

Abstract
Angiotensin II (ANG II) is a major mediator of hypertension pathogenesis. In addition, there are well-documented differences in expression of the renin-angiotensin system (RAS) components and ANG II responses between males and females, which may explain sex differences in blood pressure (BP) and hypertension epidemiology. We previously showed that type 1A angiotensin (AT1A) receptors in vascular smooth muscle cells (VSMCs) play a critical role in BP regulation and hypertension pathogenesis, but these studies were carried out in male mice. Therefore, the major goal of the current studies was to examine the impact of VSMC AT1A receptors on BP and hypertension pathogenesis in female mice. We found that elimination of VSMC AT1A receptors in female mice reduced (≈8 mmHg) baseline BP without altering sodium sensitivity. The severity of ANG II-induced hypertension was diminished (≈33% reduction in BP), particularly during the last 2 wk of chronic ANG II infusion, compared with controls, but natriuresis was not altered during the first 5 days of ANG II infusion. Urinary norepinephrine levels were enhanced in female SMKO compared with control mice. There was a virtually complete elimination of ANG II-induced kidney hemodynamic responses with attenuation of acute vasoconstrictor responses in the systemic vasculature. These findings demonstrate that direct vascular actions of AT1A receptors play a prominent role in BP control and hypertension pathogenesis in female mice.
Keywords: angiotensin, blood pressure, sex differences, sodium homeostasis
INTRODUCTION
The hormone angiotensin II (ANG II), through its action on type 1 angiotensin (AT1) receptors, has been implicated as a major mediator of hypertension pathogenesis and resultant end-organ damage, such as kidney disease, heart failure, and stroke (32). Pharmacological blockade of this system in humans with angiotensin-converting enzyme inhibitors or angiotensin receptor blockers is a mainstay therapy for hypertension (25) and has been shown to reduce major clinical end points, such as end-stage kidney disease (6, 19a, 28) and heart failure (13, 23). The advent of tissue-specific knockout systems, such as Cre-LoxP, is further elucidating the primary cellular mechanisms for these effects (10, 21, 43). The importance of vascular function in blood pressure (BP) pathogenesis has been highlighted in multiple studies targeting vascular smooth muscle cells (VSMCs), such as α1-adrenergic subtypes (49), mineralocorticoid (31), and peroxisome proliferator-activated receptor-γ (9). The major murine AT1 receptor isoform (mice have AT1A and AT1B receptors) mediating the classical physiological action of ANG II is the AT1A receptor (24). We previously demonstrated that AT1A receptors in proximal tubule cells of the kidney promote hypertension and sodium retention, whereas AT1A receptors in intercalated cells limit the hypertensive response to ANG II (21). Furthermore, we demonstrated that VSMC AT1A receptors play a prominent role in determination of basal BP, sodium sensitivity, and hypertension pathogenesis (44). These studies showed that kidney vascular responses (as opposed to systemic responses) to ANG II were absent in mice lacking VSMC AT1A receptors and, thus, suggest that the vascular mechanism of ANG II-induced hypertension involves regulation of sodium excretion by the kidney by impacting kidney blood flow (44). However, male mice were used for the aforementioned studies utilizing cell-specific AT1A receptor deletion; therefore, the goal of the current studies is to examine the vascular effects of the renin-angiotensin system (RAS) in female mice.
Females have historically been underrepresented or even excluded from both animal (55) and clinical (33) studies. Therefore, the results of clinical and basic studies that have been performed only in males cannot be generalized to females. Several investigations have demonstrated important differences in BP regulation between men and women (19, 30, 37). Basal BP is significantly lower in women before menopause than in men (29). Antihypertensive medication use is higher in women (12, 20), but women are less likely than men to achieve BP control (20, 27). Women also exhibit a greater rate of increase in BP with age (17). It is believed that hormones such as estrogen have a role in BP determination, exemplified by increased BP and prevalence of hypertension in postmenopausal compared with premenopausal women (46).
The mechanisms underlying sex differences in hypertension pathophysiology remain unclear. One area of focus is differences in the levels of RAS components between men and women (34, 47). Animal models have elucidated some of these differences: males [Wistar Kyoto (WKY) and spontaneously hypertensive rats (SHR)] express higher levels of angiotensinogen mRNA in the kidney (11, 16), more AT1 receptor mRNA in the kidney and vasculature (SHR) (41), and higher AT1 receptor binding in the glomeruli (Sprague-Dawley rats and C57BL/6J mice) (1, 38). On the other hand, females demonstrate higher levels of AT2 receptor mRNA in the renal medulla, greater AT2 receptor binding in the inner medulla (C57BL/6J mice) (1), increased angiotensin-converting enzyme type 2 in the kidney (Sprague-Dawley rats) (39), and increased ANG-(1–7) in the renal cortex (SHR and WKY) (5, 48).
Clinically, blockade of the RAS is an important pharmacological intervention for both women and men with hypertension, diabetes, and heart failure. Therefore, in this study we aim to elucidate whether VSMC AT1A receptors in female mice play a role in BP determination, salt sensitivity, and hypertension pathogenesis. We hypothesized that vascular AT1A receptors play a prominent role in BP determination and ANG II-induced hypertension pathogenesis in female mice.
METHODS
Generation of mice with deletion of AT1A receptors from VSMCs.
A mouse line with a conditional Agtr1a allele was generated using homologous recombination in embryonic stem cells, as previously described (43). To remove the AT1A receptor from VSMCs, we crossed an inbred C57BL/6 transgenic mouse line expressing Cre recombinase specifically in smooth muscle cells under the control of the Sm22α promoter (KISm22α-Cre, strain B6.129S6-Taglntm2(cre)Yec/J; stock no. 006878, Jackson Laboratory, Bar Harbor, ME) with C57BL/6-Agtr1aflox/flox mice. We previously confirmed robust expression of Cre recombinase in large arteries and resistance vessels in the mTmG dual-reporter mouse line KISm22a-Cre-mTmG via robust green fluorescent protein fluorescence (42).
To document sufficient elimination of AT1A receptors, we measured mRNA levels of AT1A receptors by real-time RT-PCR in female control (n = 5) and SMKO (n = 4) mice, as previously reported (44). AT1A receptor mRNA expression, normalized to GAPDH, was efficiently extinguished from the aorta (1 ± 0.1 and 0.07 ± 0.03 arbitrary units in control and SMKO, respectively, P = 0.001, by unpaired t-test).
All animal studies were approved by the Durham Veterans Affairs Health Care System Institutional Animal Care and Use Committee and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animals had free access to standard rodent chow and water unless specified. All studies were carried out on adult [10- to 30-wk-old (average 20 ± 0.6 wk old)] female mice. Experimental groups within each experiment were age-matched to control and SMKO littermates.
BP measurements in conscious mice with manipulation of dietary sodium intake.
