

Abstract
Familial hyperkalemic hypertension is caused by mutations in with-no-lysine kinases (WNKs) or in proteins that mediate their degradation, kelch-like 3 (KLHL3) and cullin 3 (CUL3). Although the mechanisms by which WNK and KLHL3 mutations cause the disease are now clear, the effects of the disease-causing CUL3Δ403–459 mutation remain controversial. Possible mechanisms, including hyperneddylation, altered ubiquitin ligase activity, decreased association with the COP9 signalosome (CSN), and increased association with and degradation of KLHL3 have all been postulated. Here, we systematically evaluated the effects of Cul3Δ403–459 using cultured kidney cells. We first identified that the catalytically active CSN subunit jun activation domain-binding protein-1 (JAB1) does not associate with the deleted Cul3 4-helix bundle domain but instead with the adjacent α/β1 domain, suggesting that altered protein folding underlies the impaired binding. Inhibition of deneddylation with JAB1 siRNA increased Cul3 neddylation and decreased KLHL3 abundance, similar to the Cul3 mutant. We next determined that KLHL3 degradation has both ubiquitin ligase-dependent and -independent components. Proteasomal KLHL3 degradation was enhanced by Cul3Δ403–459; however, autophagic degradation was also upregulated by this Cul3 mutant. Finally, to evaluate whether deficient substrate adaptor was responsible for the disease, we restored KLHL3 to wild-type (WT) Cul3 levels. In the absence of WT Cul3, WNK4 was not degraded, demonstrating that Cul3Δ403–459 itself cannot degrade WNK4; conversely, when WT Cul3 was present, as in diseased humans, WNK4 degradation was restored. In conclusion, deletion of exon 9 from Cul3 generates a protein that is itself ubiquitin-ligase defective but also capable of enhanced autophagocytic KLHL3 degradation, thereby exerting dominant-negative effects on the WT allele.
Keywords: cullin-RING ubiquitin ligase, deneddylation, JAB1, neddylation
INTRODUCTION
With-no-lysine kinases (WNKs) control blood pressure and potassium homeostasis, predominantly by regulating membrane expression and activity of the thiazide-sensitive NaCl cotransporter in the distal convoluted tubule. These kinases signal via serine/threonine protein kinase 39 and oxidative stress-response 1, which directly phosphorylate and activate the transport protein (19, 30). The human Mendelian disease familial hyperkalemic hypertension (FHHt; also called pseudohypoaldosteronism type 2 or Gordon syndrome) results from activation of this signaling pathway in the distal nephron (21). Patients with FHHt exhibit hyperkalemia, metabolic acidosis, and hypertension, symptoms that largely disappear during treatment with thiazide diuretics (13). FHHt can be caused by mutations in WNK1 or WNK4 (32) or in the cullin-RING ligase (CRL) proteins cullin 3 (Cul3; 4) and kelch-like 3 (KLHL3; 28). CUL3 is part of an E3 ubiquitin ligase complex that regulates protein degradation. CRLs do not degrade proteins directly but instead attach strings of ubiquitin moieties to a protein, thereby targeting it for degradation, typically within the proteasome. Cullin acts as a scaffold protein for the other CRL subunits. It is now clear that WNKs are targets for CRLs. The substrate adaptor KLHL3 binds both WNKs and Cul3, bringing the WNK into proximity to the catalytic region of the CRL, thereby permitting ubiquitylation (18, 25). The WNK4 or KLHL3 mutations that cause disease do so by disrupting these binding reactions, permitting WNKs to accumulate (31). The CUL3 mutations that cause FHHt, however, do not decrease substrate adaptor binding (27, 31); although mouse models indicate that WNKs are not degraded normally (24), the precise mechanisms involved remain controversial.
An important feature of CRL activity is the attachment of neuronal precursor cell expressed developmentally downregulated protein 8 (NEDD8) through a process called neddylation. NEDD8 attachment is required to activate CRLs by increasing the flexibility of the cullin-ring structure, allowing the transfer of ubiquitin from the RING protein to the target substrate (3, 20, 23). The reverse process, deneddylation, is facilitated by the multisubunit COP9 signalosome (CSN) complex. The CSN interacts with CRLs and removes NEDD8 through its catalytically active CSN5 subunit, jun activation domain-binding protein-1 (JAB1; 6). Although a simple model originally suggested that neddylated Cul3 is active, whereas unneddylated Cul3 is inactive, inhibition of CSN paradoxically increases rather than decreases the abundance of substrate proteins in vivo (22). It appears instead that, although neddylation of cullins is indeed essential to activate them, it also makes them unstable and prone to degradation (35). Thus, the effects of neddylation on cullin activity and abundance are complex.
All known FHHt-causing CUL3 mutations cause deletion of exon 9, resulting in a mutant protein that lacks 57 amino acid residues (Cul3Δ403–459; 4). Previous work by our group (14), and confirmed by others (24), showed that Cul3Δ403–459 expressed in cells is hyperneddylated compared with wild type (WT), suggesting either that it is more susceptible to neddylation or that it is resistant to deneddylation. Schumacher et al. (24) confirmed that Cul3Δ403–459 had impaired deneddylation and compromised CSN binding, suggesting that the deleted segment was responsible. Exon 9 of CUL3 encodes the 4-helix bundle (4HB) domain. Min et al. (15) demonstrated that the CSN binding site for the closely related cullin 1 protein was located within the 4HB and α/β1 domains. However, the CSN binding site for Cul3 has yet to be determined.
Cul3Δ403–459 has an increased association with bric-a-brac, tramtrack, broad-complex (BTB)-Kelch substrate adaptor proteins, including KLHL3 (10, 14, 24). The FHHt Cul3 mutant strongly ubiquitylated KLHL3 (14, 24) leading to decreased abundance in vitro (14). However, administration of the nonspecific neddylation inhibitor MLN4924, which prevents NEDD8 conjugation and presumably inactivates CRLs, only partially normalized KLHL3 abundance. Additionally, the enhanced interaction of KLHL3 with Cul3Δ403–459 remained in the absence of neddylation. The results indicate that Cul3Δ403–459 may have neddylation-dependent and neddylation-independent effects. Here, we examined the mechanisms and consequences of Cul3Δ403–459 hyperneddylation and identified novel ligase-dependent and ligase-independent mechanisms for the human disease.
MATERIALS AND METHODS
Antibodies.
Antibodies used are described in Table 1.
Table 1.
Antibodies used for Western blot
| Figure | Target | Antibody Name | Source, cat. no. | 1° Dilution | 2° Dilution |
|---|---|---|---|---|---|
| 1E, 2 | JAB1 | JAB1 FL-334 | Santa Cruz, SC-9074 | 1:1,000, 1 h at RT | 1:5,000 |
| 1, C and D | JAB1 | JAB1 6C3.38 | Thermo Fisher Scientific | 1:2,000, 1 h at RT | 1:5,000 |
| 1D | GST | GST B-14 | Santa Cruz, SC-138 | 1:1,000, 1 h at RT | 1:2,500 |
| 1, 2, 3, 4, 5, 6, 7, 8 | Myc | c-Myc | Sigma-Aldrich, M5546 | 1:5,000, 1 h at RT | 1:10,000 |
| 1E, 2, 4A | NEDD8 | NEDD8 19E3 | Cell Signaling, 2754 | 1:1,000, o/n at 4°C | 1:2,500 |
| 2, 4D | Cul3 | Cul3 | Cell Signaling, 2759 | 1:1,000, o/n at 4°C | 1:2,500 |
| 2, 8 | β-actin | β-actin | Abcam, Ab8227 | 1:5,000, 1 h at RT | 1:2,500 |
| 1, 4, 5, 6, 7, 8, 9 | FLAG | FLAG M2 | Sigma-Aldrich, F3165 | 1:10,000, 1 h at RT | 1:10,000 |
| 4C | HA | HA.11 | Covance, MMS-101P | 1:1,000, 1 h at RT | 1:10,000 |
| 5, 6 | GAPDH | GAPDH | Santa Cruz, SC-20357 | 1:1,000, 1 h at RT | 1:2,500 |
| 9 | Keap1 | Keap1 | Abcam, Ab139729 | 1:1,000, o/n at 4°C | 1:2,500 |
| 9 | Nrf2 | Nrf2 H-300 | Santa Cruz, SC-13032 | 1:1,000, o/n at 4°C | 1:2,500 |
| 9 | Cyclin E | Cyclin E HE12 | Santa Cruz, SC-247 | 1:1,000, o/n at 4°C | 1:2,500 |
Cul3, cullin 3; GST, glutathione S-transferase; HA, hemagglutinin; JAB1, jun activation domain-binding protein-1; Keap1, kelch-like ECH-associated protein 1; NEDD8, neuronal precursor cell expressed developmentally downregulated protein 8; Nrf2, nuclear factor erythroid 2-related factor 2; o/n, overnight; RT, room temperature.
