysine as variable modifications. Protein group (i.e., group of proteins that are indistinguishable by LC-MS/MS) identifications with at least two distinct peptide identifications were further filtered to overall FDR of 1%.
Next, protein group intensities between a pair of experimental groups of interest were compared to find differentially expressed proteins as follows: filtered protein group intensities were log2-transformed (to achieve normality) and normalized to remove batch effects using a software approach described in detail elsewhere (2). Briefly, the normalized intensities for each protein group were modeled using a Gaussian-linked generalized linear model, and ANOVA contrasts were utilized to assess statistical significance of differential expression. Protein groups with an absolute log2 fold change ≥0.5 (where 0.0 signifies no change) and an adjusted differential expression P ≤ 0.05 were considered candidates for further analyses. Relative expression of protein posttranslational modification (PTM; at the sample level) between two groups was determined using peptide intensities as previously described (20) and then displayed as percent difference between diet groups. Raw data files are available at the MassIVE repository (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp).
Gene set enrichment and pathway analysis.
We used gene set enrichment analysis (GSEA) to identify pathways that were differentially regulated between LFD and HFD. Protein lists were ranked based on differential expression fold change and statistical significance and then processed using WebGestalt (42). Protein lists were generated for differentially expressed proteins between diet groups and processed by Ingenuity Pathway Analysis (Qiagen) to determine canonical pathways and upstream regulators. Pathways and regulators with an enriched adjusted P ≤ 0.05 were considered for interpretation.
Quantitative PCR.
Total mRNA was extracted from gastrocnemius muscles to determine transcriptional activation of peroxisome proliferator-activated receptor (PPAR) signaling. mRNA concentration and contamination were determined by spectrophotometry (NanoDrop, Thermo Fisher Scientific), and then 1 μg was reverse-transcribed to cDNA. Quantitative PCR was performed in triplicate in 384-well clear plates using SYBR green reagents (Thermo Fisher Scientific), ~20 ng of cDNA, and 100 nM primers in 20-μl reaction volumes. Thermocycler conditions were as follows: 10 min at 60°C, 40 cycles of denaturation for 15 s (95°C) and annealing/extension for 60 s (60°C), and a melt curve. Relative quantification was performed using a five-point standard curve that spanned 3 log dilutions. Amplification efficiencies were similar between target and reference genes, which were analyzed on a single plate along with no-template controls (see sequence information in Table 2). Melt curves revealed single peaks for each primer set.
Table 2.
Primer sequence information
| Name | Gene Name | Reference Sequence | Sequence |
|---|---|---|---|
| Pyruvate dehydrogenase kinase 4 | Pdk4 | NM_013743.2 | F: GCCTAGGTGGGCGTCAGGATGA R: CCAGCTGCACCGAAGGAGTGT |
| Mitochondrial carnitine acylcarnitine translocase | Slc25a20 | NM_020520.3 | F: GAGAACGGATCAAATGCTTACTG R: TCACTGACACTCTTTCCCTC |
| Heat shock protein 90ab1 | Hsp90 | NM_008302 | F: CATCATGGACAGCTGTGACG R: AGTTCTCCTTGTCCTCAGCC |
| Cyclophilin A | Ppia | NM_008907.1 | F: CTTCTTGCTGGTCTTGCCATTCCT R: GGATGGCAAGCATGTGGTCTTTG |
F, forward; R, reverse.
Lipid extraction.
Quadriceps muscle (~15 mg) was dissected under a microscope to remove any contaminating lipid or adipose tissue. Dissected muscles were homogenized in 950 μl of ice-cold ultrapure water (ddH2O). A 750-μl sample was transferred to glass screw-cap tubes, and 900 μl of methanol, 3 ml of methyl tert-butyl ether, and 40 μl of internal standard were added. Samples were vortexed and rotated to extract lipids and centrifuged at 2,500 g for 5 min to separate phases. The upper phase containing lipids was collected. The lower phase was extracted again and combined with the first lipid extract. Nitrogen gas and low heat (~37°C) were used to dry the combined extracts. Dried lipids were transferred to autosampler vials and dried again. Lipids were resuspended in 200 μl of 95:5:0.1 hexane-dichloromethane-acetic acid for analysis using a high-performance LC (model 1100, Agilent) connected to a triple-quadrupole LC-MS/MS system (model API 2000, MX, Sciex, Framingham, MA). Specific multiple-reaction monitoring was used for each molecular species of lipids. Ten percent of the lipid extract was injected onto a 3 × 100 m hydrophilic interaction LC column. A gradient from 15% B to 80% B of the mobile phase, where A is 3:2:0 hexane-isopropanol-water and 0.056% acetic acid and B is 3:4:0.12 hexane-isopropanol-water and 0.1% acetic acid, both 10 mM in ammonium acetate, was used to separate the polar lipids. Concentration of each lipid molecular species in the samples was determined by comparison of area ratio (peak area of analyte ÷ peak area of its internal standard) with standard curves. Lipid species within the linear range were quantified using MultiQuant software (Sciex).
Mitochondrial respiratory efficiency.
We determined O2 consumed during a subsaturating pulse of 113.6 nmol of ADP [ratio of ATP production to O2 consumption (P:O)] using high-resolution respirometry analysis (Oroboros Instruments, Innsbruck, Austria). The P:O assumes that ADP consumption and ATP production occur in a 1:1 molar ratio. Respirometry was performed at 37°C on mitochondria that were isolated from quadriceps muscle using differential centrifugation, as previously reported (27). The ADP pulse was delivered during titrations of substrates, providing electrons through β-oxidation (5 μM palmitoylcarnitine + 2 mM malate) or NADH-linked respiration (10 mM glutamate + 2 mM malate). The respiration buffer was MiR05 (0.5 mM EGTA, 3 mM MgCl2·6H2O, 60 mM lactobionic acid, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM sucrose, and 1 g/l bovine serum albumin).
Statistics.
Unpaired t-tests were used to compare univariate data, including individual PTM, lipid species, and P:O, with statistical significance set at 0.05. Group sizes were based on initial power calculations based on variance of a mitochondrial PTM (mitochondrial deamidation) of 636 ± 62 determined from seven C57BL/6J mice following a stimulus for mitochondrial damage [48 h of insulin deprivation (32)]. With the assumption of a standard deviation of 62 and α = 0.05, a sample size of n = 8 per group would have power to detect a difference of 93 (at 1 − β = 0.8) or 65 (at 1 − β = 0.5). We anticipated that n = 8 per diet would have statistical power to detect physiologically relevant (>10%) differences in protein abundance and PTM.