Radiotelemetry (model PA-C10, Data Science International, St. Paul, MN) was used to measure BP in conscious unrestrained SMKO and control mice, as described previously (44). Arterial BP data were collected, stored, and analyzed using Dataquest ART software (version 4.0, Data Sciences International). Mice were allowed to recover for 7 days after placement of the telemetry implant to reestablish normal circadian rhythms (7). After recovery, telemetry data were collected continuously, with sampling every 5 min for 10-s intervals. Baseline measurements were recorded for 7 consecutive days while mice ingested a normal-sodium (0.4% Na+) diet (Harlan Teklad, Indianapolis, IN). On day 8, mice were fed a sodium-deficient (<0.02% Na+) diet (Harlan Teklad) for 1 wk and then a high-sodium (6% Na+) diet (Harlan Teklad) for 1 wk. Data were analyzed by averaging the first 5 days starting at time of sodium diet change. At the end of the salt challenge and during ingestion of the normal-sodium diet, ANG II (1,000 ng·kg−1·min−1; Sigma-Aldrich) was infused via subcutaneously implanted osmotic minipump (Alzet, Cupertino, CA), and BP measurements continued for 30 days after minipump implantation (44).
Determination of urinary norepinephrine and creatinine content.
Norepinephrine was extracted from the urine and quantitated by enzyme immunoassay (norepinephrine ELISA, catalog no. 17-NORHU-E01.1, ALPCO Diagnostics, Salem, NH) according to the manufacturer’s instructions. Creatinine content in urine was determined using the creatinine companion kit (Exocell, Philadelphia, PA).
Determination of serum estradiol levels.
Estradiol content in the serum was measured by enzyme immunoassay (mouse/rat estradiol ELISA; catalog no. ES180S-100, Calbiotech, El Cajon, CA) according to the manufacturer’s instructions (22).
Metabolic balance studies.
A separate group of mice were individually placed in metabolic cages (Hatteras Instruments, Cary, NC) and fed a gel diet (Nutra-Gel;10 g/day) containing nutrients, water, and 0.1% (wt/wt) sodium (catalog no. S5769-TRAY, Bio-Serv, Frenchtown, NJ). After 5 days of baseline collections, osmotic minipumps (model no. 2004, Alzet), which infused ANG II at 1,000 ng·kg−1·min−1, were implanted, and the mice were returned to metabolic cages for 5 more days. Urine was collected daily, and urinary sodium content was measured daily using a dual-channel flame photometer (model 02655-10, Cole-Parmer, Vernon Hills, IL). Sodium balance was determined by subtraction of the total amount of sodium ingested daily from the total amount of sodium excreted in the urine over a 24-h period (44).
Assessment of kidney cortical tissue perfusion and acute vasoconstrictor responses.
We examined acute vasoconstrictor and kidney cortical tissue perfusion responses to ANG II (Sigma-Aldrich, reconstituted in sterile saline) in mice anesthetized with 2% isoflurane, as described previously (2, 4, 43). A catheter (PE-50) was inserted into the left jugular vein for administration of basal fluids and vasoconstrictors. A second catheter (pulled-down PE-50) attached to a pressure transducer (model MLT844, ADInstruments, Colorado Springs, CO) was placed in the left carotid artery. Intra-arterial BP was recorded continuously through the carotid catheter using the PowerLab data acquisition system and LabChart software (ADInstruments). The kidney was isolated in a specially designed Lucite cup to prevent movement during measurements. A laser-Doppler needle probe (18 gauge; Transonic Systems, Ithaca, NY) connected to a tissue perfusion monitor (model BLF22, Transonic Systems) was placed on the renal cortex during continuous measurement of tissue perfusion. At 5-min intervals, increasing doses (0.03, 0.1, 0.3, and 1 µg/kg) of ANG II were injected intravenously into the internal jugular vein at a volume of 1 µl/g body wt (20–25 µl total volume) followed immediately by a 25-µl bolus of saline. Before injection of vasoactive agents, each mouse received an equivalent volume (20–25 µl) of saline (1 µl/g body wt) as a vehicle control. Intra-arterial pressures were continuously monitored.
Statistical analysis.
Values for each parameter within a group are means ± SE.
Experimental design.
All experiments are performed in a blinded fashion. For the physiological studies, all genotyping information was excluded from the cage cards and no genotype assignment was entered thereafter. The surgeon and/or technician randomly selected mice from cages (all mice were eventually used), and the physiological study (e.g., chronic BP, acute BP, sodium balance) was performed. Data were analyzed only after the entire experiment was completed (with a predetermined number of mice based on a power calculation) by an individual with no knowledge of genotype. Genotype key was used only after all data for that particular experiment were complete and analyzed. To ensure correct genotype, each animal’s ear was clipped before and after experiments were performed, and the clipped tissue was sent for regenotyping.
RESULTS
Deletion of AT1A receptors from VSMCs contribute to basal BP determination but not salt sensitivity.
To demonstrate the physiological effect of AT1A receptors in the vasculature on BP control, radiotelemetry units were implanted into female SMKO and control mice to continuously measure intra-arterial pressure in their conscious and unrestrained state. As depicted in Fig. 1A, female SMKO mice show a significant reduction in baseline mean arterial pressure (MAP) compared with control mice fed a normal-sodium (0.4% Na+) diet (108 ± 2 vs. 116 ± 1 mmHg). To determine the contribution of VSMC AT1A receptors to salt sensitivity, we measured BP while altering dietary sodium content. During low-sodium (0.02% Na+) feeding, the difference in MAP between SMKO and control mice was maintained (108 ± 2 and 115 ± 1 mmHg, respectively). Similarly, during high-sodium (6% Na+) feeding, BP increased in both groups of mice, and the difference between groups was sustained (113 ± 2 and 120 ± 1 mmHg in SMKO and control, respectively). The change in BP upon transfer from the low- to the high-sodium diet was used to measure salt sensitivity. BP increased significantly in control and SMKO mice between the low- and the high-salt diet, and there was no difference in the extent of BP increase between the groups (Fig. 1B). Heart rate did not differ between control and SMKO mice at baseline or during low- or high-salt feedings (Fig. 1C). Heart rate in both groups increased with the low-salt diet and decreased with the high-salt diet. The change in heart rate from the low- to the high-salt diet did not differ between control and SMKO mice (Fig. 1D). Thus, VSMC AT1A receptors play an important role in determination of basal BP in female mice. However, elimination of VSMC AT1A receptors does not further accentuate salt-sensitive BP responses in female C57BL/6 mice.
Fig. 1.