Cell culture, plasmids, and transfections.
For cell culture experiments, HEK293 cells were used unless otherwise stated. CRISPR-Cas9-edited Cul3 knockdown HEK293T cells (HEK293TCul3-KO) were previously reported (10). Cells were maintained in DMEM supplemented with 10% FBS, 25 mM HEPES, 100 units/ml penicillin, and 100 µg/ml streptomycin. Cells were transiently transfected using Lipofectamine 2000 (Ambion, Foster City, CA; Invitrogen). Cul3 constructs were made by amplifying FLAG-Cul3 WT DNA using Phusion Hot Start II DNA Polymerase (Thermo Fisher Scientific, Boston, MA) with the appropriate primers, purified with the PureLink PCR Purification Kit (Invitrogen) and properly digested. The products were then extracted using the UltraClean GelSpin DNA Extraction Kit (MoBio Laboratories, Inc., Carlsbad, CA) and ligated into the N-terminal glutathione S-transferase tag mammalian plasmid, pSF-CMV-Puro-NH2-GST (Oxford Genetics, Oxford, UK), with T4 DNA Ligase (New England BioLabs, Ipswich, MA). Ligated constructs were transformed using DH5α-competent cells (Thermo Fisher Scientific) and plasmid DNA was purified with either the QIAprep Spin Miniprep Kit or HiSpeed Plasmid Midi Kit (Qiagen, Hilden, Germany). Sanger sequencing was performed for all constructs.
For siRNA experiments, either 40 nM of COPS5 siRNA (Ambion) or control siRNA was transfected along with DNA plasmids. Cells were harvested at 36 h posttransfection.
For cycloheximide chase experiments, cycloheximide was added 36 h after transfection at a concentration of 100 µg/ml, and the cells were lysed at the time points indicated.
For MG132, chloroquine, 3-methyadenine, MLN4924, and tBHQ experiments, the respective drug was added to cells 18 h before harvesting at the concentrations given.
For ubiquitin assay experiments, cells were cotransfected with ubiquitin DNA plasmid. Cells were lysed 48 h after transfection in cell lysis buffer containing 10 mM N-ethylmaleimide. Immunoprecipitation and Western blotting were carried out as below.
Immunoprecipitation and Western blotting.
Transfected cells were harvested in 0.5% Triton X-100 in PBS cell lysis buffer containing enzyme inhibitors. For immunoprecipitation, cell lysate was precleared with protein A-sepharose beads for 1–2 h. Cell lysate was then incubated with Glutathione Sepharose 4B medium (GE Healthcare, Piscataway, NJ) for 2 h at room temperature, and primary antibody and Protein A-Sepharose 4B medium (GE Healthcare) or anti-FLAG Affinity Gel (Biotool, Houston, TX) overnight at 4°C. Protein samples were separated by electrophoresis on 4%–12% NuPAGE bis-tris polyacrylamide gels (Thermo Fisher Scientific) or 4%–15% Criterion TGX stain-free gels (Bio-Rad Laboratories, Hercules, CA) and transferred to Immobilon-P PVDF membranes (EMD Millipore, Billerica, MA). For all experimental conditions performed in triplicate, each well represents a unique transfection. Stain-free imaging was used as a total protein loading control, unless otherwise stated. Membranes were blocked with 5% milk in PBS for 1 h at room temperature before incubation with primary antibody in blocking buffer for 1 h at room temperature or overnight at 4°C. Appropriate horseradish peroxidase-conjugated secondary antibody in blocking buffer was added to membranes for 1 h at room temperature. Membranes were developed using enhanced chemiluminescence, Western Lightning Plus–ECL (Perkin Elmer, Waltham, MA); proteins were visualized using PXi digital imaging system (Syngene, Frederick, MA).
Statistics.
Data are presented as individual values as well as means ± SE. Differences between two groups were determined using two-tailed unpaired Student’s t-test, and differences between multiple groups were analyzed using one-way ANOVA followed by Tukey’s multiple comparisons test. A P value of less than 0.05 was considered significant. Statistical analysis was performed using GraphPad Prism 7 software (GraphPad Software, San Diego, CA).
RESULTS
CSN binds to Cul3 at the α/β1 domain.
CRLs are activated by NEDD8 attachment, but for full functionality, they must also be deneddylated. The deneddylation of Cul3 appears to be disrupted in Cul3Δ403–459 FHHt, as the mutant protein is more highly neddylated than WT, at least when expressed in cultured cells (14, 24). Schumacher et al. (24) reported that Cul3Δ403–459 exhibits decreased interaction with JAB1, suggesting that the deleted domain may include the CSN-binding site. The crystal structure of CSN with the highly homologous cullin 1 (12) and cullin 4A (5) showed that the CSN2 subunit interacts directly with the C-terminal domain. Additionally, Min et al. (15) determined that the CSN-binding site for cullin 1 (which is structurally similar to Cul3) was located specifically within the C-terminal domains 4HB and α/β1. Inspection of the Cul3 gene revealed that exon 9 (deleted in FHHt-causing CUL3 mutations) encodes the 4HB domain (see Fig. 1A). To determine sites of CSN interaction with Cul3, we generated Cul3 deletion constructs (Fig. 1A). We first confirmed that compared with WT Cul3, there was minimal JAB1 precipitation by Cul3Δ403–459 (Fig. 1B). Similarly, the Cul3 construct containing N-terminal residues 1–402, which lacks the 4HB domain and the adjacent α/β1 domain showed nominal precipitation of JAB1 (Fig. 1C). Surprisingly, inclusion of the 4HB domain with the N-terminal region also showed low binding to JAB1 (Fig. 1C, Cul3 1–459). These results indicate clearly that Cul3 amino acids 403–459 (containing the 4HB domain) are not sufficient for binding to the CSN. This suggests that the Cul3Δ403–459 mutation does not impair Cul3-CSN binding directly but rather disrupts protein folding within a site C-terminal to the 4HB domain; this suggestion is consistent with structural modeling of wild type and mutant Cul3 (24).
Fig. 1.

CSN binds to Cul3 at the α/β1 domain. A: Diagram of Cul3 domain structure and schematic of the Cul3 constructs. B: Coimmunoprecipitation was performed with HEK293 cells transfected with myc-JAB1 and FLAG-tagged WT Cul3 or Cul3Δ403–459 and analyzed by immunoblot. Cul3Δ403–459 exhibited a decreased interaction with JAB1 compared with WT Cul3. C: Effects of Cul3Δ403–459 on JAB1 binding was determined by coimmunoprecipitation of HEK293 cells with N-terminal domain Cul3 constructs using anti-FLAG and analyzed by immunoblot. Coimmunoprecipitation of N-terminal domain Cul3 constructs with (1–459) and without (1–402) the 4HB domain showed no binding to JAB1. D: Segments of the Cul3 protein were generated with a GST tag and cotransfected with myc-tagged JAB1 in HEK293 cells. Coimmunoprecipitation was performed using glutathione sepharose beads. Immunoblotting for JAB1 showed binding to 4HB:α/β1 and α/β1 Cul3 constructs but not to 4HB, WH-A:α/β:WH-B, or R1:R2:R3 Cul3 constructs. E: Coimmunoprecipitation was performed in HEK293 cells with myc-JAB1 and FLAG-tagged WT Cul3 or Cul3Δ461–586 constructs. Cul3Δ461–586 demonstrated less binding to JAB1 protein compared with WT Cul3. Immunoblotting for NEDD8 showed enhanced neddylation of the Cul3Δ461–586 construct compared with WT Cul3. *Nonspecific band. 4HB, 4-helix bundle; Cul3, cullin 3; CSN, COP9 signalosome; GST, glutathione S-transferase; HEK, human embryonic kidney; IP, immunoprecipitation; JAB1, jun activation domain-binding protein-1; NEDD8, neuronal precursor cell expressed developmentally downregulated protein 8; WT, wild type.
