

Abstract
Our laboratory previously reported that agonists of the 5-hydoxytryptamine 1F (5-HT1F) receptor induce renal mitochondrial biogenesis (MB) and that stimulation of the 5-HT1F receptor following ischemia/reperfusion (I/R)-induced acute kidney injury (AKI) accelerated the recovery of renal function in mice. The goal of this study was to examine the contribution of the 5-HT1F receptor in the regulation of renal mitochondrial homeostasis and renal function in naïve and injured mice. Although 5-HT1F receptor knockout (KO) mice were healthy and fertile, and did not exhibit renal dysfunction, renal mitochondrial DNA copy number and mitochondrial fission gene expression increased at 10 wk of age. The 5-HT1F receptor KO mice exhibited greater proximal tubular injury and diminished renal recovery after I/R-induced AKI compared with wild-type mice. These findings were associated with persistent suppression of renal cortical MB and ATP levels after injury. In summary, the 5-HT1F receptor is a component of physiological MB regulation in the kidney, and its absence potentiates renal injury and impedes recovery.
Keywords: acute kidney injury, gene expression, mitochondria, serotonin
INTRODUCTION
Mitochondrial dysfunction is a hallmark pathophysiological mediator of many acute and chronic diseases. As such, loss of ATP-dependent cellular functions and increased reactive oxygen species propagate injury and subsequent tissue and organ dysfunction (16, 27). For example, our laboratory demonstrated that persistent mitochondrial dysfunction and suppression of MB corresponds to worsened renal function after ischemia/reperfusion (I/R)- and folic acid-induced acute kidney injury (AKI; 11, 15, 28).
Mitochondrial biogenesis (MB) has received increasing attention as a therapeutic target to promote restoration of normal cellular and organ function, and several studies have reported pharmacological induction of MB as a treatment for acute organ injury and cardiovascular disease (7, 13, 21, 24). MB is defined as the dynamic process of generating new, functional mitochondria and is a highly regulated process controlled by the central mediator and transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) (23). Several groups have developed strategies designed to increase the expression and activity of PGC-1α and subsequent induction of MB, as evidenced by increased mitochondrial number and function (2, 3, 25, 27). For example, we reported that a 5-hydroxytryptamine 1F (5-HT1F) receptor agonist LY344864 induced MB in vitro and in vivo, restored MB, and accelerated the recovery of renal function following I/R-induced AKI in mice (11). Furthermore, knockdown of the 5-HT1F receptor in renal proximal tubular cells resulted in decreased mitochondrial proteins, suggesting that the 5-HT1F receptor may be regulating MB under basal conditions. These findings implicate the 5-HT1F receptor as a potential regulator of MB in the kidney.
Based on these findings, we utilized a recently developed 5-HT1F receptor knockout (KO) mouse to understand further the contribution of the 5-HT1F receptor in the regulation of renal mitochondrial homeostasis. In addition, we tested the hypothesis that the absence of the 5-HT1F receptor potentiates AKI in mice.
METHODS
I/R-induced AKI model.
Male and female 5-HT1F receptor KO mice (B6N(Cg)-Htr1ftm1.1(KOMP)Vlcg/J, stock no. 024269) and age-matched wild-type (WT) female and male C57BL/6NJ (WT, stock no. 005304) were purchased from the Jackson Laboratory (Bar Harbor, ME). A 5-HT1F receptor KO breeding colony was generated from heterozygous mutants and were housed in temperature-controlled conditions under a light/dark photocycle with unrestricted food and water supplied ad libitum. Male and female mice were weighed periodically. Changes in 5-HT1F KO mice were evaluated at 10 wk of age.
Ten-week-old male WT and KO mice underwent I/R surgery using bilateral renal pedicle clamping for 18 min as described previously (10). Briefly, the renal artery and vein were isolated, and blood flow was occluded with a vascular clamp for 18 min while body temperature was maintained at 36°C–37°C. Sham mice were treated the same as I/R mice without clamping of the renal pedicles. Mice were euthanized 24 h or 144 h after surgery, and blood and kidneys (flash-frozen in liquid nitrogen) were collected for analysis. Studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All protocols were approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina and the University of Arizona, and all efforts were made to minimize animal suffering.
Agarose gel electrophoresis.
Genomic DNA was isolated from mice tail clippings using Viagen Direct PCR DNA Lysis Reagent (Los Angeles, CA) containing 0.4 mg/ml proteinase K. PCR amplification was performed using Promega PCR Master Mix (Madison, WI). PCR products were separated on a 2% agarose gel in 1× TBE (89 mM Tris, 89 mM boric acid, 2 mM EDTA) and 0.5 µg/ml ethidium bromide. Gels were visualized using the GE ImageQuant LAS-4000 (GE Life Sciences, Pittsburgh, PA). Primer pairs annealing with the WT allele and the mutant allele were obtained from Jackson Laboratory and are listed in Table 1.
Table 1.