RESULTS
Metabolic phenotyping of these mice demonstrated insulin resistance with a HFD, as determined by glucose and insulin tolerance tests, along with increased whole body lipid oxidation and mitochondrial oxidation capacity that was specific to lipids (27). In mice consuming a HFD, abundance of β-hydroxyacyl-CoA dehydrogenase was greater, but there was no difference in subunits for mitochondrial respiratory complexes when measured by immunoblotting. The same mice were used for the current proteomics analysis to further understand mitochondrial alterations to a HFD. A mitochondrial-enriched fraction was prepared from gastrocnemius muscle of LFD and HFD mice, and high-resolution MS was used to identify 2,625 proteins, of which 658 were specific to mitochondria (see Supplemental Material for this article available online at the Journal website). Using a fold change cutoff of 0.5 and P ≤ 0.05, a total of 131 proteins (40 mitochondrial) were increased with the HFD, while 103 proteins (14 mitochondrial) were lower in HFD than LFD mice (Fig. 2).
Fig. 2.
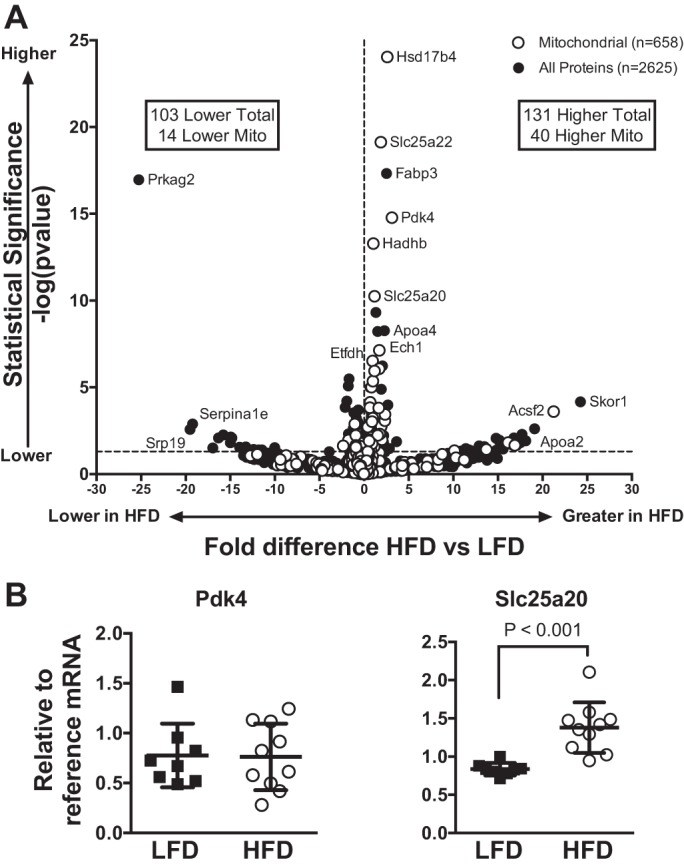
A: volcano plot displaying fold differences in individual proteins between mice consuming the low-fat diet (LFD) and mice consuming the high-fat diet (HFD) for 12 wk. Proteins were identified by high-resolution mass spectrometry of mitochondrial-enriched samples prepared from gastrocnemius muscle. Gene symbols are displayed for select proteins: Apoa, apolipoprotein A; Ascl2, achaete-scute family basic helix-loop-helix transcription factor 2; Ech1, enoyl-CoA hydratase 1; Etfdh, electron transfer flavoprotein dehydrogenase; Fabp3, fatty acid-binding protein 3; Hadhb, hydroxyacyl-CoA dehydrogenase trifunctional enzyme subunit β; Hsd17b4, hydroxysteroid 17β dehydrogenase 4; Prkag2, protein kinase AMP-activated noncatalytic subunit γ2; Serpina1e, serpin family A member 1e; Skor1, Sk1 family transcriptional corepressor 1; Slc25a, solute carrier family 25a; Srp19, signal recognition particle 19. Horizontal dashed line represents statistical significance of P = 0.05 with n = 8 per diet group. B: relative quantification of mRNA by real-time polymerase chain reaction for select genes expressed relative to cyclophilin [for pyruvate dehydrogenase kinase 4 (Pdk4)] and heat shock protein 90 [for solute carrier family 25a member 20 (Slc25a20)] and compared by unpaired t-test. Individual data points are displayed along with mean (SD) (n = 8 per diet group).
The mitochondrial proteins that increased with the HFD compared with the LFD were grouped according to their role in mitochondrial metabolism (Fig. 3A). There was an overall increase in proteins related to β-oxidation, and there were fewer changes in proteins related to electron transfer or oxidative phosphorylation. Of the 54 mitochondrial proteins that were altered with the HFD, 30 were related to lipid oxidation, including β-oxidation proteins, lipid transporters (e.g., carnitine-palmitoyl transferase 2), or proteins regulating β-oxidation (e.g., acetyl-CoA carboxylase-β). Seven proteins were related specifically to the electron transfer system, while 17 were related to other functions, including structure (e.g., mitochondrial elongation factor 2) or regulation of the tricarboxylic acid cycle (e.g., pyruvate dehydrogenase kinase 2). The overall remodeling of the mitochondrial proteome with the HFD revealed greater changes in the abundance of β-oxidation proteins than proteins of other mitochondrial pathways.
Fig. 3.