Type 1A angiotensin receptors in vascular smooth muscle cells contribute to basal blood pressure (BP), but not response to Na+. A: mean arterial pressure (MAP) in control (n = 8) and SMKO (n = 9) mice fed normal (0.4% Na+)-, low (0.02% Na+)-, and high (6% Na+)-salt diets over 5 days. Values are means ± SE. Data were analyzed by 2-way repeated-measures ANOVA with Sidak’s post hoc test for multiple comparisons. ANOVA table for genotype: F(1,15) = 15.7, P < 0.001; Sidak’s post hoc test: *P = 0.002, **P = 0.001, #P = 0.002. B: Na+ sensitivity, measured as change in MAP from high- to low-salt diet, was not significantly different (by unpaired t-test) between control (n = 8) and SMKO (n = 9) mice. Values are means ± SE. C: heart rate, averaged over each of the 5-day periods of salt diet feeding (as described in A), in control (n = 8) and SMKO (n = 9) mice. Heart rates increased during low-salt feeding and decreased during high-salt feeding. There was no significant difference between groups. D: average change in heart rate from high- to low-salt diet was not significantly different (by unpaired t-test) between control (n = 8) and SMKO (n = 9) mice. Values are means ± SE.
VSMC AT1A receptors contribute to ANG II-induced hypertension.
To assess the contribution of VSMC AT1A receptors to hypertension pathogenesis in female mice, we infused ANG II (1,000 ng·kg−1·min−1 sc) via osmotic minipumps in female control and SMKO mice over 4 wk (Fig. 2A). Chronic ANG II resulted in a substantial elevation of MAP in female control and SMKO mice from baseline during the first 2 wk (averaged from days 1–14); hence, there was no significant difference between the two groups (Fig. 2C). In contrast, during the final 2 wk of treatment, the ANG II-induced increase in MAP was significantly less (~33%) in female SMKO than control mice (20 ± 2 vs. 30 ± 3 mmHg; Fig. 2D). Overall, MAP was significantly lower (145 ± 4 vs. 130 ± 3 mmHg) and heart rate was significantly higher (591 ± 9 vs. 621 ± 5 beats/min) in SMKO than control mice during 4 wk of ANG II infusion. Thus, in VSMC, AT1A receptors play a role in the hypertensive response to chronically infused ANG II in female mice, and this response is most pronounced over the later stages (weeks 2–4) of hypertension development.
Fig. 2.

Type 1A angiotensin receptors in vascular smooth muscle cells promote the hypertensive response to high-dose (1,000 ng·kg−1·min−1) angiotensin II (ANG II)-induced hypertension in conscious female mice. A: mean arterial pressure (MAP) in control (n = 7) and SMKO (n = 9) mice during high-dose ANG II infusion over 4 wk. Arrow at day 1 represents start of ANG II infusion. Values are means ± SE. Data were analyzed by 2-way repeated-measures ANOVA. ANOVA table for genotype: F(1,13) = 9.7, P = 0.008. B: heart rate in control (n = 7) and SMKO (n = 9) mice during high-dose ANG II infusion over 4 wk. Values are means ± SE. Data were analyzed by 2-way repeated-measures ANOVA. ANOVA table for genotype: F(1,13) = 8.9, P = 0.01. C: change in MAP from baseline over the first 2 wk (averaged from days 1–14) was not significantly different (by unpaired t-test) between control (n = 7) and SMKO (n = 9) mice. Values are means ± SE. D: MAP increase from baseline to the last 2 wk (averaged from days 15–27) in control (n = 7) and SMKO (n = 9) mice. Values are means ± SE. #P = 0.01 (by unpaired t-test).
Development of ANG II-induced cardiac hypertrophy.
Since hypertension plays a dominant role in the development of cardiac hypertrophy, we examined the effect of VSMC AT1A receptors on development of cardiac hypertrophy (14). We measured heart weight-to-body weight ratio in the SMKO and control groups following 4 wk of ANG II infusion, as well as in their respective uninfused controls. Heart weight-to-body weight ratio was similar at baseline in SMKO and control mice. However, ANG II-induced cardiac hypertrophy was significantly attenuated in SMKO mice compared with controls (Fig. 3).
Fig. 3.
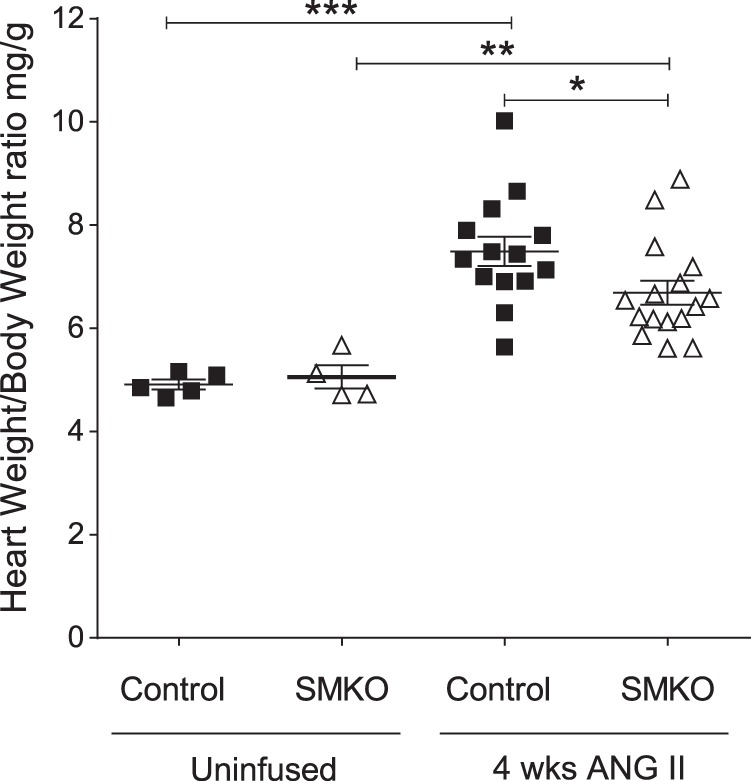
Attenuated cardiac hypertrophy in response to high-dose (1,000 ng·kg−1·min−1) angiotensin II (ANG II)-induced hypertension in female SMKO mice. Heart weight-to-body weight ratio in control and SMKO female mice was determined before and after 4 wk of ANG II infusion. Baseline heart weight-to-body weight ratio did not differ between control (n = 5) and SMKO (n = 4) mice before infusion. There was significant cardiac hypertrophy in ANG II-treated control (n = 14) and SMKO (n = 16) mice compared with their respective baseline groups. Values are means ± SE. Data were analyzed by 2-way ANOVA with Sidak’s post hoc test for multiple comparisons: **P = 0.004 for SMKO + ANG II (6.7 ± 0.2 mg/g) vs. baseline (5.0 ± 0.4 mg/g); ***P < 0.0001 for control + ANG II (7.5 ± 0.3 mg/g) vs. baseline (4.9 ± 0.1 mg/g). After ANG II infusion, heart weight-to-body weight ratio was significantly lower in SMKO than control mice (6.7 ± 0.2 vs. 7.5 ± 0.3 mg/g). *P = 0.04.