Mouse and human primer pairs used for qRT-PCR
| Gene | Primer Sequence | Accession No. |
|---|---|---|
| Mouse | ||
| PGC-1α | Sense 5′-AGGAAATCCGAGCGGAGCTGA-3′ | NM_008904.2 |
| Antisense 5′-GCAAGAAGGCGACACATCGAA-3′ | ||
| NDUFS1 | Sense 5′-AGATGATTTGGGAACAACAG-3′ | NM_001160038.1 |
| Antisense 5′-TAAGGCTTAGAGGTTAGGGC-3′ | ||
| ATP Synthase β | Sense 5′-CTATGTGCCTGCTGATGACC-3′ | NM_016774.3 |
| Antisense 5′-GGATAGATGCCCAACTCAGC-3′ | ||
| COXI | Sense 5′-ACCATCATTTCTCCTTCTCC-3′ | ENSMUSG00000064351.1 |
| Antisense 5′-GGTGGGTAGACTGTTCATCC-3′ | ||
| ND1 | Sense 5′-TAGAACGCAAAATCTTAGGG-3′ | ENSMUSG00000064341.1 |
| Antisense 5′-TGCTAGTGTGAGTGATAGGG-3′ | ||
| NRF1 | Sense 5′-TCGGGCATTTATCCCAGAGATGCT-3′ | NM_001164226.1 |
| Antisense 5′-TACGAGATGAGCTATACTGTGTGT-3′ | ||
| NRF2 | Sense 5′-CCTCTGTCACCAGCTCAAGG-3′ | NM_010902.4 |
| Antisense 5′-TTCTGGGCGGCGACTTTATT-3′ | ||
| TFAM | Sense 5′-GCTGATGGGTATGGAGAAG-3′ | NM_009360.4 |
| Antisense 5′-GAGCCGAATCATCCTTTGC-3′ | ||
| DRP1 | Sense 5′-GCCTCAGATCGTCGTAGTGG-3′ | NM_001276340.1 |
| Antisense 5′-TCTGCTTCAACTCTCCAGCTC-3′ | ||
| Pink1 | Sense 5′-TTGCAATGCCGCTGTGTATG-3′ | NM_026880.2 |
| Antisense 5′-TGGAGGAACCTGCCGAGATA-3′ | ||
| SOD2 | Sense 5′-CAAGGGAGATGTTACAACTCAGG-3′ | NM_013671.3 |
| Antisense 5′-CTTAGGGCTCAGGTTTGTCCA-3′ | ||
| UCP2 | Sense 5′-GAGATACCAGAGCACTGTCG-3′ | NM_011671.5 |
| Antisense 5′-GTCATCTGTCATGAGGTTGG-3′ | ||
| 5-HT1B | Sense 5′-AGACAGGGGTACCTCTCACCAACC-3′ | NM_010482.1 |
| Antisense 5′-ATGAGCGCCAACAAAGCAACCAGC-3′ | ||
| 5-HT1D | Sense 5′-AAACCAGTCCCTAGAAGGCCTTCC-3′ | NM_008309.5 |
| Antisense 5′-GCCAGTGTGATGACGGACAGCAC-3′ | ||
| 5-HT1F | Sense 5′-GCCGTGATGATGAGTGTGTC-3′ | NM_008310.3 |
| Antisense 5′-ATCATCCGACTCGCTTGTCT-3′ | ||
| 5-HT2A | Sense 5′-GCTGAGCCGACAGCTAATGA-3′ | NM_172812.2 |
| Antisense 5′-CATCCGGTCCATCACACACA-3′ | ||
| 5-HT2B | Sense 5′-TCTTCAATAAGACATTTCGGGA-3′ | NM_008311.2 |
| Antisense 5′-GAATGGTTGAACTTCGGAGC-3′ | ||
| 5-HT2C | Sense 5′-AGATATTTCTGCCCCGTCTG-3′ | NM_008312.4 |
| Antisense 5′-GCCTTAGTCCGCGAATTGAA-3′ | ||
| β-Actin | Sense 5′-GGGATGTTTGCTCCAACCAA-3′ | NM_007393.5 |
| Antisense 5′-GCGCTTTTGACTCAGGATTTAA-3′ | ||
| Human | ||
| 5-HT1B | Sense 5′-ACGCCGTGGAGTACTCAGCTAAAAG-3′ | NM_000863.2 |
| Antisense 5′-GGAGTAGACCGTGTAGAGGATGTGG-3′ | ||
| 5-HT1D | Sense 5′-TGCCGTGGTCCTTTCCGTC-3′ | NM_000864.4 |
| Antisense 5′-GGTGATGGTATAGGCGATGCTG-3′ | ||
| 5-HT1F | Sense 5′-GCTATAGCTTTGGATCGGTATCGAG-3′ | NM_001322209.1 |
| Antisense 5′-CAATCCTACTTGCTTGTCTCTTGTG-3′ | ||
| 5-HT2A | Sense 5′-TCGCCATCCAGAATCCCATCCACC-3′ | NM_001165947 0.2 |
| Antisense 5′-GATGGACTGCATAGTCCTCCTGCC-3′ | ||
| 5-HT2B | Sense 5′-ACGCCTAACATGGTTGACTGTGTC-3′ | NM_000867.4 |
| Antisense 5′-TGAGGCTCTCTGTCCGTTGGAA-3′ | ||
| 5-HT2C | Sense 5′-TAACACTGAAGCAATCATGG-3′ | NM_001256761.2 |
| Antisense 5′-GACTGTGCTGTTCTTTCTCACAC-3′ | ||
| β-Actin | Sense 5′-CTGGAACGGTGAAGGTGACA-3′ | NM_001101.3 |
| Antisense 5′-GAAGGGACTTCCTGTAACAATGCA-3′ | ||
5-HT, 5-hydroxytryptamine; COX1, cytochrome c oxidase subunit 1; Drp1, dynamin-related protein 1; ND1, NADH dehydrogenase 1; NDUFS1, NADH dehydrogenase (ubiquinone) FE-S protein 1; Nrf, nuclear respiratory factor; PGC-1α, peroxisome proliferator-activated receptor γ coactivator-1α; PINK1, PTEN-induced putative kinase 1; qRT-PCR, quantitative real-time PCR; TFAM, mitochondrial transcription factor A; UCP, uncoupling protein.