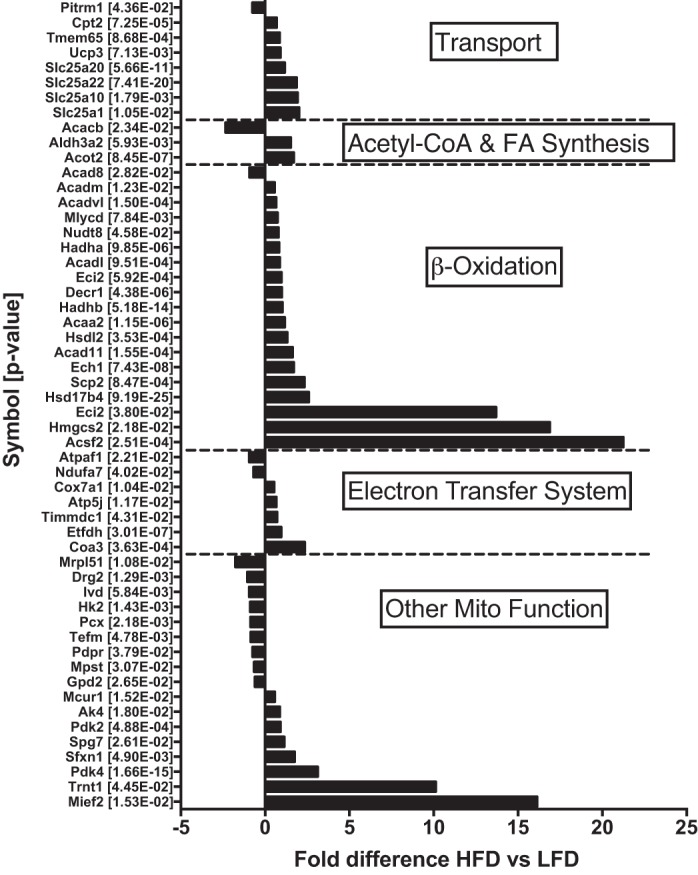
Individual mitochondrial proteins with fold difference ≥0.5 and P ≤ 0.05 were grouped according to function and are displayed as fold difference between mice consuming the low-fat diet (LFD) and mice consuming the high-fat diet (HFD). Proteins are displayed with gene symbol and exact P value for unpaired t-test of fold difference between diet groups (n = 8 per diet group). FA, fatty acid; Mito, mitochondrial.
We considered the upstream regulators of the proteins that increased with the HFD to determine potential signals for coordinated upregulation of groups of proteins within the mitochondrial proteome. Analysis of the upstream regulators identified transcription factors regulating lipid oxidation (e.g., PPARα and PPARδ) and targets of lipid-lowering drugs (Table 3). An increase in mRNA abundance with the HFD was determined for select PPAR-regulated genes (Fig. 3B). PPARγ coactivator (PGC)-1α was identified as an upstream regulator for proteins that were decreased with the HFD.
Table 3.
Upstream regulators identified from proteins that were increased or decreased in mice fed the HFD compared with mice fed the LFD
| Upstream regulators of proteins | P Value |
|---|---|
| Increased with HFD | |
| Pirinixic acid | 1.09 × 10–23 |
| Bezafibrate | 1.71 × 10–22 |
| PPARα | 5.33 × 10–21 |
| PPARδ | 5.16 × 10–20 |
| Fenofibrate | 1.41 × 10–17 |
| Decreased with HFD | |
| EBF1 | 2.42 × 10–4 |
| Interleukin 15 | 4.96 × 10–4 |
| TRPC4AP | 5.00 × 10–4 |
| Huntingtin | 3.27 × 10–3 |
| PGC-1α | 3.88 × 10–3 |
HFD, high-fat diet; LFD, low-fat diet; PPAR, peroxisome proliferator-activated receptor; EBF1, early B cell factor 1; TRPC4AP, transient receptor potential cation channel subfamily C member 4-associated protein; PGC1α, peroxisome proliferator-activated receptor-γ coactivator 1α.
Pathways that changed with the HFD were identified using GSEA, which takes into account the magnitude and significance of the changes in expression along with the relative contribution to a given pathway. GSEA revealed multiple pathways related to β-oxidation and lipid metabolism that were upregulated with the HFD (Table 4). The most enriched pathways that were identified included metabolism of lipids and lipoproteins and fatty acid, triacylglycerol, and ketone body metabolism. Pathways that were downregulated with the HFD included those that were not specific to the mitochondria, such as membrane trafficking and vesicle-mediated transport, as well as pathways related to activation of PGC-1α, import of lipids to mitochondria, and energy metabolism (Table 5). GSEA revealed that HFD increased expression pathways enriched with mitochondrial proteins that were related to lipid oxidation.
Table 4.
GSEA of proteins that decreased in skeletal muscle mitochondrial fractions from mice fed the HFD compared with mice fed the LFD for 12 wk
| GSEA Identification | Pathway | No. of Genes | FDR |
|---|---|---|---|
| R-MMU-2151209 | Activation of PGC-1α by phosphorylation | 3 | 4.65e-02 |
| R-MMU-200425 | Import of palmitoyl-CoA into mitochondrial matrix | 3 | 4.65e-02 |
| R-MMU-199991 | Membrane trafficking | 12 | 4.65e-02 |
| R-MMU-109703 | Protein kinase B-mediated events | 4 | 4.65e-02 |
| R-MMU-165159 | mTOR signaling | 4 | 4.65e-02 |
| R-MMU-5653656 | Vesicle-mediated transport | 12 | 4.97e-02 |
| R-MMU-5628897 | Tumor protein 53 regulates metabolic genes | 4 | 4.97e-02 |
| R-MMU-163685 | Integration of energy metabolism | 5 | 8.87e-02 |
| R-MMU-1592230 | Mitochondrial biogenesis | 3 | 8.87e-02 |
| R-MMU-163680 | AMPK inhibits chREBP transcriptional activation activity | 2 | 9.78e-02 |
GSEA, gene set enrichment analysis; HFD, high-fat diet; LFD, low-fat diet; FDR, false discovery rate; PGC1α, peroxisome proliferator-activated receptor-γ coactivator 1α; mTOR, mechanistic target of rapamycin; AMPK, AMP-activated protein kinase; chREBP, carbohydrate-responsive element-binding protein.
Table 5.