Systemic vascular responses to acute ANG II.
We next examined the acute peripheral vasoconstrictor responses to intravenous ANG II administration in female mice with specific deletion of the AT1A receptor in VSMCs. Bolus injection of ANG II caused acute vasoconstriction in control mice, with a marked and dose-dependent increase in systolic BP from baseline (Fig. 4A) with subsequent increases in ANG II concentration (0.1, 0.3, and 1 μg/kg). However, the vasoconstrictor response in female SMKO mice was significantly attenuated (Fig. 4A). The vasoconstrictor response to exogenously administered ANG II was significantly reduced (60–75%) during 0.3 and 1.0 μg/kg ANG II infusions in female SMKO compared with control mice (3.3 ± 1.8 vs. 12.1 ± 1.3 mmHg at 0.3 μg/kg and 8.5 ± 1.9 vs. 19.4 ± 2.0 mmHg at 1.0 μg/kg). Conversely, heart rates diminished with increasing doses of ANG II in control mice (Fig. 4B). This diminution of heart rate was not seen in SMKO mice, where BP was likewise attenuated compared with controls. Thus the absence of VSMC AT1A receptors in female mice markedly reduces peripheral acute vasoconstrictor responses and, concomitantly, alters heart rate after an acute ANG II bolus.
Fig. 4.

Attenuated systemic vascular responses and attenuated vasoconstriction in renal tissue perfusion to angiotensin II (ANG II) infusion in female SMKO mice. A: average change in blood pressure after acute administration of sequential ANG II doses (0.10, 0.30, and 1.00 μg/kg) to anesthetized control (n = 11) and SMKO (n = 8) mice. Values are means ± SE. Data were analyzed by 2-way repeated-measures ANOVA with Sidak’s post hoc test for multiple comparisons. ANOVA table for genotype: F(1,17) = 23.15, P < 0.001; Sidak’s post hoc test: P = not significant for 0.10 μg/kg, ***P < 0.001 for 0.30 and 1.00 μg/kg. B: average change in heart rate after acute administration of sequential ANG II doses in anesthetized control (n = 11) and SMKO (n = 8) mice. Values are means ± SE. Data were analyzed by 2-way repeated-measures ANOVA with Sidak’s post hoc test for multiple comparisons. ANOVA table for genotype: F(1,17) = 16.98, P = 0.0007; Sidak’s post hoc test: P = not significant for 0.10 μg/kg, ***P = 0.0008 for 0.30 μg/kg, *P < 0.01 for 1.00 μg/kg. C: baseline kidney tissue perfusion in control (n = 11) and SMKO (n = 8) mice (P = 0.053). Values (means ± SE) are shown as tissue perfusion units (TPUs). Data were analyzed by unpaired t-test. D: reduction in mean kidney tissue perfusion was compared with respective baseline values in response to increasing concentrations (0.03, 0.1, and 0.30 μg/kg) of ANG II. Baseline changes were compared between SMKO (n = 8) and control (n = 11) mice. Values are means ± SE. Data were analyzed by 2-way repeated-measures ANOVA with Sidak’s post hoc test for multiple comparisons. ANOVA table for genotype: F(1,17) = 48.28, P < 0.0001; Sidak’s post hoc test: P = not significant for 0.03 μg/kg, ***P < 0.0001 for 0.10 and 0.30 μg/kg.
Kidney tissue perfusion responses to acute ANG II.
We next examined the effect of VSMC AT1A receptors on kidney tissue perfusion in response to ANG II. We used a laser-Doppler needle probe to measure renal cortical capillary blood flow (2, 4). Secondary to anatomic limitations, we were unable to measure renal artery blood velocity using an ultrasound probe in female mice. There was a difference in baseline renal tissue perfusion between control and SMKO mice (1.9 ± 0.1 and 2.6 ± 0.4 tissue perfusion units, respectively); however, this was not significant (Fig. 4C). Similar to reductions in the acute peripheral vasoconstrictor response to ANG II, SMKO mice demonstrated significantly reduced changes in cortical kidney tissue perfusion compared with control mice. More specifically, the percent decrease in tissue perfusion from baseline during 0.1 and 0.3 μg/kg ANG II infusions was significantly different between the two groups (18.8 ± 2.6 and 1.3 ± 1.7 in control and SMKO, respectively, for 0.1 μg/kg and 41.1 ± 4.4 and 4.9 ± 3 in control and SMKO, respectively, for 0.3 μg/kg; Fig. 4D), indicating that the physiological response of ANG II to reduce blood flow to the kidney was absent in female SMKO mice. Therefore, AT1A receptors in VSMCs are critically important mediators of kidney vascular responses to ANG II in female mice.
Increased urinary catecholamine excretion in female SMKO mice.
We previously demonstrated elevated urinary norepinephrine levels at baseline and during ANG II-induced hypertension in male SMKO mice (44). Therefore, we measured urinary norepinephrine excretion in female control and SMKO mice at baseline and after 5 days of chronic ANG II administration (1,000 ng·kg−1·min−1), during which mice were housed in metabolic cages and fed a gel diet. As shown in Fig. 5A, baseline urinary norepinephrine excretion was higher, but not significantly, in SMKO than control mice (58 ± 25 vs. 27 ± 6 pg/24 h). After 5 days of ANG II infusion, norepinephrine excretion was enhanced in control (from 27 ± 6 to 99 ± 25 pg/24 h) and SMKO (from 58 ± 25 to 246 ± 38 pg/24 h) mice. The difference in urinary norepinephrine excretion between SMKO and control mice was magnified by chronic ANG II infusion (246 ± 38 vs. 99 ± 25 pg/24 h). After 5 days of ANG II infusion, 24-h urine volumes (2.8 ± 0.2 vs. 2.5 ± 0.1 ml/24 h) and food consumption (9.4 ± 0.1 vs. 7.3 ± 0.4 g/24 h) diminished in control mice after 5 days of ANG II infusion. However, urine volumes (3.1 ± 0.2 and 3.3 ± 0.2 ml/24 h at baseline and day 5 of ANG II, respectively) and food consumption (9.1 ± 0.2 and 8.6 ± 0.1 g/24 h at baseline and day 5 of ANG II, respectively) remained the same in SMKO mice after 5 days of ANG II infusion. Thus, at day 5 both urine volume (2.5 ± 0.1 and 3.3 ± 0.2 ml/24 h in control and SMKO, respectively) and food consumption (7.3 ± 0.4 and 8.6 ± 0.1 g/24 h in control and SMKO, respectively; Fig. 5, C and D) increased in SMKO compared with control mice. To adjust for differences in 24-h urine quantity, urinary norepinephrine content was expressed as norepinephrine-to-creatinine ratio, and the results were similar, particularly after 5 days of ANG II infusion (87 ± 20 vs. 201 ± 30 pg norepinephrine/mg creatinine; Fig. 5B). These findings suggest that deletion of AT1A receptors from VSMCs is associated with enhancement of urinary norepinephrine in female mice after ANG II-induced hypertension.