Complete blood count, blood urea nitrogen, and serum creatinine evaluation.
Following euthanasia by isoflurane inhalation, peripheral blood was collected via cardiac puncture using EDTA as an anticoagulant and complete blood counts were obtained using a HEMAVET Multispecies Hematology Analyzer. Blood was collected via retro-orbital bleed following isoflurane inhalation. Blood urea nitrogen (BUN) was determined using the QuantiChrom Urea Assay kit (BioAssay Systems, Hayward, CA), and serum creatinine was determined using the Creatinine Enzymatic Reagent Kit (Pointe Scientific, Canton, MI) based on the manufacturer’s directions.
Protein isolation and immunoblot analysis.
Mouse kidney cortex tissue was homogenized in protein lysis buffer (1% Triton X 100, 150 mM NaCl, 10 mM Tris-HCl, pH 7.4; 1 mM EDTA; 1 mM EGTA; 2 mM sodium orthovanadate; 0.2 mM phenylmethylsulfonylfluoride; 1 mM HEPES pH 7.6; 1 µg/ml leupeptin; and 1 µg/ml aprotinin) using a polytron homogenizer. The homogenate was stored on ice for 10 min and then centrifuged at 7,500 g for 5 min at 4°C. The supernatant was collected and protein determined using a bicinchoninic acid kit (Sigma Chemical Co., St. Louis, MO) with bovine serum albumin as the standard. Proteins (50 µg) were separated on 4%–20% gradient sodium dodecyl sulfate polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were blocked in 5% dried milk in Tris-buffered saline-Tween 20 (0.1% Tween 20 in 1× Tris-buffered saline) for 1 h and incubated with antibodies for kidney injury molecule-1 (KIM-1) (1:1,000; R&D systems, Minneapolis, MN), neutrophil gelatinase-associated lipocalin (NGAL), PGC-1α, mitochondrial transcription factor A (TFAM), cytochrome c oxidase subunit 1 (COX1), and NADH dehydrogenase (ubiquinone) FE-S protein 1 (NDUFS1), overnight at 4°C. After incubation for 2 h at room temperature with donkey anti-goat secondary antibody (1:2,000; Santa Cruz Biotechnology Inc., Dallas, TX), goat anti-rabbit (1: 2,0000; Abcam), or rabbit anti-mouse (1:20,000, Abcam) conjugated with horseradish peroxidase before visualization using enhanced chemiluminescence (Thermo Scientific, Waltham, MA) and the GE ImageQuant LAS-4000 (GE Life Sciences, Pittsburgh, PA).Optical density was determined using the National Institutes of Health ImageJ software (version 1.46). Each target protein was normalized to β-actin loading control.
Histology.
Kidney sections (5 microns) from WT and 5-HT1F receptor KO mice at 24 and 144 h after sham or I/R surgery were stained with periodic acid-Schiff staining. Each section was scored based on morphologic changes in a blinded fashion by a renal pathologist (J. M.). Tubular necrosis and brush border damage were chosen as an indication of morphologic damage to the kidney. These measures were evaluated on a scale from 0 to 4, which ranged from not present, 0; mild, 1; moderate, 2; severe, 3; and very severe, 4.
Quantitative real-time polymerase chain reaction analysis of mRNA expression.
Total RNA was isolated from renal cortical tissue with TRIzol reagent (Life Technologies, Grand Island, NY). The iScript Advanced cDNA Synthesis Kit for quantitative real-time polymerase chain reaction (qRT-PCR; Bio-Rad Laboratories, Hercules, CA) was used to produce a cDNA library from 1 μg total RNA according to the manufacturer’s protocol. For human experiments, we obtained human renal cDNA from Takara Bio USA (Mountain View, CA). We performed qRT-PCR with the generated cDNA using the SsoAdvanced Universal SYBR Green Supermix reagent (Bio-Rad Laboratories). The relative mRNA expression of all genes was determined by the 2−ΔΔCt method, and β-actin was used as a reference gene for normalization. Primer pairs used for PCR are listed in Table 1.
Analysis of mitochondrial DNA content.
Total DNA was isolated from renal cortex tissue using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA) as described by manufacturer’s protocol. Extracted DNA was quantified, and 5 ng was used for qRT-PCR. Relative DNA content was assessed by the mitochondrial-encoded NADH dehydrogenase subunit 1 and was normalized to nuclear-encoded β-actin. Primer sequences are listed in Table 1.
Determination of ATP content.
Renal cortical ATP was measured using the ATP Assay Kit (Abcam). Briefly, mouse renal cortex was homogenized in ice-cold 2 M perchloric acid using a polytron homogenizer. Homogenate was stored on ice for 45 min and then centrifuged at 13,000 × g for 2 min at 4°C. The supernatant was collected and diluted in the ATP Assay buffer. For each sample, pH was measured and adjusted to 6.5–8 using 0.1 M KOH or perchloric acid. Samples were then centrifuged at 13,000 × g for 15 min at 4°C and supernatant was collected. ATP was measured using a Tecan Spark 10M.
Statistics.
Data are expressed as means ± SE for all the experiments. Multiple comparisons of normally distributed data were analyzed using one-way ANOVA with an appropriate post hoc test; group means were compared using Holm-Sidak’s posttest. Single comparisons were analyzed by Student’s t-test where appropriate. The criterion for statistical differences was P ≤ 0.05 for all comparisons.