To identify the specific binding site for the CSN and to confirm that the 4HB does not bind JAB1, we developed individual Cul3 domain constructs for 4HB and α/β1. Additionally, we generated a construct containing both the 4HB and α/β1 domains (4HB:α/β1), an N-terminal construct containing cullin repeat sequences (R1:R2:R3) and a C-terminal construct containing domains WH-A, α/β2, and WH-B (WH-A:α/β2:WH-B). JAB1 immunoprecipitated with the Cul3 construct containing both the 4HB and α/β1 domains and with α/β1 alone but not with 4HB alone (Fig. 1D). JAB1 was not precipitated with Cul3 constructs that lacked the α/β1 domain, R1:R2:R3 and WH-A:α/β2:WH-B. The results indicate that the first α/β domain of Cul3 is the binding site for JAB1 and therefore the CSN.
To verify that the α/β1 domain is the binding site for the CSN we developed a full-length Cul3 construct in which the α/β1 was deleted (Cul3Δ461–586). JAB1 was not precipitated with Cul3Δ461–586 (Fig. 1E). As expected, Cul3Δ461–586 also showed enhanced neddylation compared with WT Cul3. These results further confirm that the CSN-binding site is contained within the α/β1 domain of Cul3.
CSN inhibition enhances Cul3 neddylation and reduces KLHL3 and WNK4 abundance.
Neddylation of cullins has paradoxical effects, with both neddylation and deneddylation being required for normal CRL function. Because Cul3Δ403–459 exhibits decreased interaction with JAB1 (24), we examined the effects of inhibiting JAB1 activity on Cul3 in HEK293 cells. Cells were transfected with siRNA to reduce endogenous JAB1. Western blotting for endogenous Cul3 exhibited a more neddylated Cul3 (as detected by an increase in the higher molecular weight Cul3 band) when JAB1 was reduced, compared with control (Fig. 2). Similarly, probing the blot with an antibody against NEDD8 showed a greater abundance of neddylated Cul3 when JAB1 was knocked down, as the antibody recognized a product at the molecular weight of Cul3. The effects of JAB1 inhibition on the protein abundance of the Cul3 substrate adaptor KLHL3 and substrate WNK4 were also determined. Abundance of overexpressed KLHL3 and WNK4 were lower in cells transfected with JAB1 siRNA compared with control siRNA, indicating that JAB1 knockdown and increased neddylation activates Cul3 in transfected cells.
Fig. 2.
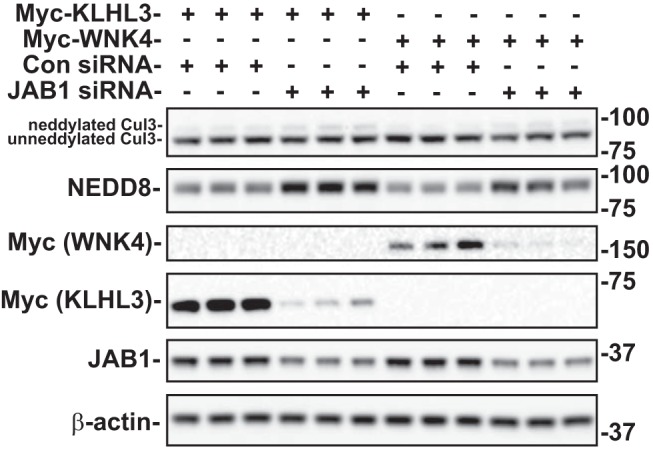
Effects of JAB1 inhibition on Cul3 neddylation and substrate protein abundance. Myc-tagged KLHL3 or WNK4 was cotransfected into HEK293 cells with either JAB1 siRNA or control siRNA. The proteins were examined by immunoblot in cells with endogenous WT Cul3. JAB1 siRNA decreased JAB1, KLHL3, and WNK4 abundance and increased NEDD8 abundance and the neddylated form of Cul3 (top band). β-actin was used as a loading control. Con, control; Cul3, cullin 3; HEK, human embryonic kidney; JAB1, jun activation domain-binding protein-1; KLHL3, kelch-like 3; NEDD8, neuronal precursor cell expressed developmentally downregulated protein 8; WNK, with-no-lysine kinase; WT, wild type.
Cul3Δ403–459 causes decreased stability of KLHL3.
We showed previously that KLHL3 protein abundance was lower when coexpressed with Cul3Δ403–459 than with WT Cul3 in HEK293 cells (14), suggesting that degradation was more rapid in the presence of mutant Cul3. To test this, we measured the stability of KLHL3 using the cycloheximide chase assay. KLHL3 abundance was significantly lower 24 h after cycloheximide treatment when cotransfected with Cul3Δ403–459 compared with WT Cul3, but the apparent degradation rates at other time points were not different (Fig. 3A). As KLHL3 cotransfected with WT Cul3 was stable at least 24 h after cycloheximide treatment [the long stability of KLHL3 has been previously published (17)], which can lead to anomalous results in cycloheximide chase experiments (37), we decided to examine a canonical CRL substrate, WNK4, using a similar approach. Similar to the Cul3-KLHL3 experiments, WNK4 was stable when cycloheximide was introduced in the absence of KLHL3; however, when cotransfected together with KLHL3, WNK4 abundance was strikingly reduced (Fig. 3B). When we compared the effects of KLHL3 on WNK4 abundance with those of Cul3Δ403–459 on KLHL3 abundance, the results are remarkably similar (Fig. 3C). In fact, when other groups have examined the effects of KLHL3 on WNK4 abundance, they also noted remarkably reduced WNK4 abundance in the presence of KLHL3 (31). Thus, although we cannot prove that the Cul3Δ403–459 and WT Cul3 have differential effects on KLHL3 synthesis, the current results, when coupled with those shown below suggest that KLHL3 is degraded more rapidly by the Cul3 mutant.
Fig. 3.

Cul3Δ403–459 decreases the stability of KLHL3. A: Cycloheximide chase assay was performed with HEK293 cells cotransfected with myc-tagged KLHL3 and either FLAG-WT Cul3 or FLAG-Cul3Δ403–459. Because of a robust decrease in KLHL3 by the Cul3Δ403–459 that prevented quantification, the amount of Cul3Δ403–459 transfected was reduced to half of WT Cul3. Cycloheximide (100 µg/ml) was added 36 h posttransfection, and cells were lysed at 0, 1, 2, 4, 8, and 24 h time points. KLHL3 protein abundance was more rapidly degraded in cells coexpressing Cul3Δ403–459. Right, quantitative analysis of KLHL3 protein abundance. Stain-free imaging was used as a loading control. Linear regression was used to determine the slope of each group. The differences between the slopes were significantly different (P < 0.001). Data represent mean values ± SE relative to the 0 h time point. Statistical differences were examined using two-tailed unpaired Student’s t-test. *P = 0.01 vs. WT. B: Cycloheximide chase assay was performed with HEK293 cells cotransfected with myc-tagged WNK4 in the presence or absence of KLHL3. Cycloheximide (100 µg/ml) was added 36 h posttransfection and cells were lysed at 0, 2, 4, and 6 h time points. Stain-free imaging was used as a loading control. C: Left, quantitative analysis of KLHL3; all data points are relative to WT Cul3 0 h time point. Right, quantitative analysis of WNK4 protein abundance; all data points are relative to WNK4 without KLHL3 0 h time point. The effects of Cul3Δ403–459 on KLHL3 abundance is similar to the effects of KLHL3 on WNK4 abundance. Cul3, cullin 3; KLHL3, kelch-like 3; WNK, with-no-lysine kinase; WT, wild type.
Ligase-dependent and -independent effects of Cul3Δ403–459 on KLHL3 and WNK4.