Fig. 5.

Urinary norepinephrine levels in female mice lacking type 1A angiotensin receptors in vascular smooth muscle cells. A: at baseline, urinary norepinephrine excretion was increased in SMKO (n = 8) compared with control (n = 8) mice. Norepinephrine excretion was increased in control and SMKO mice after 5 days of angiotensin II (ANG II) infusion compared with baseline. Urinary norepinephrine excretion was significantly greater in SMKO mice after 5 days of ANG II infusion. ANOVA table for genotype: F(1,14) = 12.23, P = 0.004; Sidak’s post hoc test: P = not significant for baseline, ***P = 0.0008 for day 5 of ANG II. B: to account for differences in urine volume at day 5, norepinephrine is expressed as ratio of urinary norepinephrine to creatinine in SMKO (n = 8) and control (n = 8) female mice at baseline and day 5 of ANG II infusion. ANOVA table for time: F(1, 14) = 9.9, P = 0.007; Sidak’s post hoc test: P = not significant for baseline, *P = 0.02 for day 5 of ANG II. C: 24-h urine excretion was increased in SMKO (n = 8) compared with control (n = 8) mice at day 5 of ANG II infusion. ANOVA table for genotype: F(1,14) = 8.21, P = 0.01; Sidak’s post hoc test: P = not significant for baseline, **P = 0.006 for day 5 ANG II. D: 24-h food consumption was increased in SMKO (n = 8) compared with control (n = 8) mice at day 5 of ANG II infusion. ANOVA table for genotype: F(1,14) = 6.03, P = 0.03; Sidak’s post hoc test: P = not significant for baseline, ***P = 0.002 for day 5 of ANG II. Values are means ± SE. Data were analyzed by 2-way repeated-measures ANOVA with Sidak’s post hoc test for multiple comparisons.
Estrogen levels in SMKO mice.
To determine if sex hormones were perturbed by elimination of VSMC AT1A receptors, we measured serum estradiol using a validated commercially available ELISA kit (22). Serum levels were measured at baseline in adult control (n = 11) and SMKO (n = 6) female mice. We did not detect a difference in serum estradiol levels between control (2.9 ± 0.3) and SMKO (2.9 ± 0.6) mice. Furthermore, we measured uterine weights in control (n = 7) and SMKO (n = 6) female mice and did not detect a difference (3.6 ± 0.3 and 3.9 ± 0.4 mg/g for control and SMKO, respectively). Thus, differences in baseline BP are not correlated with alteration in estradiol.
Natriuresis in SMKO mice during development of hypertension.
To assess how VSMC AT1A receptors impact natriuresis during ANG II-induced hypertension in females, we performed a sodium balance study in control and SMKO female mice. Mice were placed in metabolic cages for daily urine collection and fed a gel diet (10 g/day) to ensure consistency in both water and solute intake. After 2 days of acclimation, baseline values were obtained, and, on day 5, osmotic minipumps filled with ANG II were implanted into both groups. Daily urinary sodium excretion was calculated and subtracted from daily sodium intake. As shown in Fig. 6, there was no significant difference in sodium excretion between control and SMKO female mice before or after ANG II-induced hypertension (0.12 ± 0.03 and 0.14 ± 0.03 mmol in control and SMKO, respectively), consistent with their similar levels of salt sensitivity (see above). This indicates that vascular AT1A receptors do not impact the early natriuretic effect of chronic ANG II administration in female mice.
Fig. 6.

No difference in natriuresis in control and SMKO female mice at baseline and during development of angiotensin II (ANG II)-induced hypertension. A: daily sodium balance in control (n = 8) and SMKO (n = 8) mice at baseline (3 days before) and in response to (5 days after) high-dose (1,000 ng·kg−1·min−1) ANG II-induced hypertension. There was no significant difference in natriuresis during any of the 8 days or in cumulative data. Values are means ± SE. Data were analyzed by 2-way repeated-measures ANOVA with Sidak’s post hoc test for multiple comparisons. B: cumulative sodium balance for 5 days following ANG II infusion were not different between control (n = 8) and SMKO (n = 7) mice: 0.12 ± 0.03 and 0.14 ± 0.03 mmol Na for control and SMKO, respectively (P = not significant, by unpaired t-test). Values are means ± SE.
DISCUSSION
Actions of ANG II on the AT1 receptor contribute significantly to cardiovascular morbidity and mortality (32). However, the ubiquitous pattern of AT1 receptor expression has not allowed for precise understanding of their tissue-specific actions (8, 54). Conditional gene targeting in mice has allowed for a more granular understanding of the relative contributions of distinct cellular AT1 receptor pools to determine basal BP, the development of hypertension, and detrimental end-organ sequelae. However, a majority of these studies utilized only male mice. Therefore, these findings cannot be extrapolated fully to females. Mounting evidence in both humans and animal models suggests that sex has a substantial impact on BP determination and hypertension pathogenesis (18, 19, 30, 37). Therefore, it is imperative to study how sex impacts the vascular function of the RAS.
First, these studies demonstrated that removal of VSMC AT1A receptors in female mice led to an ~8-mmHg decrease in baseline BP. Along with reduced baseline BP, the severity of ANG II-induced hypertension in the later phase was reduced by ~33% in female SMKO mice. This effect was most pronounced during the final 2 wk of the 4-wk ANG II infusion period. Consistent with reduced BP during ANG II-induced hypertension, cardiac hypertrophy was reduced in female mice. Concomitantly, heart rate increased ~30 beats/min in female SMKO mice. Thus, female SMKO mice are protected from ANG II-induced hypertension and subsequent cardiac hypertrophy, but not to the same extent as male mice (45). The influence of sex hormones on hemodynamic responses to ANG II has been varied. Studies in women suggest that levels of 17-estradiol affect renal plasma flow, specifically in response to ANG II (34); however, Dayton et al. showed that there is no significant difference in MAP during phases of the estrous cycle in rats fed both normal- and high-salt diets (15). Moreover, Veiras et al. found no correlation between the phase of the estrous cycle and the abundance or activity of transporter proteins in the cortex and medulla of rat kidneys (51). We did not observe differences in serum estrogen levels or uterine weights between SMKO and control female mice at baseline.