RESULTS
Absence of the 5-HT1F receptor gene does not induce renal injury.
To verify the absence of the 5-HT1F receptor, we measured 5-HT1F receptor mRNA in the renal cortex. Renal 5-HT1F mRNA was decreased 50% in heterozygous (HET) mice and absent in 5-HT1F receptor KO mice (Fig. 1A).
Fig. 1.

Absence of the 5-HT1F receptor gene does not induce renal injury. Expression of renal cortical 5-HT1F receptor was measured at the mRNA level in WT, HET, and KO mice (A). Immunoblot analysis of renal cortical KIM-1 and NGAL expression was used to assess tubular injury in 10-wk old male 5-HT1F receptor WT and KO mice (B). Data are reported as mean ± SE, n = 6. a,b,cStatistically significant differences (P < 0.05). 5-HT, 5-hydroxytryptamine; HET, heterozygous; KIM-1, kidney injury molecule-1; KO, knockout; NGAL, neutrophil gelatinase-associated lipocalin; WT, wild type.
We evaluated BUN in WT and KO mice. There was no difference in BUN levels at 10 wk of age (WT: 26 ± 3 mg/dl, KO: 25 ± 2 mg/dl). To assess further if the absence of the 5-HT1F receptor induced renal tubular injury, KIM-1 and NGAL were evaluated in the renal cortex of WT and KO mice. At 10 wk of age, KIM-1 was not detected and NGAL was unchanged in KO mice compared with WT mice (Fig. 1B).
It is important to mention that HET × HET matings yielded Mendelian ratios of 1:2:1 of WT, HET, and KO. In addition, KO × KO matings produced healthy, viable mice with similar increases in body weight over time and no differences in blood counts and serum chemistries (data not shown). Taken together, these data suggest that the KO of the 5-HT1F receptor alone does not cause renal injury at 10 wk of age.
Absence of the 5-HT1F receptor potentiates I/R-induced AKI at 24 h.
Although the absence of the 5-HT1F receptor did not cause renal injury, it is possible that under stress the absence of the 5-HT1F receptor could potentiate renal injury. To test this hypothesis, we subjected 10-wk-old WT and KO mice to renal I/R injury. Renal function was assessed 24 h after I/R-induced AKI and an increase in serum creatinine in WT and KO AKI mice was observed compared with sham controls (Fig. 2A). However, there was no statistical difference in serum creatinine between genotypes. We also measured two markers of renal injury by immunoblot analysis, KIM-1 and NGAL, in the renal cortex at 24 h. KIM-1 and NGAL increased three- and twofold, respectively, in 5-HT1F receptor KO mice subjected to I/R injury compared with WT mice subjected to I/R injury (Fig. 2, B–D). It is important to note that 5-HT1F receptor mRNA decreased by ~50% in WT mice subjected to I/R (Fig. 2E).
Fig. 2.

Absence of the 5-HT1F receptor potentiates I/R-induced AKI at 24 h. Serum creatinine was assessed 24 h after renal I/R injury in WT and 5-HT1F receptor KO mice (A). Immunoblot analysis of renal cortical KIM-1 and NGAL expression was used to assess tubular injury (B). Densitometry analysis of KIM-1 (C) and NGAL (D). Total RNA was harvested from renal cortex of WT and 5-HT1F receptor KO mice and expression of 5-HT1F receptor mRNA was measured (E). Data are reported as mean ± SE; Sham: n = 4; KO: n = 6. a,b,cStatistically significant differences (P < 0.05). 5-HT, 5-hydroxytryptamine; AKI, acute kidney injury; I/R, ischemia/reperfusion; KIM-1, kidney injury molecule-1; KO, knockout; NGAL, neutrophil gelatinase-associated lipocalin; WT, wild type.
Renal histopathology was assessed using periodic acid-Schiff staining. Kidneys from both WT and 5-HT1F receptor KO mice subjected to I/R injury exhibited proximal tubule necrosis and brush border damage. However, the absence of the 5-HT1F receptor did not potentiate tubular necrosis or the loss of the brush border at 24 h (Fig. 3). These data indicate that I/R-induced AKI 1) is greater in mice lacking the 5-HT1F receptor, 2) results in depletion of 5-HT1F receptor mRNA, and 3) suggests that the 5-HT1F receptor is renal protective under stress.
Fig. 3.

Absence of the 5-HT1F receptor has no effect on renal necrosis in I/R-induced AKI at 24 h. Representative slides of the outer stripe of the outer medulla stained with periodic acid-Schiff (PAS) at 24 h (magnification, ×100) following sham or I/R-induced AKI in WT and 5-HT1F receptor KO mice (A). Tubular necrosis scoring (B). Loss of brush border scoring (C). Data are reported as mean ± SE; WT I/R: n = 5; KO I/R: n = 6. 5-HT, 5-hydroxytryptamine; AKI, acute kidney injury; I/R, ischemia/reperfusion; KO, knockout; WT, wild type.
Absence of the 5-HT1F receptor reduces the recovery of renal function.