The Cul3Δ403–459 mutation has altered ubiquitin ligase activity as shown by increased ubiquitylation of KLHL3 and decreased ubiquitylation of WNK4 (14, 24). Yet, Cul3Δ403–459 also has ligase-independent effects, such as enhanced binding to BTB-Kelch adaptors and decreased binding to CSN subunits and cullin-associated and NEDD8-dissociated protein 1 (10, 14, 24). To try to understand the anomalous effects of the Cul3Δ403–459 protein better, we generated a neddylation-deficient Cul3Δ403–459 double mutant, Cul3Δ403–459 K712R. Neddylation is generally considered to be necessary to activate CRLs. The RING subunit utilizes specific E1 and E2 enzymes to covalently attach a NEDD8 protein to lysine 712 of Cul3 (33). Cul3Δ403–459 K712R includes a point mutation at the neddylation site preventing NEDD8 attachment and rendering the construct ligase-deficient. Immunoprecipitation of FLAG-tagged Cul3Δ403–459 K712R showed an almost complete loss of neddylation (Fig. 4A), indicating that the vast majority of Cul3Δ403–459 neddylation occurs at the lysine 712 residue. Coimmunoprecipitation of the different Cul3 constructs and KLHL3 showed that similar to Cul3Δ403–459, Cul3Δ403–459 K712R bound to more KLHL3 protein compared with WT Cul3 (Fig. 4B), confirming that neddylation, and therefore ubiquitin ligase activity, is not required for increased protein binding.
Fig. 4.

Ligase-deficient Cul3Δ403–459 K712R double mutant blunts the effects of Cul3Δ403–459 on KLHL3 and WNK4. A: FLAG-tagged Cul3 constructs were cotransfected into HEK293 cells and immunoprecipitated using FLAG antibody. Immunoblotting for NEDD8 showed no neddylation of the K712R mutant for both WT Cul3 and Cul3Δ403–459. B: Coimmunoprecipitation was performed with HEK293 cells transfected with myc-KLHL3 and FLAG-tagged WT Cul3, Cul3Δ403–459, Cul3Δ403–459 K712R, or empty vector. Pull-down with FLAG antibodies showed that KLHL3 had more binding to Cul3Δ403–459 and Cul3Δ403–459 K712R proteins. C: Ubiquitin assay was performed for KLHL3 in HEK293 cells by cotransfecting FLAG-tagged Cul3 constructs with myc-KLHL3 and HA-tagged ubiquitin. Immunoprecipitation was performed using anti-myc antibody and polyubiquitylation of KLHL3 was visualized by immunoblotting for anti-HA. Cul3Δ403–459 K712R double mutant attenuated the higher abundance of KLHL3 ubiquitylation shown with Cul3Δ403–459. D: Top, abundance of myc-tagged KLHL3 and WNK4 protein was examined by immunoblot in Cul3 knockdown HEK293T (HEK293TCul3-KO) cells cotransfected with different FLAG-tagged Cul3 constructs. KLHL3 and WNK4 expression was higher and lower, respectively, in Cul3Δ403–459 K712R compared with Cul3Δ403–459. Bottom, quantitative analysis of KLHL3 and WNK4 protein abundance. Stain-free imaging was used as a loading control. Data represent individual values as well as means ± SE relative to control. Statistical differences were examined by one-way ANOVA with Tukey’s post hoc analysis. Cul3, cullin 3; HA, hemagglutinin; HEK, human embryonic kidney; IB, immunoblot; IP, immunoprecipitation; KLHL3, kelch-like 3; Ub, ubiquitin; WNK, with-no-lysine kinase; WT, wild type.
Ubiquitylation of the BTB-adaptor KLHL3 (Fig. 4C) was greater in cells transfected with Cul3Δ403–459, compared with WT Cul3, as reported previously (14). As expected, this greater KLHL3 ubiquitylation was not apparent when the neddylation deficient Cul3Δ403–459 K712R was transfected. Yet the current results also suggest that ubiquitin ligase activity is not fully responsible for the anomalous Cul3Δ403–459 activity. In cells that lack Cul3 (HEK293TCul3-KO) that were transfected with both KLHL3 and WNK4, the abundance of KLHL3 was lower, when Cul3Δ403–459 was present than with WT Cul3, consistent with increased substrate adaptor ubiquitylation and degradation by the mutant protein (Fig. 4D). Cul3Δ403–459 also led to more WNK4 abundance than did WT Cul3 (Fig. 4D), as we, and others, have reported previously. When the ligase-deficient Cul3Δ403–459 K712R construct was transfected; however, the effects on KLHL3 and WNK4 were reduced compared with Cul3Δ403–459 toward normal (WT Cul3) levels, (Fig. 4D). Unexpectedly, the effects of Cul3Δ403–459 on KLHL3 were not completely removed by the ligase-deficient Cul3Δ403–459 K712R double mutant, indicating that the effects of Cul3Δ403–459 is only partially dependent on ubiquitin ligase activity.
Cul3Δ403–459-mediated KLHL3 degradation is both proteasome and autophagy dependent.
As shown above, when ubiquitin ligase activity is lacking, as in the Cul3Δ403–459 K712R double mutant there is less abundance of KLHL3; however, the amount of KLHL3 abundance retained by the construct (ligase-independent degradation) is still significant. Because the neddylation-deficient construct should block the ubiquitin-proteasome degradation pathway, the remaining Cul3Δ403–459-mediated KLHL3 degradation could be autophagy-dependent. The proteasome is the canonical pathway for degrading ubiquitylated proteins, and it has been shown to contribute to WNK degradation (14); yet there is also evidence that KLHL3 and WNK4 can be degraded via autophagy (16). To determine the pathways involved in Cul3Δ403–459-mediated KLHL3 degradation, we used the inhibitors MG132 and chloroquine. Proteasomal inhibition with 10 μM MG132 had no effect on the control group but partially suppressed Cul3Δ403–459-mediated KLHL3 degradation (Fig. 5). Inhibition of autophagy with 100 µM chloroquine, however, had a small but significant effect on the control group, indicating that KLHL3 is constitutively degraded via autophagy (Fig. 6A). Incubation of Cul3Δ403–459 with chloroquine also resulted in a partial suppression of KLHL3 degradation. The percent change in KLHL3 protein abundance because of chloroquine administration was significantly different between control and Cul3Δ403–459 (Fig. 6B), which indicates an increased autophagic degradation of KLHL3 mediated by the Cul3 mutant. To confirm these results, we treated cells with another autophagy blocker, 3-methylandenine, an inhibitor of autophagosome formation (Fig. 6C). The results closely resembled chloroquine administration further indicating autophagic KLHL3 degradation. Treatment of the cells with both MG132 and chloroquine completely abolished Cul3Δ403–459-mediated KLHL3 degradation (Fig. 6D). The data suggest that, under the conditions provided, KLHL3 is degraded by Cul3Δ403–459 through both the ubiquitin-proteasome pathway and the autophagy pathway. Thus, WT Cul3 and Cul3Δ403–459 degrade WNK4 and KLHL3, respectively, via two different pathways. WNK4 is ubiquitylated and degraded through the proteasomal pathway. KLHL3 is degraded by both the proteasome and the autophagy pathway.
Fig. 5.

Effects of proteasome inhibition on Cul3Δ403–459-mediated KLHL3 degradation. The pathway for degradation of KLHL3 by the Cul3Δ403–459 mutant was examined by inhibiting the proteasomal pathway with the drug MG132. HEK293 cells were cotransfected with myc-KLHL3 and either no Cul3 or FLAG-Cul3Δ403–459. The cells were incubated with vehicle or 10 μM MG132 for 18 h before harvesting. Immunoblot analysis showed that inhibition of the proteasomal pathway partially blocked Cul3Δ403–459-mediated KLHL3 degradation. Right, quantitative analysis of KLHL3 protein abundance. GAPDH was used as a loading control. Data represent individual values as well as means ± SE relative to control. Statistical differences were examined by one-way ANOVA with Tukey’s post hoc analysis. Con, control; Cul3, cullin 3; HEK, human embryonic kidney; KLHL3, kelch-like 3; NS, not significant; Veh, vehicle.
Fig. 6.