GSEA of proteins that increased in skeletal muscle mitochondrial fractions from mice fed the HFD compared with mice fed the LFD for 12 wk
| GSEA Identification | Pathway | No. of Genes | FDR |
|---|---|---|---|
| R-MMU-556833 | Metabolism of lipids and lipoproteins | 32 | 5.65e-12 |
| R-MMU-1430728 | Metabolism | 45 | 3.93e-09 |
| R-MMU-535734 | Fatty acid, triacylglycerol, and ketone body metabolism | 14 | 2.82e-08 |
| R-MMU-77289 | Mitochondrial fatty acid β-oxidation | 6 | 1.04e-05 |
| R-MMU-390918 | Peroxisomal lipid metabolism | 6 | 1.33e-04 |
| R-MMU-77286 | Mitochondrial fatty acid β-oxidation of saturated fatty acids | 4 | 1.85e-04 |
| R-MMU-77346 | β-Oxidation of decanoyl-CoA to octanoyl-CoA-CoA | 3 | 1.93e-03 |
| R-MMU-77348 | β-Oxidation of octanoyl-CoA to hexanoyl-CoA | 3 | 1.93e-03 |
| R-MMU-174800 | Chylomicron-mediated lipid transport | 4 | 1.64e-02 |
| R-MMU-6809371 | Formation of the cornified envelope | 6 | 1.64e-02 |
GSEA, gene set enrichment analysis; HFD, high-fat diet; LFD, low-fat diet; FDR, false discovery rate.
We considered if the changes in protein abundance, with smaller changes in electron transfer system proteins, would have a functional consequence on mitochondria. 1) There was the potential for a buildup of intermediates of lipid oxidation due to the increase in proteins related to β-oxidation relative to the electron transfer system. 2) There were increases to specific acylcarnitines species, such as 18:0 (+87%, P < 0.05), with less change (P = 0.1) in 16:0 and 18:2, in HFD mice (Fig. 4A). Such increases may be a result of lower respiratory efficiency of lipids. However, the mitochondrial P:O measured during palmitoylcarnitine- or glutamate-malate-supported respiration was not different between diet groups (Fig. 4B). 3) There was a potential for accumulation of ROS damage, given that mitochondrial proteins are in close proximity to O2 and electron flow through the electron transfer system. A relative imbalance of protein abundance may impact efficient electron flow. We determined oxidation and deamidation as markers of irreversible damage to mitochondrial proteins and did not detect differences between the LFD and HFD (Fig. 5). Overall, these results indicate that the buildup of specific, but not all, lipid intermediates occurred without major defects in the efficiency of the mitochondrial respiratory chain system or accumulation of ROS damage.
Fig. 4.
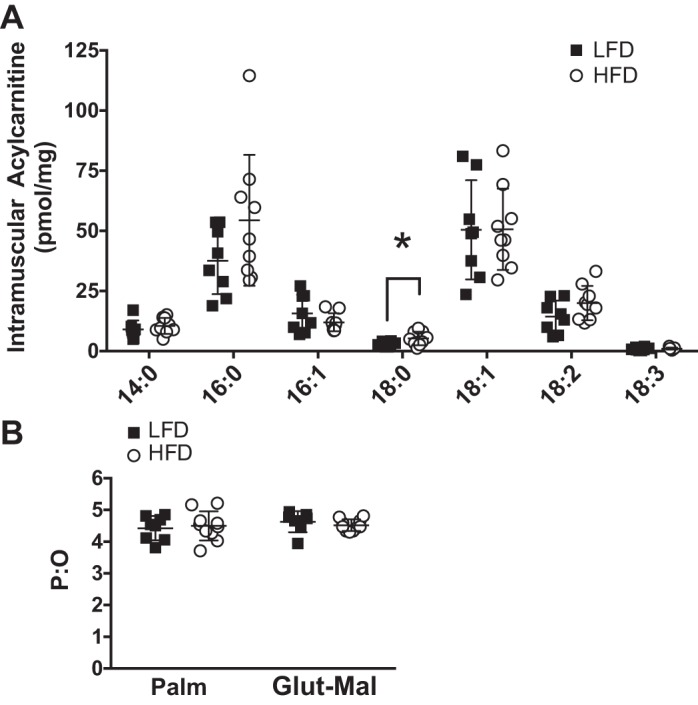
A: skeletal muscle acylcarnitines identified by liquid chromatography paired with mass spectrometry and classified based on saturation and chain length. B: mitochondrial phosphorylation efficiency [ratio of ATP production to O2 consumption (P:O)] of mice consuming the low-fat diet (LFD) and mice consuming the high-fat diet (HFD) for 12 wk determined by a subsaturating pulse of ADP during high-resolution respirometry using substrates of palmitoylcarnitine (Palm) and glutamate-malate (Glu-Mal). Bars represent mean (SD) (n = 8 per diet group). *P ≤ 0.05, LFD vs. HFD (by unpaired t-test).
Fig. 5.

Accumulation of oxidative or deamidation modification to amino acid residues of proteins identified by high-resolution mass spectrometry of mitochondrial-enriched samples collected from the gastrocnemius muscle of mice following 12 wk of low-fat diet (LFD) or high-fat diet (HFD) consumption. Oxidative damage and deamidation were compared between all proteins that were identified (A and B) and also separated to include only mitochondrial proteins (C and D). Bars represent mean (SD) (n = 8 per diet group). Differences between diet groups were compared by unpaired t-test. AU, arbitrary units.
DISCUSSION
Our results indicate remodeling of the skeletal muscle mitochondrial proteome toward greater abundance of proteins involved in β-oxidation, but not of all mitochondrial pathways, in mice consuming a HFD for 12 wk. Such adaptations may favor the initiation of lipid oxidation. The changes in protein abundance did not alter the respiratory efficiency of lipid substrates or mitochondrial protein damage, as measured by amino acid residues with oxidative or deamidation modifications. Overall, remodeling of the mitochondrial proteome with high-fat feeding was consistent with greater initiation of lipid oxidation without evidence of intrinsic respiratory impairments.
We tested the overall hypothesis that high-fat feeding would globally increase proteins involved with lipid oxidation as a mechanism to explain changes in respiratory phenotypes with high-fat feeding. Interestingly, the differences in protein abundance were predominantly in β-oxidation and less in other pathways, specifically the electron transfer system. The increase in β-oxidation proteins is in agreement with increases in mitochondrial content and respiration following high-fat feeding and insulin resistance (13, 24). Such changes are consistent with our previous report of increased lipid respiration and electron transfer system capacity in these high-fat-fed mice (27). Other proteomic comparisons also showed that differences in protein abundance are not uniform across all pathways. For example, in human muscle homogenates from adults with obesity or type 2 diabetes, expression of mitochondrial proteins specific to the electron transfer system was lower (16). Abundance of lipid oxidation proteins was greater in leptin-deficient obese than lean mice, and a shift toward oxidative fiber types was reported in leptin-deficient obese compared with lean mice (38). Overall, the proteomic changes may represent a greater increase in β-oxidation proteins than in the electron transfer system. These changes do not support a loss of mitochondria with insulin resistance but indicate greater initiation of lipid oxidation and possible incomplete oxidation of lipids.