A previous study demonstrated that the stimulation of MB though the activation of the 5-HT1F receptor accelerates the recovery of renal function after AKI (11). Thus, we postulated that the absence of the 5-HT1F receptor would reduce recovery of renal function following injury (11). Renal function and tubular injury were assessed at 24 h and 144 h following I/R-induced AKI in WT and KO mice. All I/R mice had equal initial injury with an overall average serum creatinine of 0.7 ± 0.2 mg/dl at 24 h. However, 5-HT1F receptor KO and WT mice failed to recover renal function as demonstrated by persistent overall elevated serum creatinine of 0.5 ± 0.2 mg/dl at 144 h (Fig. 4, A and C). To explore renal recovery further in 5-HT1F receptor KO mice, renal function improvement was calculated as change in serum creatinine (Δ serum creatinine) at 144 h compared with 24 h following AKI in each mouse. Using this analysis, serum creatinine decreased ~0.25 mg/dl in AKI WT mice compared with no decrease in serum creatinine in AKI KO mice (Fig. 4B). It is noteworthy that tubular necrosis and loss of brush border remained elevated and to a similar degree at 144 h following I/R injury (Fig. 5).
Fig. 4.

Absence of the 5-HT1F receptor reduces the recovery of renal function. Serum creatinine (A) and Δ serum creatinine (B) were assessed 144 h after renal I/R injury in WT and 5-HT1F receptor KO mice. Immunoblot analysis of renal cortical KIM-1 and NGAL expression was used to assess tubular injury (C). Densitometry analysis of KIM-1 (D) and NGAL (E). Data are reported as mean ± SE; Sham: n = 4; I/R: n = 6. a,b,cStatistically significant differences (P < 0.05). 5-HT, 5-hydroxytryptamine; I/R, ischemia/reperfusion; KIM-1, kidney injury molecule-1; KO, knockout; NGAL, neutrophil gelatinase-associated lipocalin; WT, wild type.
Fig. 5.

Absence of the 5-HT1F receptor has no effect on renal necrosis in I/R-induced AKI at 144 h. Representative slides of the outer stripe of the outer medulla stained with periodic acid-Schiff (PAS) at 144 h (magnification, ×100) following sham or I/R-induced AKI in WT and 5-HT1F receptor KO mice (A). Tubular necrosis scoring (B). Loss of brush border scoring (C). Data are reported as mean ± SE; WT I/R: n = 5; KO I/R: n = 6. 5-HT, Δ serum creatinine, change in serum creatine; 5-hydroxytryptamine; AKI, acute kidney injury; I/R, ischemia/reperfusion; KO, knockout; WT, wild type.
To assess renal tubular injury, renal cortical KIM-1 and NGAL levels were measured by immunoblot analysis. KIM-1 and NGAL were upregulated two- and threefold, respectively, in 5-HT1F receptor KO mice subjected to I/R injury compared with injured WT mice (Fig. 4, C–E). Taken together, these findings provide strong evidence that the loss of the 5-HT1F receptor potentiates AKI and decreases renal recovery.
Absence of the 5-HT1F receptor suppresses MB during I/R- induced AKI.
To determine whether greater tubular injury in the 5-HT1F receptor KO mice was associated with suppressed MB, we assessed renal cortical PGC-1α, the master regulator of MB; electron transport chain (ETC) proteins, nuclear-encoded NDUFS1, mitochondrial-encoded COX1, and TFAM protein at 24 and 144 h in mice subjected to I/R. PGC-1α and TFAM protein were reduced ~50%–60% in KO mice after I/R-induced AKI compared with WT and KO sham mice at 24 h (Fig. 6, A, C, F). No difference in PGC-1α and TFAM protein was detected in I/R injured WT mice (Fig. 6, A, C, F). We assessed the same end points at 144 h following I/R injury. Renal cortical PGC-1α, NDUFS1, COX1, and TFAM proteins were reduced ~50%, 75%, 50%, 60%, respectively, in 5-HT1F receptor KO mice compared with sham animals of both genotypes at 144 h (Fig. 6, B, G–J). It is important to note that NDUFS1 protein was reduced at 144 h in WT mice following injury and to a further extent in 5-HT1F receptor KO mice subjected with I/R (Fig. 6, B and H). Similar findings were observed following mRNA analysis of these end points (Fig. 7). These findings suggest that MB is persistently suppressed following AKI and to a greater magnitude in the absence of the 5-HT1F receptor.
Fig. 6.

Absence of the 5-HT1F receptor suppresses MB in I/R-induced AKI. Total protein was harvested from the renal cortex of WT and 5-HT1F receptor KO mice following I/R-induced AKI. Immunoblot analysis of renal cortical PGC-1α, NDUFS1, COX1, and TFAM protein expression was used to assess MB at 24 h (A) and 144 h (B). Densitometry analysis of PGC-1α (C), NDUFS1 (D), COX1 (E), and TFAM (F) at 24 h and PGC-1α (G), NDUFS1 (H), COX1 (I), and TFAM (J) at 144 h following I/R injury. Data are reported as mean ± SE; Sham: n = 4; I/R: n = 6. a,b,cStatistically significant differences (P < 0.05). 5-HT, 5-hydroxytryptamine; AKI, acute kidney injury; COX1, cytochrome c oxidase subunit 1; I/R, ischemia/reperfusion; KO, knockout; MB, mitochondrial biogenesis; NDUFS1, NADH dehydrogenase (ubiquinone) FE-S protein 1; PGC-1α, peroxisome proliferator-activated receptor γ coactivator-1α; TFAM, mitochondrial transcription factor A; WT, wild type.
Fig. 7.