Effects of autophagy inhibition on Cul3Δ403–459-mediated KLHL3 degradation. The pathway for degradation of KLHL3 by the Cul3Δ403–459 mutant was examined by inhibiting the autophagy pathway with the drugs chloroquine or 3-methyladenine (3-MA). HEK293 cells were cotransfected with myc-KLHL3 and either no Cul3 or FLAG-Cul3Δ403–459. The cells were incubated with vehicle or 100 μM chloroquine (A) or 5 mM 3-MA (C) for 18 h before harvesting. Immunoblot analysis showed that inhibition of autophagy with chloroquine or 3-MA partially blocked Cul3Δ403–459-mediated KLHL3 degradation, whereas administration of the drugs together completely eliminated KLHL3 degradation. Right, quantitative analysis of KLHL3 protein abundance. B: Bar graph depicting the percent change in KLHL3 protein abundance caused by autophagy inhibition from chloroquine administration between control and Cul3Δ403–459 groups. D: HEK293 cells were incubated with both the proteasomal inhibitor MG132 and autophagy inhibitor chloroquine, simultaneously. Administration of the drugs together completely eliminated KLHL3 degradation. GAPDH was used as a loading control. Data represent individual values as well as means ± SE relative to control. Statistical differences were examined by one-way ANOVA with Tukey’s post hoc analysis. Con, control; Cul3, cullin 3; HEK, human embryonic kidney; KLHL3, kelch-like 3; Veh, vehicle.
Increasing KLHL3 expression normalizes Cul3Δ403–459-mediated inhibition of WNK4 degradation in the presence of WT Cul3.
We suggested previously that the increased activity of Cul3Δ403–459 toward KLHL3 reduced the availability of KLHL3 to participate in degrading WNKs (14). Alternatively, others have suggested that the increased association of Cul3Δ403–459 with KLHL3 may sequester it and accomplish the same effect. If the low level of KLHL3 contributes to the increase in WNK4 in Cul3Δ403–459 patients, then increasing the amount of KLHL3 should reduce WNK4. By increasing the amount KLHL3 DNA transfected into the cells we were able to increase KLHL3 protein abundance. In HEK293 cells that lack Cul3 (HEK293TCul3-KO), an increase in KLHL3 protein levels caused only a slight reduction in WNK4 abundance (28%); WNK4 was still substantially higher than in cells transfected with WT Cul3, even though KLHL3 protein abundance was similar (compare the first and last three lanes in Fig. 7A). However, in HEK293 cells that contained endogenous WT Cul3 (Fig. 7B), increasing KLHL3 protein levels in cells transfected with Cul3Δ403–459 caused a striking reduction in WNK4 protein abundance (64%), to a value that was not significantly different from WT Cul3-transfected cells. The data show that Cul3Δ403–459 itself is unable to degrade WNK4 substantially, even when KLHL3 is normalized, but when Cul3Δ403–459 and Cul3 are both present in cells, as they are in heterozygous humans, the addition of KLHL3 normalizes degradation of WNK4. This suggests that in vitro Cul3Δ403–459 degrades KLHL3, preventing WT Cul3 from binding to WNK4.
Fig. 7.

Increased expression of KLHL3 can overcome effects of Cul3Δ403–459 on WNK4 in the presence of WT Cul3. HEK293TCul3-KO cells (A) or HEK293 cells (B) were transfected with myc-WNK4 and either FLAG-tagged WT Cul3 or Cul3Δ403–459 along with increasing amounts of myc-tagged KLHL3 and analyzed by immunoblot. The increased KLHL3 expression only slightly decreased WNK4 protein abundance in HEK293TCul3-KO cells, however, HEK293 cells had a larger decrease in WNK4 which was not significantly different from WT Cul3. Bar graphs depict quantification of KLHL3 and WNK4 protein abundance. Stain-free imaging was used as a loading control. Data represent relative individual values as well as means ± SE. Statistical differences were examined by one-way ANOVA with Tukey’s post hoc analysis. Cul3, cullin 3; HEK, human embryonic kidney; KLHL3, kelch-like 3; WNK, with-no-lysine kinase; WT, wild type.
Cul3Δ403–459 exhibits a dominant effect in cells.
It has been suggested that Cul3Δ403–459 causes FHHt by inducing functional Cul3 haploinsufficiency (10, 24). This suggestion derives from the observation that the abundance of Cul3Δ403–459 is very low in a knock-in mouse model of FHHt and also that introducing a 1:1 molar ratio of WT Cul3 to Cul3Δ403–459 did not inhibit ubiquitylation of WNK4 (24). Yet Uchida and colleagues (2) noted that mice with functional Cul3 haploinsufficiency do not exhibit signs of FHHt, and as noted above, Cul3Δ403–459 may either degrade and/or sequester KLHL3, thereby exerting a dominant negative effect (10, 14). To determine whether Cul3Δ403–459 has dominant effects in cells, we transfected both WT Cul3 and Cul3Δ403–459 together at different ratios. In the presence of constant WT Cul3, increasing Cul3Δ403–459 reduced KLHL3 abundance and increased WNK4 abundance (Fig. 8), and increasing WT Cul3 in the presence of Cul3Δ403–459 increased KLHL3 and decreased WNK4. Thus, Cul3Δ403–459 clearly exerts a dominant effect in cultured cells; this is consistent with the autosomal dominant inheritance of FHHt type 4.
Fig. 8.

WT Cul3 and Cul3Δ403–459 compete for KLHL3. Myc-tagged KLHL3 and WNK4 were co-transfected with different amounts of FLAG-tagged WT Cul3 and Cul3Δ403–459 into HEK293 cells. The ratio of FLAG-WT Cul3 to Cul3Δ403–459 was adjusted as labeled and analyzed by immunoblot. β-actin was used as a loading control. Increasing the ratio of Cul3Δ403–459 to WT Cul3 decreased KLHL3 and increased WNK4 protein expression. The opposite was observed when increasing the ratio of WT Cul3 to Cul3Δ403–459. Bar graphs are a summary of the densitometry analysis of the blot. Cul3, cullin 3; KLHL3, kelch-like 3; WNK, with-no-lysine kinase; WT, wild type.
Cul3Δ403–459 does not affect Keap1 and cyclin E protein abundance.
Because Cul3 can interact with and ubiquitinate multiple substrates through many different substrate adaptors, then it would be expected that the Cul3Δ403–459 mutant would affect other proteins besides KLHL3 and WNK4. To understand the effects of the Cul3Δ403–459 mutant better, we examined three other proteins that interact with Cul3. The oxidative stress response protein, nuclear factor erythroid 2-related factor 2 (Nrf2), is another substrate of Cul3 and interacts through the BTB-Kelch substrate adaptor protein kelch-like ECH-associated protein-1 (Keap1). Unlike KLHL3, Keap1 showed no change in protein abundance when HEK293TCul3-KO cells were transfected with Cul3Δ403–459 or Cul3Δ403–459 K712R (Fig. 9). Although Keap1 was unchanged, its substrate Nrf2 showed an increase in protein abundance in Cul3Δ403–459-transfected cells. The amplified Nrf2 was also observed in cells transfected with Cul3Δ403–459 K712R. Cyclin E, which is a canonical Cul3 substrate and is involved in cell cycle regulation, was unchanged when transfected with Cul3Δ403–459 or Cul3Δ403–459 K712R. The results demonstrate that the Cul3Δ403–459 mutant has differential effects on its substrates and substrate adaptors.
Fig. 9.

Effects of Cul3Δ403–459 on Keap1, Nrf2, and cyclin E. Top, abundance of endogenous Keap1, Nrf2, and cyclin E protein was examined in HEK293TCul3-KO cells cotransfected with different FLAG-tagged Cul3 constructs and analyzed by immunoblot. Keap1 and cyclin E showed no difference in protein abundance between the groups. Nrf2 protein levels were higher in Cul3Δ403–459 and Cul3Δ403–459 K712R transfected cells. Stain-free imaging was used as a loading control. Bottom, quantitative analysis of Keap1, Nrf2, and cyclin E protein abundance. Data represent relative individual values as well as means ± SE. Statistical differences were examined by one-way ANOVA with Tukey’s post hoc analysis. Cul3, cullin 3; HEK, human embryonic kidney; Keap1, kelch-like ECH-associated protein-1; Nrf2, nuclear factor erythroid 2-related factor 2.