The identification of specific proteins that changed with the HFD provides mechanisms contributing to increases in mitochondrial lipid oxidation. Indeed, proteins with the greatest changes in abundance were major regulators that promote fatty acid oxidation, including acetyl-CoA carboxylase-β, isobutyryl-CoA dehydrogenase 8, and mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase 2. Enoyl-CoA-δ-isomerase 2, a protein involved in β-oxidation of unsaturated lipids (18), was increased to a large extent in HFD-fed mice. Such an increase may contribute to differences in acylcarnitine content that varied between degrees of saturation, including increases in saturated acylcarnitine species (e.g., 18:0 and a tendency for 16:0), but varied less for unsaturated species (e.g., 18:1 and 16:1). The acylcarnitine species of 14–18 carbon chains were within the linear range of quantification. We cannot exclude the possibility of changes in short-chain species with very low abundance. Acetyl-CoA synthetase family member 2 catalyzes the formation of acetyl-CoA from acetate and coenzyme A and was increased more in HFD- than LFD-fed mice. These increases in individual proteins were consistent with greater lipid oxidation capacity with the HFD.
A consideration is how specific proteins increased with the HFD while others did not. The common upstream regulators that increased with the HFD were members of the PPAR family of lipid-sensitive transcription factors. We measured increases in protein and mRNA abundance for targets activated by PPAR, particularly PPARα. Such activation may support mitochondrial protein synthesis, which is regulated in part by insulin concentrations when sufficient amino acids are available (3, 34). We previously reported that mitochondrial protein synthesis rates were increased with a HFD, regardless of improvements in insulin sensitivity with pioglitazone treatment (27). Such measures are an average rate based on all mitochondrial proteins, which can vary between individual mitochondrial proteins (17). The selective increase in mitochondrial proteins may be a combined response to increased activation of lipid-responsive genes and protein synthesis supported by hyperinsulinemia.
We used pathway analysis, specifically, GSEA, that considers the magnitude and significance of proteins contributing to a pathway. This approach allows identification of pathways based on multiple features with changes that are small and coordinated and decreases the potential for pathways to be identified by a few specific proteins that change with large magnitude. GSEA identified multiple pathways for lipid metabolism (9 of the top-10 pathways identified) that were increased with the HFD. Collectively, both GSEA and the identification of individual proteins demonstrated increases in major regulatory pathways and proteins of lipid metabolism in mice consuming the HFD. These increases did not indicate that defects in lipid metabolism occurred in a mouse model of skeletal muscle insulin resistance.
Koves et al. (22) provided evidence that incomplete oxidation of lipids and lack of metabolic flexibility may have a key role in development of insulin resistance. Such results imply that substrate availability and mitochondrial inefficiencies, and not necessarily decreased capacity, contribute to accumulation of intermediates of lipid oxidation. A potential contributor is inefficient oxidation of lipids, yet we did not detect differences in mitochondrial respiratory efficiency between mice consuming the LFD and mice consuming the HFD as measured by P:O. The finding that P:O was similar between palmitoylcarnitine- and glutamate-malate-based respiration, both of which contribute electrons through NADH-linked substrates, further supports the notion that there were no major respiratory impairments following the HFD. We used the C57BL/6J strain that has a recognized deficiency in nicotinamide nucleotide transhydrogenase that can alter mitochondrial responses to a HFD, particularly responses of proteins involved with redox balance and ROS production, compared with a LFD (10). Our proteomic results cannot exclude an influence of nicotinamide nucleotide transhydrogenase deficiency on insulin action, which may impact protein turnover relative to C57BL/6NJ mice. Overall, our current data do not indicate that mitochondrial content or efficiency was impaired in mice previously reported to be insulin-resistant (27).
Intramuscular lipid content is influenced by dietary lipid availability, and the predominance of 16- and 18-carbon fatty acid species is likely related to the high content of these lipids in the lard-based diet, which has a higher lipid content than a “Western” diet. Unsaturated lipids can protect against accumulation of saturated acylcarnitines and are associated with improvements in insulin sensitivity in mouse models (24). In humans, a greater intake of unsaturated lipids (oleic acid) was negatively correlated with medium-chain acylcarnitines and insulin sensitivity, suggesting a beneficial response to more highly unsaturated lipids (21). It is possible that unsaturated lipids may have a beneficial impact on mitochondrial lipid metabolism and improvements to glucose metabolism following exercise training (29).
Previous work showed a decrease in mitochondrial content and respiratory capacity in obese adults and patients with type 2 diabetes (31, 39). Such declines in mitochondria are not consistently detected. For example, studies of permeabilized fibers reported similar rates of mitochondrial oxidation in lean and obese adults, along with similar protein abundance of subunits for mitochondrial complexes when detected by Western blotting (11). Such analysis cannot exclude the possibility that specific proteins are altered with obesity. Interestingly, the lower mitochondrial respiration in patients with type 2 diabetes may not be an intrinsic dysfunction of the respiratory system but, instead, a result of decreased mitochondrial content (5). DNA microarray analysis identified lower content of genes for oxidative phosphorylation in patients with type 2 diabetes than age-matched adults with normal glucose tolerance (25). Declines in mitochondria are implicated in the accumulation of intramuscular lipids that may interfere with insulin signaling (30), although high intramuscular lipid content can occur without detriment to insulin signaling (12, 43). Given the long-term progression of insulin resistance, it is possible that mitochondrial content or function may decline over time. Our model was high-fat feeding for 12 wk, while the progression of insulin resistance in humans occurs over several years. Gradual declines in mitochondrial content and function may be secondary to alterations in protein turnover with decreased insulin signaling (28) or ROS emission that could impact function of proteins (1).