Absence of the 5-HT1F receptor disrupts mitochondrial homeostasis following I/R-induced AKI. Total RNA was extracted from renal cortical tissue. Gene expression of PGC-1α (A), NDUFS1 (B), COX1 (C), TFAM (D) at 24 h and PGC-1α (E), NDUFS1 (F), COX1 (G), TFAM (H) at 144 h were measured to assess mitochondrial homeostasis following I/R-induced renal injury. Data are reported as mean ± SE; Sham: n = 4; I/R: n = 6. a,b,cStatistically significant differences (P < 0.05). 5-HT, 5-hydroxytryptamine; AKI, acute kidney injury; COX1, cytochrome c oxidase subunit 1; I/R, ischemia/reperfusion; KO, knockout; MB, mitochondrial biogenesis; NDUFS1, NADH dehydrogenase (ubiquinone) FE-S protein 1; PGC-1α, peroxisome proliferator-activated receptor γ coactivator-1α; TFAM, mitochondrial transcription factor A; WT, wild type.
5-HT1F receptor KO mice exhibit a persistent decrease in ATP levels following I/R-induced renal injury.
To assess whether the absence of the 5-HT1F receptor alters mitochondrial ATP following I/R-induced AKI, cortical ATP levels were measured. ATP was reduced by ~50% following I/R injury in both genotypes at 24 h (Fig. 8A). However, WT mice exhibited partial recovery in ATP content by 144 h, which was absent in the 5-HT1F receptor KO mice (Fig. 8B).
Fig. 8.
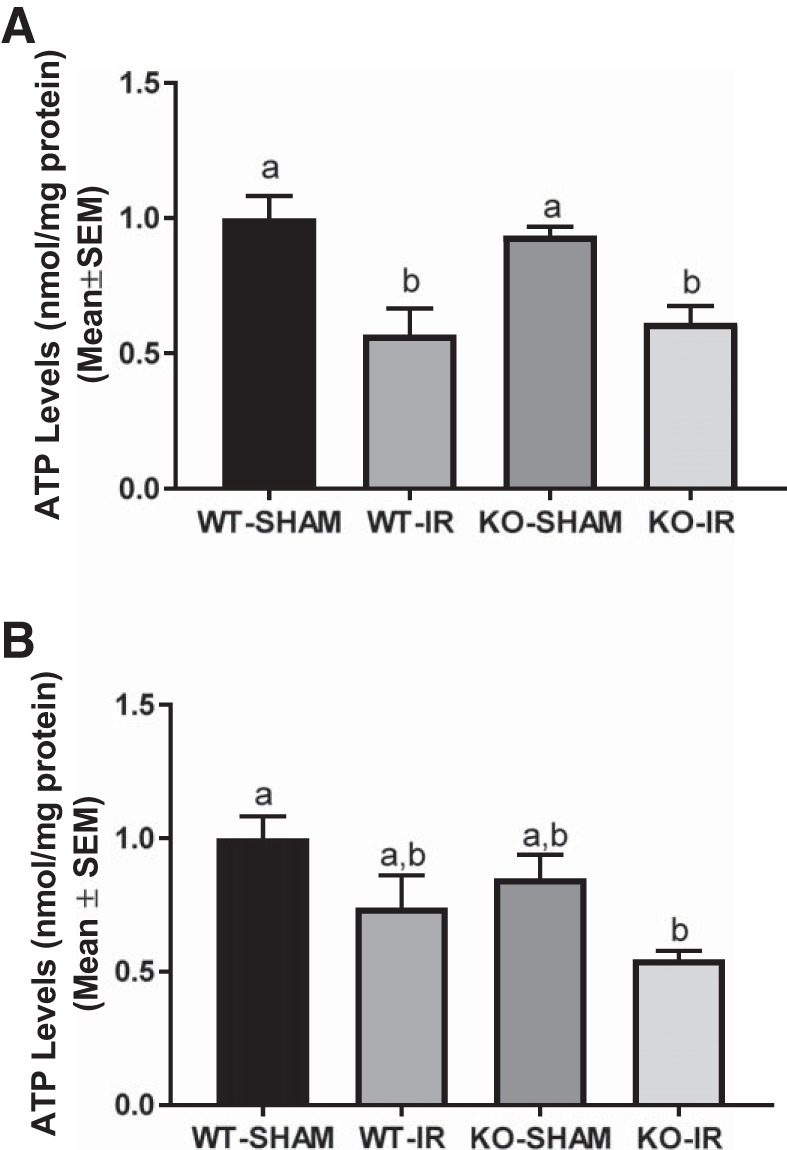
5-HT1F receptor KO mice exhibit persistent decreased ATP following I/R-induced renal injury. Renal cortical ATP was measured in WT and 5-HT1F receptor KO mice 24 h (A) and 144 h (B) following renal I/R injury and then normalized to protein. Data are reported as mean ± SE; Sham: n = 4; I/R: n = 6. a,b,cStatistically significant differences (P < 0.05). 5-HT, 5-hydroxytryptamine; I/R, ischemia/reperfusion; KO, knockout; WT, wild type.
Renal physiological mitochondrial homeostasis is disrupted in 5-HT1F receptor KO mice.
Our laboratory demonstrated that knockdown of the 5-HT1F receptor in renal proximal tubular cells resulted in a decrease in mitochondrial proteins (11). Thus, we hypothesized that the absence of the 5-HT1F receptor would lead to a reduction in mitochondrial markers in the renal cortex.
We assessed mRNA expression of MB proteins and components of the ETC, NDUFS1, ATP synthase β, and COX1 and NADH dehydrogenase 1 (ND1). Additionally, we assessed PGC-1α and transcription factors, nuclear respiratory factor-1, nuclear respiratory factor-2 and TFAM at 10 wk of age. Transcript levels of nuclear respiratory factor-2, TFAM, NDUFS1, ATP synthase β, and COX1 were increased (1.4-, 1.4-, 1.7-, 1.3-, and 1.6-fold, respectively) at 10 wk and these increases corresponded with elevated mitochondrial DNA copy number (Fig. 9, A–C). These results reveal the loss of the 5-HT1F receptor leads to increased MB genes in the kidney.