In global male AT1A receptor knockout mice, BP is markedly reduced and sodium sensitivity is enhanced (36). This finding was attributed to a combination of vascular relaxation and enhanced urinary sodium excretion, and sodium sensitivity was ascribed to replenishment of sodium losses. The result was recapitulated in male mice with deletion of AT1A receptors, specifically in VSMCs (44). However, in female SMKO mice, while basal BP was reduced, we did not observe a further exaggeration of sodium sensitivity compared with controls. However, wild-type C57BL/6 female mice demonstrated a more pronounced effect on BP upon transition from a low-salt to a high-salt diet compared with males. This would suggest that female C57BL/6 mice are more salt-sensitive than male C57BL/6 mice; however VSMC AT1A receptors do not further augment this response.
The classical actions of ANG II to induce vasoconstriction and increase BP are linked to discrete cellular actions of AT1A receptors. We previously reported a largely preserved acute vasoconstrictor response (~20% reduction) in male SMKO mice (44). This preservation of acute vasoconstriction was linked to enhanced adrenergic tone, as evidenced by increased urinary norepinephrine levels and attenuation of acute vasoconstriction with the -andrenergic blocker phentolamine (44). In contrast, in female SMKO mice the attenuation of the acute vasoconstrictor response to ANG II was more dramatic (~50–75%). This is supported in humans by the findings of Schmitt et al., who reported a blunted reduction of BP following phentolamine treatment in women compared with men (40). Xue et al. also reported that, during ANG II-induced hypertension in C57/BL6J mice, ganglionic blockade with hexamethonium led to a greater reduction in BP in males than females, suggesting an increased contribution of sympathetic activity in BP in male mice (53). However, we did demonstrate increased urinary norepinephrine levels after ANG II-induced hypertension in female SMKO mice. Thus, in female mice, VSMC AT1A receptors predominate in the peripheral acute vasoconstrictor response; in male mice, however, α-adrenergic tone synergizes this response.
The kidney circulation is exquisitely sensitive to ANG II. We used a laser-Doppler probe to assess kidney tissue perfusion in the kidney cortex. Kidney tissue perfusion is preserved after ANG II administration in female SMKO mice. However, in contrast to our previously reported finding in male mice (44), we did not detect a significant difference in sodium excretion between female SMKO and control mice during the first 5 days of ANG II-induced hypertension. The largest difference in BP between control and SMKO mice occurs during the last 2 wk of ANG II-induced hypertension. Thus, it is possible that natriuresis develops later in females, corresponding to the larger difference in BP seen in weeks 2–4. Female SHR have been shown to express reduced AT1A receptor mRNA in the kidney and vasculature compared with males (41); downregulation of AT1A receptors in cultured VSMCs of female WKY rats in response to estrogen has also been reported (35). Therefore, the natriuretic response elicited by increased ANG II may not be as effective in females as in males. For instance, it has been shown that the location of sodium transporters in the kidney among female murine models differs from that in males (51, 52). Female rats are reported to have a blunted increase in sodium-chloride cotransporter in response to ANG II infusion compared with males (50). Veiras et al. suggest that female murine models have lower proximal tubule reabsorption, expediting elimination of salt, compared with males (51).
Also, the physiological differences between male and female SMKO mice could be due to differences in experimental design, housing, or other experimental conditions. These physiological differences could also be explained by random genetic variation by the introduction of spontaneous mutations over time; it has been speculated that such mutations have occurred in other mouse models (26). Lastly, it is possible to make definitive direct comparisons only if both sexes are studied simultaneously.
Our studies further extend the importance of vascular ANG II responses in BP regulation and hypertension pathogenesis in female mice. The action of ANG II directly on VSMCs has a pronounced effect on mediating peripheral vasoconstriction in female mice. Lastly, the powerful effect of VSMC AT1 receptors on kidney vascular blood flow does not translate to a powerful natriuretic response, at least in the early phase of ANG II-induced hypertension. Thus, vascular actions of ANG II play a prominent role in BP determination, hypertension pathogenesis, cardiac hypertrophy, and acute BP responses in female mice.
GRANTS
This work was supported by Career Development Award IK2BX002240 from the Biomedical Laboratory Research and Development Service of the Department of Veterans Affairs Office of Research and Development (to M. A. Sparks). Research reported in this publication was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant P30 DK-096493.
DISCLAIMERS
The views expressed in this article are those of the authors and do not necessarily represent the policy or position of the US Department of Veterans Affairs or the US Government.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.A.S. and E.W. conceived and designed research; M.A.S., E.W., E.J.D., A.N.H., T.A.H., H.A.A., K.M.B., and R.C.G. performed experiments; M.A.S. and E.W. analyzed data; M.A.S. and E.W. interpreted results of experiments; M.A.S. and E.W. prepared figures; M.A.S. and E.W. drafted manuscript; M.A.S., E.W., E.J.D., T.A.H., A.N.H., H.A.A., K.M.B., and R.C.G. approved final version of manuscript; M.A.S. and E.W. edited and revised manuscript.
ACKNOWLEDGMENTS
We acknowledge Elizabeth Hauser for critical review of the statistical methods.