Understanding the regulation of specific classes of mitochondrial proteins helps identify how the mitochondrial proteome is adapted to diverse stimuli, such as nutrition or exercise. High-fat feeding appears to increase lipid oxidation capacity, while exercise is a potent stimulus to increase mitochondrial respiratory capacity, which is not limited to β-oxidation pathways (33). Mitochondrial adaptations to exercise may therefore stimulate regulators that upregulate multiple pathways. For example, the PGC-1α family of transcriptional coactivators can regulate mitochondrial content along with other pathways, such as muscle hypertrophy (35). Oxidative phosphorylation genes that are regulated by PGC-1α have been shown to be lower in adults with type 2 diabetes (25). Our proteomic results extend such genetic data and demonstrate that high-fat feeding resulted in a lower abundance of proteins with PGC-1α as an upstream regulator. GSEA further identified pathways involving PGC-1α that were also decreased in HFD-fed mice. Other major regulators of the electron transfer system, such as mitochondrial transcription factor A and p53, were not primarily identified in our current analysis but have been shown to be altered with exercise training (36).
Liver mitochondria may be more responsive than skeletal muscle to obesity. For example, hyperphagic and high-fat-feeding models of obesity resulted in greater changes in mitochondrial protein abundance in liver extracts than skeletal muscle (26). Isolation of individual proteins revealed increased glycolytic enzymes following a HFD in brown adipose tissue but declines in a subunit for ATP synthase (ATP5A1) and β-enolase in muscle (37). Such findings indicate a shift in energy balance pathways in select tissues in response to high energy surplus. Our proteomics analysis of isolated mitochondria also indicates a shift in energy metabolism pathways toward greater lipid oxidation in skeletal muscle in response to high dietary lipids.
Our current proteomic analysis includes a few considerations. 1) Single proteins overlap in multiple pathways, and the influence of a single protein on pathway function may not be clear. GSEA takes into account multiple targets that contribute to a single pathway, yet the pathways can overlap. We used a combination of manual curation and published databases to classify proteins according to location and function. Similar approaches have also revealed distinct classes of mitochondrial changes, such as decreased oxidative phosphorylation with insulin resistance (25) and decreased proteins of β-oxidation following insulin deprivation (44). 2) Differences in the mitochondrial proteome may alter mitochondrial metabolism between different types of tissues (19). We isolated mitochondria from the gastrocnemius muscle, which includes both red and white heads, while respiration was studied in mitochondria from the quadriceps muscle. We cannot differentiate the effects of high-fat feeding between different fiber types: responses of mitochondrial respiration to obesity can differ between oxidative and glycolytic fibers (15). 3) The current analysis was not powered for lipid species as a primary outcome.
Our mitochondrial preparation did not include additional steps to liberate the intermyofibrillar fraction, and the proteomic analysis is predominantly of the subsarcolemma fraction. The extraction procedures exclude other fractions that may influence mitochondria with obesity, such as sarcoplasmic signaling or structural proteins (16). The subsarcolemma population of mitochondria appears to be predominantly altered by insulin resistance in humans (31) or rodent models (23). The subsarcolemma mitochondria also appear to respond to high-fat feeding with increases in lipid transport and oxidative capacity (14). Our preparation has previously been shown to have little influence on cytochrome c titration as a membrane integrity test during high-resolution respirometry (27), but the test cannot exclude the possibility that proteins in the outer membrane space were lost during isolation procedures. Perhaps loss of proteins due to leak or gel extraction procedures contributed to our detection of 658 proteins compared with the 1,158 mitochondrial proteins included in the MitoCarta 2.0 mouse database (7).
In summary, high-fat feeding associated with insulin resistance induced a remodeling of the mitochondrial proteome toward increased proteins involved with β-oxidation, but such changes were not uniformly distributed across all mitochondrial processes. Our results do not support the idea that declines in mitochondrial respiration occurred along with high-fat feeding. Instead, remodeling of the mitochondrial proteome may be a result of activation of signaling pathways related to lipid surplus.
GRANTS
This study was funded by National Institute of Diabetes and Digestive and Kidney Diseases Grant K01 DK-103829 (M. M. Robinson). This project was supported in part through the Mayo Clinic Medical Genome Facility Proteomics Core and its supporting grant from the National Cancer Institute (5P30 CA-15083-43C1). Research reported in this publication was supported in part by National Institutes of Health Grant KL2 TR-002370 (S. A. Newsom) awarded through the Oregon Clinical and Translational Research Institute by National Center for Advancing Translational Sciences under Grant UL1 TR-0002369.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.M.R. conceived and designed research; M.M.R. performed experiments; S.D., S.A.N., S.E.E., H.D.S., and M.M.R. analyzed data; S.D., S.A.N., S.E.E., H.D.S., and M.M.R. interpreted results of experiments; S.A.N., H.D.S., and M.M.R. prepared figures; M.M.R. drafted manuscript; S.D., S.A.N., S.E.E., H.D.S., and M.M.R. edited and revised manuscript; S.D., S.A.N., S.E.E., H.D.S., and M.M.R. approved final version of manuscript.
Supplemental Data
ACKNOWLEDGMENTS
We thank Dr. Sreekumaran Nair, Dr. Ian Lanza, Dr. Manjunatha Shankarappa, Kate Klaus, and Dawn Morse (Mayo Clinic) and Emily Burney and Bergen Sather (Oregon State University) for skilled assistance and contributions; Carrie Jo Heppelmann, Michael Holmes, and Dr. Bob Bergen 3rd (Proteomics Core at Mayo Clinic); Dr. Bryan Bergman and Dr. Kathleen Harrison (University of Colorado Denver) for assistance with lipidomics analysis; Felix Morales-Palomo (University of Castilla-La Mancha) for technical support; and Dr. Donald Jump (Oregon State University) for generously providing primers for quantitative PCR analysis.