Fig. 9.

Altered renal mitochondrial homeostasis in 5-HT1F receptor KO mice. Total RNA was harvested from renal cortical tissue of 5-HT1F receptor WT and KO mice. Gene expression of key regulators of mitochondrial biogenesis, fission, and autophagy were measured at 10 wk of age (A–D). Relative mitochondrial DNA content in the renal cortex was determined by qRT-PCR analysis (A). Data are reported as mean ± SE, n = 6. *P < 0.05 vs. WT controls. 5-HT, 5-hydroxytryptamine; COX1, cytochrome c oxidase subunit 1; Drp1, dynamin-related protein 1; KO, knockout; ND1, NADH dehydrogenase 1; NDUFS1, NADH dehydrogenase (ubiquinone) FE-S protein 1; Nrf, nuclear respiratory factor; PGC-1α, peroxisome proliferator-activated receptor γ coactivator-1α; PINK1, PTEN-induced putative kinase 1; qRT-PCR, quantitative real-time PCR; TFAM, mitochondrial transcription factor A; WT, wild type.
Mitochondrial fission and mitophagy are critical processes in maintaining mitochondrial homeostasis (20, 29). As such, we measured markers of mitochondrial fission, dynamin-related protein 1 (Drp1) and mitophagy, mitochondrial PTEN-induced putative kinase 1 (PINK1). At 10 wk, Drp1 and PINK1 were elevated 1.7- and 1.6-fold, respectively, compared with WT mice (Fig. 9D). These results reveal that mitochondrial fission and mitophagy may be increased in response to the loss of the 5-HT1F receptor.
5-HT1 and 5-HT2 receptors are expressed in mouse and human kidney.
To determine renal 5-HT1 and 5-HT2 receptors present in mice and humans, we measured gene expression of 5-HT1 and 5-HT2 receptors. 5-HT1B, 5-HT1D, 5-HT1F, 5-HT2A, 5-HT2B, and 5-HT2C receptor gene expression was detected in human and mice renal samples (Fig. 10A).
Fig. 10.
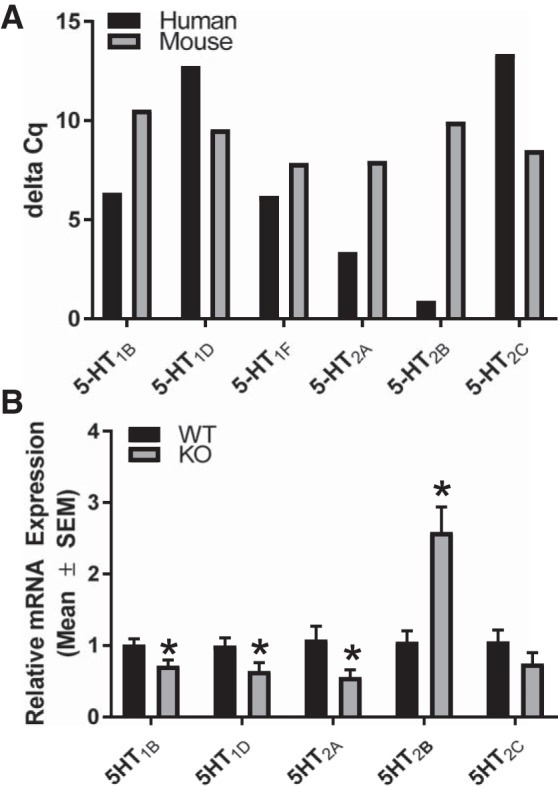
5-HT1 and 5-HT2 Receptors are expressed in mouse and human kidney and are altered in renal cortex in 5-HT1F receptor in KO mice. Total RNA was extracted from human and mouse renal tissue. Gene expression of renal 5-HT1B, 5-HT1D, 5-HT1F, 5-HT2A, 5-HT2B, and 5-HT2C receptors were measured in human and mouse (A). n = 1. Data reported as Δ Cq values. Total RNA was harvested from renal cortical tissue of 5-HT1F receptor WT and KO mice at 10 wk of age. Gene expression of 5-HT1B, 5-HT1D, 5-HT2A, 5-HT2B, and 5-HT2C receptors were measured (B). Data are reported as mean ± SE, n = 6. *P < 0.05 vs. WT controls. 5-HT, 5-hydroxytryptamine; KO, knockout; WT, wild type.
Altered renal 5-HT receptor gene expression in 5-HT1F KO mice.
We explored the possibility that the lack of the 5-HT1F receptor results in altered gene expression of other 5-HT receptors. Absence of the 5-HT1F receptor decreased renal mRNA of 5-HT1B, 5-HT1D, and 5-HT2A receptors (~30%, 35%, 45%) at 10 wk, whereas 5-HT2B receptor mRNA increased ~2.6-fold and 5-HT2C receptor mRNA did not change compared with WT (Fig. 10B). Thus, altered renal 5-HT receptor gene expression may be a compensatory mechanism in response to the absence of the 5-HT1F gene.