REFERENCES
- 1.Armando I, Jezova M, Juorio AV, Terrón JA, Falcón-Neri A, Semino-Mora C, Imboden H, Saavedra JM. Estrogen upregulates renal angiotensin II AT2 receptors. Am J Physiol Renal Physiol 283: F934–F943, 2002. doi: 10.1152/ajprenal.00145.2002. [DOI] [PubMed] [Google Scholar]
- 2.Badzyńska B, Grzelec-Mojzesowicz M, Dobrowolski L, Sadowski J. Differential effect of angiotensin II on blood circulation in the renal medulla and cortex of anaesthetised rats. J Physiol 538: 159–166, 2002. doi: 10.1113/jphysiol.2001.012921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Badzyńska B, Grzelec-Mojzesowicz M, Sadowski J. Prostaglandins but not nitric oxide protect renal medullary perfusion in anaesthetised rats receiving angiotensin II. J Physiol 548: 875–880, 2003. doi: 10.1113/jphysiol.2002.038075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhatia K, Zimmerman MA, Sullivan JC. Sex differences in angiotensin-converting enzyme modulation of Ang (1-7) levels in normotensive WKY rats. Am J Hypertens 26: 591–598, 2013. doi: 10.1093/ajh/hps088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S; RENAAL Study Investigators . Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345: 861–869, 2001. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 7.Butz GM, Davisson RL. Long-term telemetric measurement of cardiovascular parameters in awake mice: a physiological genomics tool. Physiol Genomics 5: 89–97, 2001. doi: 10.1152/physiolgenomics.2001.5.2.89. [DOI] [PubMed] [Google Scholar]
- 8.Carey RM. The intrarenal renin-angiotensin system in hypertension. Adv Chronic Kidney Dis 22: 204–210, 2015. doi: 10.1053/j.ackd.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Chang L, Villacorta L, Zhang J, Garcia-Barrio MT, Yang K, Hamblin M, Whitesall SE, D’Alecy LG, Chen YE. Vascular smooth muscle cell-selective peroxisome proliferator-activated receptor-γ deletion leads to hypotension. Circulation 119: 2161–2169, 2009. doi: 10.1161/CIRCULATIONAHA.108.815803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen D, Stegbauer J, Sparks MA, Kohan D, Griffiths R, Herrera M, Gurley SB, Coffman TM. Impact of angiotensin type 1A receptors in principal cells of the collecting duct on blood pressure and hypertension. Hypertension 67: 1291–1297, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen YF, Naftilan AJ, Oparil S. Androgen-dependent angiotensinogen and renin messenger RNA expression in hypertensive rats. Hypertension 19: 456–463, 1992. doi: 10.1161/01.HYP.19.5.456. [DOI] [PubMed] [Google Scholar]
- 12.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr, Jones DW, Materson BJ, Oparil S, Wright JT Jr, Roccella EJ; Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure, National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee . Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 42: 1206–1252, 2003. doi: 10.1161/01.HYP.0000107251.49515.c2. [DOI] [PubMed] [Google Scholar]
- 13.Cohn JN, Tognoni G; Valsartan Heart Failure Trial Investigators . A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med 345: 1667–1675, 2001. doi: 10.1056/NEJMoa010713. [DOI] [PubMed] [Google Scholar]
- 14.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci USA 103: 17985–17990, 2006. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dayton A, Exner EC, Bukowy JD, Stodola TJ, Kurth T, Skelton M, Greene AS, Cowley AW Jr. Breaking the cycle: estrous variation does not require increased sample size in the study of female rats. Hypertension 68: 1139–1144, 2016. doi: 10.1161/HYPERTENSIONAHA.116.08207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellison KE, Ingelfinger JR, Pivor M, Dzau VJ. Androgen regulation of rat renal angiotensinogen messenger RNA expression. J Clin Invest 83: 1941–1945, 1989. doi: 10.1172/JCI114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fields LE, Burt VL, Cutler JA, Hughes J, Roccella EJ, Sorlie P. The burden of adult hypertension in the United States 1999 to 2000: a rising tide. Hypertension 44: 398–404, 2004. doi: 10.1161/01.HYP.0000142248.54761.56. [DOI] [PubMed] [Google Scholar]
- 18.Gilbert JS, Nijland MJ. Sex differences in the developmental origins of hypertension and cardiorenal disease. Am J Physiol Regul Integr Comp Physiol 295: R1941–R1952, 2008. doi: 10.1152/ajpregu.90724.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gillis EE, Sullivan JC. Sex differences in hypertension: recent advances. Hypertension 68: 1322–1327, 2016. doi: 10.1161/HYPERTENSIONAHA.116.06602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19a.GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia) Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. Lancet 349: 1857–1863, 1997. doi: 10.1016/S0140-6736(96)11445-8. [DOI] [PubMed] [Google Scholar]
- 20.Gu Q, Burt VL, Paulose-Ram R, Dillon CF. Gender differences in hypertension treatment, drug utilization patterns, and blood pressure control among US adults with hypertension: data from the National Health and Nutrition Examination Survey 1999-2004. Am J Hypertens 21: 789–798, 2008. doi: 10.1038/ajh.2008.185. [DOI] [PubMed] [Google Scholar]
- 21.Gurley SB, Riquier-Brison ADM, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab 13: 469–475, 2011. doi: 10.1016/j.cmet.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haisenleder DJ, Schoenfelder AH, Marcinko ES, Geddis LM, Marshall JC. Estimation of estradiol in mouse serum samples: evaluation of commercial estradiol immunoassays. Endocrinology 152: 4443–4447, 2011. doi: 10.1210/en.2011-1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.SOLVD Investigators; Yusuf S, Pitt B, Davis CE, Hood WB, Cohn JN. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med 325: 293–302, 1991. doi: 10.1056/NEJM199108013250501. [DOI] [PubMed] [Google Scholar]
- 24.Ito M, Oliverio MI, Mannon PJ, Best CF, Maeda N, Smithies O, Coffman TM. Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proc Natl Acad Sci USA 92: 3521–3525, 1995. doi: 10.1073/pnas.92.8.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.James PA, Oparil S, Carter BL, Cushman WC, Dennison-Himmelfarb C, Handler J, Lackland DT, LeFevre ML, MacKenzie TD, Ogedegbe O, Smith SC Jr, Svetkey LP, Taler SJ, Townsend RR, Wright JT Jr, Narva AS, Ortiz E. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 311: 507–520, 2014. doi: 10.1001/jama.2013.284427. [DOI] [PubMed] [Google Scholar]
- 26.Ji H, Pai AV, West CA, Wu X, Speth RC, Sandberg K. Loss of resistance to angiotensin II-induced hypertension in the Jackson Laboratory recombination-activating gene null mouse on the C57BL/6J background. Hypertension 69: 1121–1127, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keyhani S, Scobie JV, Hebert PL, McLaughlin MA. Gender disparities in blood pressure control and cardiovascular care in a national sample of ambulatory care visits. Hypertension 51: 1149–1155, 2008. doi: 10.1161/HYPERTENSIONAHA.107.107342. [DOI] [PubMed] [Google Scholar]