DISCUSSION
Mutations in WNK1, WNK4, CUL3, and KLHL3 cause FHHt, predominantly by increasing NaCl cotransporter activity along the distal convoluted tubule, suggesting that these proteins comprise a single signaling pathway. The disease pathogenesis, in all cases, appears to result from an increase in WNK abundance, either owing to enhanced transcription or impaired degradation. Whereas WNK4 and KLHL3 mutations impair the degradative arm of this pathway by disrupting the ability of WNKs to form complexes with KLHL3 and Cul3, the mechanisms involved in CUL3 disease have been more difficult to unravel (17, 18, 25, 28, 29, 31, 34). We reported previously that the FHHt-mutant Cul3 ubiquitylates and facilitates KLHL3 degradation more actively than does WT Cul3 and suggested that the mutant exhibits dominant effects (14). The dominant nature of the Cul3Δ403–459 mutation was very recently confirmed, using mouse models (9). Here, we determined that Cul3Δ403–459 has an altered structure that decreases interaction with the CSN. Yet the results also discern a novel secondary, ligase-independent autophagocytic KLHL3 degradation pathway that appears essential for the autosomal dominant phenotype.
The current results confirm our prior work (14), indicating that that Cul3Δ403–459 is hyperneddylated when expressed in cells. Ibeawuchi and colleagues (10), in contrast, could not detect hyperneddylation of Cul3Δ403–459 and suggested that the mutant protein is neddylated less efficiently than WT. A potential resolution to this paradox is apparent from the work of Schumacher and colleagues (24), who also documented that Cul3Δ403–459 is hyperneddylated in cells but found that the neddylation process itself was less efficient. They demonstrated that the defect lies in a failure of the CSN to associate with Cul3, and therefore, a failure of deneddylation. The current work confirms that Cul3Δ403–459 does not associate normally with the CSN (in this case, the catalytically-active JAB1 subunit), but this effect is not because the deleted amino acid sequence actually binds to JAB1. FHHt-causing Cul3 mutations lead to deletion of exon 9, which encodes the 4HB domain (Fig. 1A). Min et al. (15) examined the CSN-binding domain of a homologous cullin, cullin 1, and suggested that it lies within the 4HB domain. To determine whether the same domain is relevant in Cul3 binding to CSN, we mapped the Cul3 domains that are required for association with JAB1. Using multiple Cul3 constructs, we determined that the 4HB domain is neither necessary nor sufficient for CSN binding. Instead, the α/β1 domain, which lies adjacent to the 4HB domain, was identified as essential for association with the CSN. Because the α/β1 domain is not directly altered by the Cul3Δ403–459 mutation, the results suggest that the mutation disrupts binding to the CSN through alterations in the protein folding of Cul3; this suggestion aligns with the structural modeling data of Schumacher and colleagues (24).
Because Cul3Δ403–459 does not bind efficiently to JAB1 protein we tested whether JAB1 knockdown could mimic the effects of Cul3Δ403–459 in cultured cells. Knockdown of JAB1 with siRNA decreased, rather than increased, WNK4 protein abundance indicating increased CRL activity (Fig. 2). This suggests that JAB1 knockdown alone cannot mimic Cul3Δ403–459 effects on WNK4; it should be noted, however, that CRL activity can differ between cell culture models and in vivo. The CSN positively regulates CRLs in vivo (22), whereas in vitro CRLs are negatively regulated by the CSN (36). Furthermore, in vivo, experiments are needed to understand the effects of JAB1 inhibition fully.
The Cul3Δ403–459 K712R mutant prevented NEDD8 conjugation, effectively inhibiting ubiquitin ligase activity. However, Cul3Δ403–459 K712R unexpectedly still showed significant degradation of KLHL3 (Fig. 4D), suggesting that degradation might be because of a nonligase effect of the mutant Cul3Δ403–459. The results suggest that this ligase-independent degradation of KLHL3 occurs through the autophagy pathway, as two different inhibitors of this pathway, chloroquine, and 3-methyladenine, both prevented KLHL3 degradation. Thus, as the schematic depicts in Fig. 10, Cul3Δ403–459 facilitates KLHL3 degradation through two different pathways. KLHL3 is ubiquitylated and degraded via the proteasome. Additionally, KLHL3 can be degraded via selective autophagy, which is stimulated by the Cul3 mutant in a ligase-independent manner. Furthermore, the results here suggest that degradation of KLHL3 contributes importantly to WNK4 accumulation in FHHt. As shown, in the absence of WT Cul3 (in HEK293TCul3-KO cells), Cul3Δ403–459 cannot degrade WNK4 even when KLHL3 is abundant (Fig. 7A). In cells that simultaneously express WT Cul3 with Cul3Δ403–459, however, KLHL3 abundance proves limiting for WNK4 degradation (Fig. 7B), and thus it is the ability of the mutant Cul3 to drive KLHL3 degradation that proves essential. These observations provide substantial insight into the mechanisms of the disease, which is characterized by the presence of one WT and one mutant CUL3 allele. In this case, the protein generated from Cul3Δ403–459 cannot degrade WNK4, creating functional haploinsufficiency, as suggested. Yet the ability of the WT Cul3 protein to facilitate WNK degradation is limited by the low abundance of KLHL3; this is maintained by enhanced proteasomal- and autophagy-driven KLHL3 degradation (Fig. 10).
Fig. 10.

Simplified model of Cul3Δ403–459 effects on KLHL3 and WNK4. KLHL3 is degraded by two separate pathways. Under normal conditions, the WT Cul3-KLHL3 ubiquitin ligase complex (left) ubiquitylates WNK4 targeting it for degradation via the proteasome. Separate from cullin-RING-ligase activity, KLHL3 is also degraded through selective autophagy. The Cul3Δ403–459 FHHt mutant (right) targets KLHL3 instead of WNK4 for ubiquitylation; causing proteasomal degradation of KLHL3 while preventing WNK4 turnover. Additionally, expression of the Cul3 mutant causes enhanced autophagic-mediated degradation of KLHL3. The lower levels of KLHL3 through both proteasomal and autophagic degradation prevent WT Cul3 from interacting with WNK4, leading to an increase in WNK4 protein abundance. Cul3, cullin 3; FHHt, familial hyperkalemic hypertension; KLHL3, kelch-like 3; WNK, with-no-lysine kinase.
The CUL3 mutation is contained within a protein that is a part of the ubiquitin-proteasome system (UPS), yet Cul3Δ403–459 causes an increase in autophagic degradation of KLHL3 (Fig. 6B). The reason for this could be because of the relationship between the two degradative pathways. The UPS and autophagy were once thought to be separate mechanisms for regulated protein turnover; however, recent work has demonstrated that the two pathways may not be independent of one another. First, inhibition of the proteasome causes an increase in autophagy (8). This is most likely due to the fact that many proteins involved in the autophagy pathway are substrates for E3 ubiquitin ligases, including CRLs, and are negatively regulated via the UPS (7). Additionally, ubiquitylated proteins can be shuttled to the autophagophore (the vesicle that ultimately binds to the lysosome) via a linker protein, such as p62 (11). These proteins connect the two pathways by binding to both ubiquitylated proteins and autophagophore-membrane proteins allowing for ubiquitylated proteins to be degraded via autophagy. p62 binds to KLHL3 and mediates its degradation via autophagy when the proteasomal pathway is inhibited (16). Thus, the Cul3 mutation could lead to upregulation of p62-mediated autophagic degradation of KLHL3; moreover, the impairment of CRL substrate degradation, as shown with the Cul3 mutant, could cause activation of autophagy because of accumulation of CRL substrates that are critical for the process.
CRLs can associate with hundreds of substrate adaptors that can target thousands of substrates. Global deletion of the Cul3 gene is embryonic lethal (26). So, the fact that Cul3Δ403–459 doesn’t produce widespread phenotypic effects has been perplexing. Here, we suggest that this paradox is resolved by dual effects of the Cul3Δ403–459 mutant protein, loss of function with respect to WNK4 degradation, and gain of function with respect to autophagocytic degradation of KLHL3. The latter effect likely contributes to the apparent tissue specificity for the disease to disrupt kidney and vascular smooth muscle. Although Cul3Δ403–459 avidly binds to and degrades KLHL3, its effects on other adaptor proteins are different. The substrate adaptors Bacurd1 and RhoBTB1 similarly showed higher levels of interaction with Cul3Δ403–459 compared with WT Cul3 (10). Yet, Cul3Δ403–459 degraded RhoBTB1 less efficiently compared with WT Cul3, and Bacurd1 showed no change in abundance. Here, Keap1, also known as Kelch-like 19, like Bacurd1, did not show a change in protein abundance because of Cul3Δ403–459 (Fig. 9). Although Cul3Δ403–459 has differential effects on these substrate adaptors, the effects of Cul3Δ403–459 on their respective substrates were similar. Analogous to WNK4, the Bacurd1 substrate RhoA, which is expressed in the vascular smooth muscle and important for arterial pressure regulation, was upregulated by Cul3Δ403–459 (1). Additionally, as shown above, the Keap1 substrate and oxidative stress response protein, Nrf2, showed increased protein abundance (Fig. 9). On the other hand, cyclin E, a protein involved in cell cycle regulation and a substrate of Cul3, was unaffected by Cul3Δ403–459. The data demonstrate that the altered structure of Cul3Δ403–459 may be sequestering adaptors in a manner that prevents normal ubiquitin ligase activity toward the substrate; yet KLHL3 may be one of only a few adaptors that undergoes active degradation, providing specificity for tissues and cell types in which this protein is highly expressed.