REFERENCES
- 1.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW 3rd, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119: 573–581, 2009. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ayers-Ringler JR, Oliveros A, Qiu Y, Lindberg DM, Hinton DJ, Moore RM, Dasari S, Choi DS. Label-free proteomic analysis of protein changes in the striatum during chronic ethanol use and early withdrawal. Front Behav Neurosci 10: 46, 2016. doi: 10.3389/fnbeh.2016.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barazzoni R, Short KR, Asmann Y, Coenen-Schimke JM, Robinson MM, Nair KS. Insulin fails to enhance mTOR phosphorylation, mitochondrial protein synthesis, and ATP production in human skeletal muscle without amino acid replacement. Am J Physiol Endocrinol Metab 303: E1117–E1125, 2012. doi: 10.1152/ajpendo.00067.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Befroy DE, Petersen KF, Dufour S, Mason GF, de Graaf RA, Rothman DL, Shulman GI. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes 56: 1376–1381, 2007. doi: 10.2337/db06-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boushel R, Gnaiger E, Schjerling P, Skovbro M, Kraunsøe R, Dela F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia 50: 790–796, 2007. doi: 10.1007/s00125-007-0594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruce CR, Hoy AJ, Turner N, Watt MJ, Allen TL, Carpenter K, Cooney GJ, Febbraio MA, Kraegen EW. Overexpression of carnitine palmitoyltransferase-1 in skeletal muscle is sufficient to enhance fatty acid oxidation and improve high-fat diet-induced insulin resistance. Diabetes 58: 550–558, 2009. doi: 10.2337/db08-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44: D1251–D1257, 2016. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cameron-Smith D, Burke LM, Angus DJ, Tunstall RJ, Cox GR, Bonen A, Hawley JA, Hargreaves M. A short-term, high-fat diet up-regulates lipid metabolism and gene expression in human skeletal muscle. Am J Clin Nutr 77: 313–318, 2003. doi: 10.1093/ajcn/77.2.313. [DOI] [PubMed] [Google Scholar]
- 9.Dykens JA, Will Y. Drug-Induced Mitochondrial Dysfunction. Hoboken, NJ: John Wiley & Sons, 2008. doi: 10.1002/9780470372531. [DOI] [Google Scholar]
- 10.Fisher-Wellman KH, Ryan TE, Smith CD, Gilliam LA, Lin CT, Reese LR, Torres MJ, Neufer PD. A direct comparison of metabolic responses to high-fat diet in C57BL/6J and C57BL/6NJ mice. Diabetes 65: 3249–3261, 2016. doi: 10.2337/db16-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fisher-Wellman KH, Weber TM, Cathey BL, Brophy PM, Gilliam LA, Kane CL, Maples JM, Gavin TP, Houmard JA, Neufer PD. Mitochondrial respiratory capacity and content are normal in young insulin-resistant obese humans. Diabetes 63: 132–141, 2014. doi: 10.2337/db13-0940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodpaster BH, He J, Watkins S, Kelley DE. Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab 86: 5755–5761, 2001. doi: 10.1210/jcem.86.12.8075. [DOI] [PubMed] [Google Scholar]
- 13.Hancock CR, Han DH, Chen M, Terada S, Yasuda T, Wright DC, Holloszy JO. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci USA 105: 7815–7820, 2008. doi: 10.1073/pnas.0802057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holloway GP, Benton CR, Mullen KL, Yoshida Y, Snook LA, Han XX, Glatz JF, Luiken JJ, Lally J, Dyck DJ, Bonen A. In obese rat muscle transport of palmitate is increased and is channeled to triacylglycerol storage despite an increase in mitochondrial palmitate oxidation. Am J Physiol Endocrinol Metab 296: E738–E747, 2009. doi: 10.1152/ajpendo.90896.2008. [DOI] [PubMed] [Google Scholar]
- 15.Holmström MH, Iglesias-Gutierrez E, Zierath JR, Garcia-Roves PM. Tissue-specific control of mitochondrial respiration in obesity-related insulin resistance and diabetes. Am J Physiol Endocrinol Metab 302: E731–E739, 2012. doi: 10.1152/ajpendo.00159.2011. [DOI] [PubMed] [Google Scholar]
- 16.Hwang H, Bowen BP, Lefort N, Flynn CR, De Filippis EA, Roberts C, Smoke CC, Meyer C, Højlund K, Yi Z, Mandarino LJ. Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and type 2 diabetes. Diabetes 59: 33–42, 2010. doi: 10.2337/db09-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaleel A, Short KR, Asmann YW, Klaus KA, Morse DM, Ford GC, Nair KS. In vivo measurement of synthesis rate of individual skeletal muscle mitochondrial proteins. Am J Physiol Endocrinol Metab 295: E1255–E1268, 2008. doi: 10.1152/ajpendo.90586.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janssen U, Stoffel W. Disruption of mitochondrial β-oxidation of unsaturated fatty acids in the 3,2-trans-enoyl-CoA isomerase-deficient mouse. J Biol Chem 277: 19579–19584, 2002. doi: 10.1074/jbc.M110993200. [DOI] [PubMed] [Google Scholar]
- 19.Johnson DT, Harris RA, Blair PV, Balaban RS. Functional consequences of mitochondrial proteome heterogeneity. Am J Physiol Cell Physiol 292: C698–C707, 2007. doi: 10.1152/ajpcell.00109.2006. [DOI] [PubMed] [Google Scholar]
- 20.Johnson ML, Irving BA, Lanza IR, Vendelbo MH, Konopka AR, Robinson MM, Henderson GC, Klaus KA, Morse DM, Heppelmann C, Bergen HR 3rd, Dasari S, Schimke JM, Jakaitis DR, Nair KS. Differential effect of endurance training on mitochondrial protein damage, degradation, and acetylation in the context of aging. J Gerontol A Biol Sci Med Sci 70: 1386–1393, 2015. doi: 10.1093/gerona/glu221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kien CL, Bunn JY, Poynter ME, Stevens R, Bain J, Ikayeva O, Fukagawa NK, Champagne CM, Crain KI, Koves TR, Muoio DM. A lipidomics analysis of the relationship between dietary fatty acid composition and insulin sensitivity in young adults. Diabetes 62: 1054–1063, 2013. doi: 10.2337/db12-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 7: 45–56, 2008. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 23.Lai N, Kummitha C, Hoppel C. Defects in skeletal muscle subsarcolemmal mitochondria in a non-obese model of type 2 diabetes mellitus. PLoS One 12: e0183978, 2017. doi: 10.1371/journal.pone.0183978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lanza IR, Blachnio-Zabielska A, Johnson ML, Schimke JM, Jakaitis DR, Lebrasseur NK, Jensen MD, Sreekumaran Nair K, Zabielski P. Influence of fish oil on skeletal muscle mitochondrial energetics and lipid metabolites during high-fat diet. Am J Physiol Endocrinol Metab 304: E1391–E1403, 2013. doi: 10.1152/ajpendo.00584.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273, 2003. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 26.Nesteruk M, Hennig EE, Mikula M, Karczmarski J, Dzwonek A, Goryca K, Rubel T, Paziewska A, Woszczynski M, Ledwon J, Dabrowska M, Dadlez M, Ostrowski J. Mitochondrial-related proteomic changes during obesity and fasting in mice are greater in the liver than skeletal muscles. Funct Integr Genomics 14: 245–259, 2014. doi: 10.1007/s10142-013-0342-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Newsom SA, Miller BF, Hamilton KL, Ehrlicher SE, Stierwalt HD, Robinson MM. Long-term rates of mitochondrial protein synthesis are increased in mouse skeletal muscle with high-fat feeding regardless of insulin-sensitizing treatment. Am J Physiol Endocrinol Metab 313: E552–E562, 2017. doi: 10.1152/ajpendo.00144.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Neill BT, Lee KY, Klaus K, Softic S, Krumpoch MT, Fentz J, Stanford KI, Robinson MM, Cai W, Kleinridders A, Pereira RO, Hirshman MF, Abel ED, Accili D, Goodyear LJ, Nair KS, Kahn CR. Insulin and IGF-1 receptors regulate FoxO-mediated signaling in muscle proteostasis. J Clin Invest 126: 3433–3446, 2016. doi: 10.1172/JCI86522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ortega JF, Morales-Palomo F, Fernandez-Elias V, Hamouti N, Bernardo FJ, Martin-Doimeadios RC, Nelson RK, Horowitz JF, Mora-Rodriguez R. Dietary supplementation with ω-3 fatty acids and oleate enhances exercise training effects in patients with metabolic syndrome. Obesity (Silver Spring) 24: 1704–1711, 2016. doi: 10.1002/oby.21552. [DOI] [PubMed] [Google Scholar]
- 30.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 350: 664–671, 2004. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 54: 8–14, 2005. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 32.Robinson MM, Dasari S, Karakelides H, Bergen HR 3rd, Nair KS. Release of skeletal muscle peptide fragments identifies individual proteins degraded during insulin deprivation in type 1 diabetic humans and mice. Am J Physiol Endocrinol Metab 311: E628–E637, 2016. doi: 10.1152/ajpendo.00175.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson MM, Dasari S, Konopka AR, Johnson ML, Manjunatha S, Esponda RR, Carter RE, Lanza IR, Nair KS. Enhanced protein translation underlies improved metabolic and physical adaptations to different exercise training modes in young and old humans. Cell Metab 25: 581–592, 2017. doi: 10.1016/j.cmet.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson MM, Soop M, Sohn TS, Morse DM, Schimke JM, Klaus KA, Nair KS. High insulin combined with essential amino acids stimulates skeletal muscle mitochondrial protein synthesis while decreasing insulin sensitivity in healthy humans. J Clin Endocrinol Metab 99: E2574–E2583, 2014. doi: 10.1210/jc.2014-2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruas JL, White JP, Rao RR, Kleiner S, Brannan KT, Harrison BC, Greene NP, Wu J, Estall JL, Irving BA, Lanza IR, Rasbach KA, Okutsu M, Nair KS, Yan Z, Leinwand LA, Spiegelman BM. A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 151: 1319–1331, 2012. doi: 10.1016/j.cell.2012.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saleem A, Hood DA. Acute exercise induces tumour suppressor protein p53 translocation to the mitochondria and promotes a p53-Tfam-mitochondrial DNA complex in skeletal muscle. J Physiol 591: 3625–3636, 2013. doi: 10.1113/jphysiol.2013.252791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmid GM, Converset V, Walter N, Sennitt MV, Leung KY, Byers H, Ward M, Hochstrasser DF, Cawthorne MA, Sanchez JC. Effect of high-fat diet on the expression of proteins in muscle, adipose tissues, and liver of C57BL/6 mice. Proteomics 4: 2270–2282, 2004. doi: 10.1002/pmic.200300810. [DOI] [PubMed] [Google Scholar]
- 38.Schönke M, Björnholm M, Chibalin AV, Zierath JR, Deshmukh AS. Proteomics analysis of skeletal muscle from leptin-deficient ob/ob mice reveals adaptive remodeling of metabolic characteristics and fiber type composition. Proteomics 18: e1700375, 2018. doi: 10.1002/pmic.201700375. [DOI] [PubMed] [Google Scholar]
- 39.Simoneau JA, Kelley DE. Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. J Appl Physiol (1985) 83: 166–171, 1997. doi: 10.1152/jappl.1997.83.1.166. [DOI] [PubMed] [Google Scholar]
- 40.Takaki M, Nakahara H, Kawatani Y, Utsumi K, Suga H. No suppression of respiratory function of mitochondrial isolated from the hearts of anesthetized rats with high-dose pentobarbital sodium. Jpn J Physiol 47: 87–92, 1997. doi: 10.2170/jjphysiol.47.87. [DOI] [PubMed] [Google Scholar]
- 41.Turner N, Bruce CR, Beale SM, Hoehn KL, So T, Rolph MS, Cooney GJ. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes 56: 2085–2092, 2007. doi: 10.2337/db07-0093. [DOI] [PubMed] [Google Scholar]
- 42.Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 45: W130–W137, 2017. doi: 10.1093/nar/gkx356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wicks SE, Vandanmagsar B, Haynie KR, Fuller SE, Warfel JD, Stephens JM, Wang M, Han X, Zhang J, Noland RC, Mynatt RL. Impaired mitochondrial fat oxidation induces adaptive remodeling of muscle metabolism. Proc Natl Acad Sci USA 112: E3300–E3309, 2015. doi: 10.1073/pnas.1418560112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zabielski P, Lanza IR, Gopala S, Holtz Heppelmann CJ, Bergen HR 3rd, Dasari S, Nair KS. Altered skeletal muscle mitochondrial proteome as the basis of disruption of mitochondrial function in diabetic mice. Diabetes 65: 561–573, 2016. doi: 10.2337/db15-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.