- 28.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD; The Collaborative Study Group . The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N Engl J Med 329: 1456–1462, 1993. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 29.Mancia G, Sega R, Bravi C, De Vito G, Valagussa F, Cesana G, Zanchetti A. Ambulatory blood pressure normality: results from the PAMELA study. J Hypertens 13: 1377–1390, 1995. doi: 10.1097/00004872-199512000-00003. [DOI] [PubMed] [Google Scholar]
- 30.Maranon R, Reckelhoff JF. Sex and gender differences in control of blood pressure. Clin Sci (Lond) 125: 311–318, 2013. doi: 10.1042/CS20130140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, Chambon P, Hill MA, Dorrance AM, Mendelsohn ME, Jaffe IZ. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med 18: 1429–1433, 2012. doi: 10.1038/nm.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292: C82–C97, 2007. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 33.Melloni C, Berger JS, Wang TY, Gunes F, Stebbins A, Pieper KS, Dolor RJ, Douglas PS, Mark DB, Newby LK. Representation of women in randomized clinical trials of cardiovascular disease prevention. Circ Cardiovasc Qual Outcomes 3: 135–142, 2010. doi: 10.1161/CIRCOUTCOMES.110.868307. [DOI] [PubMed] [Google Scholar]
- 34.Miller JA, Anacta LA, Cattran DC. Impact of gender on the renal response to angiotensin II. Kidney Int 55: 278–285, 1999. doi: 10.1046/j.1523-1755.1999.00260.x. [DOI] [PubMed] [Google Scholar]
- 35.Nickenig G, Bäumer AT, Grohè C, Kahlert S, Strehlow K, Rosenkranz S, Stäblein A, Beckers F, Smits JF, Daemen MJ, Vetter H, Böhm M. Estrogen modulates AT1 receptor gene expression in vitro and in vivo. Circulation 97: 2197–2201, 1998. doi: 10.1161/01.CIR.97.22.2197. [DOI] [PubMed] [Google Scholar]
- 36.Oliverio MI, Best CF, Smithies O, Coffman TM. Regulation of sodium balance and blood pressure by the AT1A receptor for angiotensin II. Hypertension 35: 550–554, 2000. doi: 10.1161/01.HYP.35.2.550. [DOI] [PubMed] [Google Scholar]
- 37.Reckelhoff JF. Gender differences in the regulation of blood pressure. Hypertension 37: 1199–1208, 2001. doi: 10.1161/01.HYP.37.5.1199. [DOI] [PubMed] [Google Scholar]
- 38.Rogers JL, Mitchell AR, Maric C, Sandberg K, Myers A, Mulroney SE. Effect of sex hormones on renal estrogen and angiotensin type 1 receptors in female and male rats. Am J Physiol Regul Integr Comp Physiol 292: R794–R799, 2007. doi: 10.1152/ajpregu.00424.2006. [DOI] [PubMed] [Google Scholar]
- 39.Sampson AK, Moritz KM, Jones ES, Flower RL, Widdop RE, Denton KM. Enhanced angiotensin II type 2 receptor mechanisms mediate decreases in arterial pressure attributable to chronic low-dose angiotensin II in female rats. Hypertension 52: 666–671, 2008. doi: 10.1161/HYPERTENSIONAHA.108.114058. [DOI] [PubMed] [Google Scholar]
- 40.Schmitt JA, Joyner MJ, Charkoudian N, Wallin BG, Hart EC. Sex differences in α-adrenergic support of blood pressure. Clin Auton Res 20: 271–275, 2010. doi: 10.1007/s10286-010-0061-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silva-Antonialli MM, Tostes RC, Fernandes L, Fior-Chadi DR, Akamine EH, Carvalho MH, Fortes ZB, Nigro D. A lower ratio of AT1/AT2 receptors of angiotensin II is found in female than in male spontaneously hypertensive rats. Cardiovasc Res 62: 587–593, 2004. doi: 10.1016/j.cardiores.2004.01.020. [DOI] [PubMed] [Google Scholar]
- 42.Sparks MA, Makhanova NA, Griffiths RC, Snouwaert JN, Koller BH, Coffman TM. Thromboxane receptors in smooth muscle promote hypertension, vascular remodeling, and sudden death. Hypertension 61: 166–173, 2013. doi: 10.1161/HYPERTENSIONAHA.112.193250. [DOI] [PubMed] [Google Scholar]
- 43.Sparks MA, Parsons KK, Stegbauer J, Gurley SB, Vivekanandan-Giri A, Fortner CN, Snouwaert J, Raasch EW, Griffiths RC, Haystead TA, Le TH, Pennathur S, Koller B, Coffman TM. Angiotensin II type 1A receptors in vascular smooth muscle cells do not influence aortic remodeling in hypertension. Hypertension 57: 577–585, 2011. doi: 10.1161/HYPERTENSIONAHA.110.165274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sparks MA, Stegbauer J, Chen D, Gomez JA, Griffiths RC, Azad HA, Herrera M, Gurley SB, Coffman TM. Vascular type 1A angiotensin II receptors control BP by regulating renal blood flow and urinary sodium excretion. J Am Soc Nephrol 26: 2953–2962, 2015. doi: 10.1681/ASN.2014080816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sparks MA, Stegbauer J, Chen D, Vivekanandan-Giri A, Pennathur S, Crowley SD, Gurley SB, Coffman TM Cardiac hypertrophy in angiotensin II-dependent hypertension: dominant effect of blood pressure (Abstract) Hypertension 64: A424, 2014. [Google Scholar]
- 46.Staessen J, Bulpitt CJ, Fagard R, Lijnen P, Amery A. The influence of menopause on blood pressure. J Hum Hypertens 3: 427–433, 1989. [PubMed] [Google Scholar]
- 47.Sullivan JC. Sex and the renin-angiotensin system: inequality between the sexes in response to RAS stimulation and inhibition. Am J Physiol Regul Integr Comp Physiol 294: R1220–R1226, 2008. doi: 10.1152/ajpregu.00864.2007. [DOI] [PubMed] [Google Scholar]
- 48.Sullivan JC, Bhatia K, Yamamoto T, Elmarakby AA. Angiotensin (1-7) receptor antagonism equalizes angiotensin II-induced hypertension in male and female spontaneously hypertensive rats. Hypertension 56: 658–666, 2010. doi: 10.1161/HYPERTENSIONAHA.110.153668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanoue A, Nasa Y, Koshimizu T, Shinoura H, Oshikawa S, Kawai T, Sunada S, Takeo S, Tsujimoto G. The α1D-adrenergic receptor directly regulates arterial blood pressure via vasoconstriction. J Clin Invest 109: 765–775, 2002. doi: 10.1172/JCI200214001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tiwari S, Li L, Riazi S, Halagappa VK, Ecelbarger CM. Sex and age result in differential regulation of the renal thiazide-sensitive NaCl cotransporter and the epithelial sodium channel in angiotensin II-infused mice. Am J Nephrol 30: 554–562, 2009. doi: 10.1159/000252776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Veiras LC, Girardi ACC, Curry J, Pei L, Ralph DL, Tran A, Castelo-Branco RC, Pastor-Soler N, Arranz CT, Yu ASL, McDonough AA. Sexual dimorphic pattern of renal transporters and electrolyte homeostasis. J Am Soc Nephrol 28: 3504–3517, 2017. doi: 10.1681/ASN.2017030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verlander JW, Tran TM, Zhang L, Kaplan MR, Hebert SC. Estradiol enhances thiazide-sensitive NaCl cotransporter density in the apical plasma membrane of the distal convoluted tubule in ovariectomized rats. J Clin Invest 101: 1661–1669, 1998. doi: 10.1172/JCI601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xue B, Pamidimukkala J, Hay M. Sex differences in the development of angiotensin II-induced hypertension in conscious mice. Am J Physiol Heart Circ Physiol 288: H2177–H2184, 2005. doi: 10.1152/ajpheart.00969.2004. [DOI] [PubMed] [Google Scholar]
- 54.Zhuo JL, Li XC. New insights and perspectives on intrarenal renin-angiotensin system: focus on intracrine/intracellular angiotensin II. Peptides 32: 1551–1565, 2011. doi: 10.1016/j.peptides.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zucker I, Beery AK. Males still dominate animal studies. Nature 465: 690, 2010. doi: 10.1038/465690a. [DOI] [PubMed] [Google Scholar]