All patients with FHHt reported to date who harbor mutations in CUL3 are heterozygous (4). Some have suggested that the Cul3Δ403–459 protein is unstable, leading to functional haploinsufficiency of the WT protein; according to this model, individuals with one functional CUL3 allele should exhibit the phenotype (1, 24). Yet, Uchida and colleagues generated a mouse model that lacked one Cul3 allele, expressing approximately half as much Cul3 protein as control mice; the mice, however, did not show any evidence of the FHHt phenotype (2). Furthermore, Ferdaus and colleagues (9) recently showed that mice with one Cul3 allele also lack features of FHHt, whereas mice with one mutant and one WT allele exhibit frank hyperkalemic hypertension. Although the protein derived from Cul3Δ403–459 does appear to be unstable in vivo (1, 24), the current results suggest that the FHHt phenotype requires a second, dominant-negative effect. In support of this, we found clear evidence for a dominant effect of Cul3Δ403–459 on Cul3 WT, when the ratio of WT to mutant construct was varied (Fig. 8). This is largely consistent with the model suggested by Sigmund and colleagues (10), who found that Cul3Δ403–459 exhibited enhanced interaction with substrate adaptors, such as KLHL3, and suggested that the mutant cullin might act in a dominant manner despite reduced abundance by sequestering adaptor proteins.
The results here show only a modest change in WNK4 when WT Cul3 and Cul3Δ403–459 are transfected together and increasing the ratio of WT Cul3 to Cul3Δ403–459 further diminished the effects on WNK4 abundance. This raises questions about the direct relevance of this to the human disease as all patients with FHHt are heterozygous, with one WT and one mutant allele. Additionally, in experimental disease models, the abundance of Cul3Δ403–459 is very low, relative to the WT allele. Yet, Ferdaus and colleagues (9) showed recently that Cul3Δ403–459 exerts dominant effects in vivo despite its low abundance. Thus, these limitations of using HEK293 cells imply that these hypotheses should be explored using physiologically more relevant model systems.
Thus, the current results clarify the consequences of deletion of exon 9 in Cul3 and provide novel information about how Cul3 interacts with the CSN. They suggest that this interaction requires the α/β1 domain, which lies next to, but is distinct from, the 4HB domain deleted in the human disease. Thus, although the deletion impairs binding of Cul3 to the CSN, the deleted region itself does not mediate the association. Furthermore, the results stress the importance of KLHL3 in the regulation of WNK4. Our data would be consistent with an effect of the mutant Cul3 protein to bind avidly to specific substrate adaptors, forming unstable and ineffective complexes. Additional studies will be required to evaluate this hypothesis further.
GRANTS
This work was supported by NIH grants R01-DK-51496 (to D. Ellison and C. Yang) and T32-DK-067864 (to D. H. Ellison), as well as by Merit Review Grant no. 1I01BX002228–01A1 from the Department of Veterans Affairs (to D. Ellison), and by American Heart Association grant no. 16POST3064003 and NIH grant no. F32-DK-112531 (to R. Cornelius). C. Zhang was supported by the National Natural Science Foundation of China grant. nos. 81570634 and 81770706. C. Sigmund was supported by NIH grant no. R01-HL-125603.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.J.C., C.Z., J.D.S., C.-L.Y., and D.H.E. conceived and designed research; R.J.C., C.Z., and K.J.E. performed experiments; R.J.C., C.Z., K.J.E., J.D.S., C.-L.Y., and D.H.E. analyzed data; R.J.C., C.Z., K.J.E., J.D.S., C.-L.Y., and D.H.E. interpreted results of experiments; R.J.C. prepared figures; R.J.C. and D.H.E. drafted manuscript; R.J.C., C.Z., K.J.E., L.N.A., C.D.S., J.D.S., C.-L.Y., and D.H.E. edited and revised manuscript; R.J.C., C.Z., K.J.E., L.N.A., C.D.S., J.D.S., C.-L.Y., and D.H.E. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank James McCormick for helpful discussions.
REFERENCES
- 1.Agbor LN, Ibeawuchi S, Hu C, Wu J, Davis DR, Keen HL, Quelle FW, Sigmund CD. Cullin-3 mutation causes arterial stiffness and hypertension through a vascular smooth muscle mechanism. JCI Insight 1: e91015, 2016. doi: 10.1172/jci.insight.91015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Araki Y, Rai T, Sohara E, Mori T, Inoue Y, Isobe K, Kikuchi E, Ohta A, Sasaki S, Uchida S. Generation and analysis of knock-in mice carrying pseudohypoaldosteronism type II-causing mutations in the cullin 3 gene. Biol Open 4: 1509–1517, 2015. doi: 10.1242/bio.013276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boh BK, Smith PG, Hagen T. Neddylation-induced conformational control regulates cullin RING ligase activity in vivo. J Mol Biol 409: 136–145, 2011. doi: 10.1016/j.jmb.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 4.Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, Tikhonova IR, Bjornson R, Mane SM, Colussi G, Lebel M, Gordon RD, Semmekrot BA, Poujol A, Välimäki MJ, De Ferrari ME, Sanjad SA, Gutkin M, Karet FE, Tucci JR, Stockigt JR, Keppler-Noreuil KM, Porter CC, Anand SK, Whiteford ML, Davis ID, Dewar SB, Bettinelli A, Fadrowski JJ, Belsha CW, Hunley TE, Nelson RD, Trachtman H, Cole TRP, Pinsk M, Bockenhauer D, Shenoy M, Vaidyanathan P, Foreman JW, Rasoulpour M, Thameem F, Al-Shahrouri HZ, Radhakrishnan J, Gharavi AG, Goilav B, Lifton RP. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 482: 98–102, 2012. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cavadini S, Fischer ES, Bunker RD, Potenza A, Lingaraju GM, Goldie KN, Mohamed WI, Faty M, Petzold G, Beckwith REJ, Tichkule RB, Hassiepen U, Abdulrahman W, Pantelic RS, Matsumoto S, Sugasawa K, Stahlberg H, Thomä NH. Cullin-RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature 531: 598–603, 2016. doi: 10.1038/nature17416. [DOI] [PubMed] [Google Scholar]
- 6.Chung D, Dellaire G. The role of the COP9 signalosome and neddylation in DNA damage signaling and repair. Biomolecules 5: 2388–2416, 2015. doi: 10.3390/biom5042388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cui D, Xiong X, Zhao Y. Cullin-RING ligases in regulation of autophagy. Cell Div 11: 8, 2016. doi: 10.1186/s13008-016-0022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding W-X, Ni H-M, Gao W, Yoshimori T, Stolz DB, Ron D, Yin X-M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 171: 513–524, 2007. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferdaus MZ, Miller LN, Agbor LN, Saritas T, Singer JD, Sigmund CD, McCormick JA. Mutant Cullin 3 causes familial hyperkalemic hypertension via dominant effects. JCI Insight: 2: e96700, 2017. doi: 10.1172/jci.insight.96700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ibeawuchi SR, Agbor LN, Quelle FW, Sigmund CD. Hypertension-causing mutations in Cullin3 protein impair RhoA protein ubiquitination and augment the association with substrate adaptors. J Biol Chem 290: 19208–19217, 2015. doi: 10.1074/jbc.M115.645358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 584: 1393–1398, 2010. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 12.Lingaraju GM, Bunker RD, Cavadini S, Hess D, Hassiepen U, Renatus M, Fischer ES, Thomä NH. Crystal structure of the human COP9 signalosome. Nature 512: 161–165, 2014. doi: 10.1038/nature13566. [DOI] [PubMed] [Google Scholar]
- 13.Mayan H, Vered I, Mouallem M, Tzadok-Witkon M, Pauzner R, Farfel Z. Pseudohypoaldosteronism type II: marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. J Clin Endocrinol Metab 87: 3248–3254, 2002. doi: 10.1210/jcem.87.7.8449. [DOI] [PubMed] [Google Scholar]
- 14.McCormick JA, Yang C-L, Zhang C, Davidge B, Blankenstein KI, Terker AS, Yarbrough B, Meermeier NP, Park HJ, McCully B, West M, Borschewski A, Himmerkus N, Bleich M, Bachmann S, Mutig K, Argaiz ER, Gamba G, Singer JD, Ellison DH. Hyperkalemic hypertension-associated cullin 3 promotes WNK signaling by degrading KLHL3. J Clin Invest 124: 4723–4736, 2014. doi: 10.1172/JCI76126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Min KW, Kwon MJ, Park HS, Park Y, Yoon SK, Yoon JB. CAND1 enhances deneddylation of CUL1 by COP9 signalosome. Biochem Biophys Res Commun 334: 867–874, 2005. doi: 10.1016/j.bbrc.2005.06.188. [DOI] [PubMed] [Google Scholar]
- 16.Mori Y, Mori T, Wakabayashi M, Yoshizaki Y, Zeniya M, Sohara E, Rai T, Uchida S. Involvement of selective autophagy mediated by p62/SQSTM1 in KLHL3-dependent WNK4 degradation. Biochem J 472: 33–41, 2015. doi: 10.1042/BJ20150500. [DOI] [PubMed] [Google Scholar]
- 17.Mori Y, Wakabayashi M, Mori T, Araki Y, Sohara E, Rai T, Sasaki S, Uchida S. Decrease of WNK4 ubiquitination by disease-causing mutations of KLHL3 through different molecular mechanisms. Biochem Biophys Res Commun 439: 30–34, 2013. doi: 10.1016/j.bbrc.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 18.Ohta A, Schumacher F-R, Mehellou Y, Johnson C, Knebel A, Macartney TJ, Wood NT, Alessi DR, Kurz T. The CUL3-KLHL3 E3 ligase complex mutated in Gordon’s hypertension syndrome interacts with and ubiquitylates WNK isoforms: disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem J 451: 111–122, 2013. doi: 10.1042/BJ20121903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pacheco-Alvarez D, Cristóbal PS, Meade P, Moreno E, Vazquez N, Muñoz E, Díaz A, Juárez ME, Giménez I, Gamba G. The Na+:Cl- cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem 281: 28755–28763, 2006. doi: 10.1074/jbc.M603773200. [DOI] [PubMed] [Google Scholar]
- 20.Pan Z-Q, Kentsis A, Dias DC, Yamoah K, Wu K. Nedd8 on cullin: building an expressway to protein destruction. Oncogene 23: 1985–1997, 2004. doi: 10.1038/sj.onc.1207414. [DOI] [PubMed] [Google Scholar]
- 21.Pathare G, Hoenderop JGJ, Bindels RJM, San-Cristobal P. A molecular update on pseudohypoaldosteronism type II. Am J Physiol Renal Physiol 305: F1513–F1520, 2013. doi: 10.1152/ajprenal.00440.2013. [DOI] [PubMed] [Google Scholar]
- 22.Pintard L, Kurz T, Glaser S, Willis JH, Peter M, Bowerman B. Neddylation and deneddylation of CUL-3 is required to target MEI-1/Katanin for degradation at the meiosis-to-mitosis transition in C. elegans. Curr Biol 13: 911–921, 2003. doi: 10.1016/S0960-9822(03)00336-1. [DOI] [PubMed] [Google Scholar]
- 23.Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol Cell 32: 21–31, 2008. doi: 10.1016/j.molcel.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schumacher F-R, Siew K, Zhang J, Johnson C, Wood N, Cleary SE, Al Maskari RS, Ferryman JT, Hardege I, Yasmin, Figg NL, Enchev R, Knebel A, O’Shaughnessy KM, Kurz T. Characterisation of the Cullin-3 mutation that causes a severe form of familial hypertension and hyperkalaemia. EMBO Mol Med 7: 1285–1306, 2015. doi: 10.15252/emmm.201505444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shibata S, Zhang J, Puthumana J, Stone KL, Lifton RP. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc Natl Acad Sci USA 110: 7838–7843, 2013. doi: 10.1073/pnas.1304592110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singer JD, Gurian-West M, Clurman B, Roberts JM. Cullin-3 targets cyclin E for ubiquitination and controls S phase in mammalian cells. Genes Dev 13: 2375–2387, 1999. doi: 10.1101/gad.13.18.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sohara E, Uchida S. Kelch-like 3/Cullin 3 ubiquitin ligase complex and WNK signaling in salt-sensitive hypertension and electrolyte disorder. Nephrol Dial Transplant 31: 1417–1424, 2016. doi: 10.1093/ndt/gfv259. [DOI] [PubMed] [Google Scholar]
- 28.Susa K, Sohara E, Rai T, Zeniya M, Mori Y, Mori T, Chiga M, Nomura N, Nishida H, Takahashi D, Isobe K, Inoue Y, Takeishi K, Takeda N, Sasaki S, Uchida S. Impaired degradation of WNK1 and WNK4 kinases causes PHAII in mutant KLHL3 knock-in mice. Hum Mol Genet 23: 5052–5060, 2014. doi: 10.1093/hmg/ddu217. [DOI] [PubMed] [Google Scholar]
- 29.Vidal-Petiot E, Elvira-Matelot E, Mutig K, Soukaseum C, Baudrie V, Wu S, Cheval L, Huc E, Cambillau M, Bachmann S, Doucet A, Jeunemaitre X, Hadchouel J. WNK1-related familial hyperkalemic hypertension results from an increased expression of L-WNK1 specifically in the distal nephron. Proc Natl Acad Sci USA 110: 14366–14371, 2013. doi: 10.1073/pnas.1304230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J 391: 17–24, 2005. doi: 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wakabayashi M, Mori T, Isobe K, Sohara E, Susa K, Araki Y, Chiga M, Kikuchi E, Nomura N, Mori Y, Matsuo H, Murata T, Nomura S, Asano T, Kawaguchi H, Nonoyama S, Rai T, Sasaki S, Uchida S. Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Reports 3: 858–868, 2013. doi: 10.1016/j.celrep.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 32.Wilson FH, Disse-Nicodème S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP. Human hypertension caused by mutations in WNK kinases. Science 293: 1107–1112, 2001. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 33.Wimuttisuk W, Singer JD. The Cullin3 ubiquitin ligase functions as a Nedd8-bound heterodimer. Mol Biol Cell 18: 899–909, 2007. doi: 10.1091/mbc.e06-06-0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu G, Peng J-B. Disease-causing mutations in KLHL3 impair its effect on WNK4 degradation. FEBS Lett 587: 1717–1722, 2013. doi: 10.1016/j.febslet.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu J-T, Lin H-C, Hu Y-C, Chien C-T. Neddylation and deneddylation regulate Cul1 and Cul3 protein accumulation. Nat Cell Biol 7: 1014–1020, 2005. doi: 10.1038/ncb1301. [DOI] [PubMed] [Google Scholar]
- 36.Yang X, Menon S, Lykke-Andersen K, Tsuge T, Di Xiao, Wang X, Rodriguez-Suarez RJ, Zhang H, Wei N. The COP9 signalosome inhibits p27(kip1) degradation and impedes G1-S phase progression via deneddylation of SCF Cul1. Curr Biol 12: 667–672, 2002. doi: 10.1016/S0960-9822(02)00791-1. [DOI] [PubMed] [Google Scholar]
- 37.Yewdell JW, Lacsina JR, Rechsteiner MC, Nicchitta CV. Out with the old, in with the new? Comparing methods for measuring protein degradation. Cell Biol Int 35: 457–462, 2011. doi: 10.1042/CBI20110055. [DOI] [PMC free article] [PubMed] [Google Scholar]