DISCUSSION
Mice lacking the 5-HT1F receptor gene were viable and physically similar to WT mice. Having demonstrated that 5-HT1F receptor-deficient mice did not exhibit an obvious phenotype, we investigated the effects of I/R-induced AKI on renal function and MB in 5-HT1F receptor KO and WT mice. The rationale for these experiments is supported by our previous findings that demonstrated 5-HT1F receptor stimulation accelerated renal recovery following I/R-induced AKI and rescued mtDNA copy number in mice (11).
An important role for the 5-HT1F receptor in the onset and recovery of renal injury following AKI was demonstrated by greater renal tubular injury as measured by KIM-1 and NGAL protein expression in 5-HT1F receptor KO mice 24 after I/R injury. These data are corroborated by the reduction in 5-HT1F receptor gene expression at peak renal injury following AKI in WT mice at 24 h, providing evidence that the depletion of the 5-HT1F receptor is linked to greater renal dysfunction following injury.
The absence of the 5-HT1F receptor also prevented recovery of renal function as demonstrated by persistently increased serum creatinine and potentiated KIM-1 and NGAL at 144 h. Identification of the 5-HT1F receptor as a mediator of prolonged renal dysfunction is critical for 1) better understanding of the mechanisms leading to chronic renal dysfunction and 2) better patient outcomes by developing novel therapies that target the 5-HT1F receptor to minimize renal tissue damage and tubular dysfunction. We hypothesize that the greater tubular injury is contributing to the prolonged renal dysfunction following I/R-induced AKI in 5-HT1F receptor KO mice.
The observation that renal injury was worsened in 5-HT1F receptor KO mice was also associated with persistent suppression of MB. A notable observation was the early suppression in PGC-1α and TFAM, which are both key regulators of MB. These findings were accompanied by tubular injury; thus, we predict that dysregulation of MB is at least, in part, responsible for reduced recovery of renal function following renal I/R injury. Interestingly, numerous markers of mitochondrial homeostasis, including protein expression of ETC components and ATP levels, were continuously suppressed following I/R injury in 5-HT1F receptor KO mice. These findings are supported by published results that demonstrated that PGC-1α and MB are protective following both folic-acid and I/R-induced AKI (22, 25, 26). In summary, we propose that the loss of 5-HT1F receptor-dependent MB is the mechanism responsible for diminished PGC-1α, ETC protein expression, and ATP levels, and this suppression prevents the recovery of tubular injury and renal function in the 5-HT1F receptor KO mice following injury.
Although 5-HT1F receptor KO mice did not have gross morphological alterations, nor changes in blood counts and serum chemistries, our data provide clear evidence that mitochondrial homeostasis in renal cortical tissue is disrupted following I/R renal injury. We predicted that the absence of the 5-HT1F receptor altered MB and homeostasis in naïve 5-HT1F receptor KO mice. Assimilating various mitochondrial markers revealed that the loss of the 5-HT1F receptor gene resulted in MB and increased mitochondrial DNA copy number in the naïve kidney. Associated with increased markers of MB was an increase in the mitochondrial fission protein Drp1 and mitophagy marker, PINK1. One explanation for these results is that the loss of the 5-HT1F receptor and its associated basal control of MB results in a compensatory response to increase MB. Consequently, we hypothesize that mitochondrial fission and mitophagy are increased to maintain a given level of healthy mitochondria.
It is also possible that another 5-HT receptor may compensate for the loss of the 5-HT1F receptor. This hypothesis is supported by a body of evidence indicating functional cross talk as well as direct interaction between different 5-HT receptors. For example, there is functional cross talk between 5-HT2A and 5-HT2C receptors, between 5-HT1B and 5-HT2C receptors and the 5-HT1A and 5-HT7 receptors (5, 8, 18). In this study, examination of other 5-HT receptors revealed altered gene expression in response to the absence of the 5-HT1F receptor. Only the 5-HT2B receptor was upregulated at 10 wk in the kidney of the 5-HT1F receptor KO mouse. Interestingly, overexpression of the 5-HT2B receptor leads to increased cardiac mitochondrial enzymatic protein expression and cell number and size in transgenic mice (19). Although the 5-HT1D receptor has not been linked to mitochondria, 5-HT2A,2C receptors have been linked to mitochondrial function in the kidney (14). In fact, 5-HT2A receptor agonists induce MB in renal proximal tubular cells (14). Other possibilities exist, and future studies are needed to identify the mechanism by which MB is induced in the absence of the 5-HT1F receptor and the potential role of the other 5-HT receptors.
It is important to note that there is limited knowledge regarding the signaling mechanisms linking 5-HT receptors to MB. To this end, our group recently reported that the 5-HT1F receptor induces MB through dual mechanisms in renal proximal tubule cells (12). Specifically, following activation of the 5-HT1F receptor, G beta-gamma complex mediates the induction of the Akt, endothelial nitric oxide, cyclic guanosine-monophosphate, protein kinase G, and PGC-1α pathway, while simultaneously repressing the extracellular signal-regulated kinase 1/2 and forkhead box O3 pathway (12). The 5-HT1F receptor-induced MB pathways provide insight into how MB is regulated downstream of 5-HT receptors. Elucidation of the signaling mechanisms leading to increased gene expression of renal MB markers in the absence of the 5-HT1F receptor may reveal additional strategies to target MB following acute organ injuries.
This study provides insight for the role of 5-HT1F receptor in modulating MB and mitochondrial homeostasis. Mitochondrial dysfunction is a critical pathophysiological mediator in various disease states, including acute organ injury and cardiovascular disease (1, 4, 6, 9, 27). The induction of MB to restore mitochondria number and function increases the energy supply needed for tissue repair. Thus, MB is a promising therapeutic strategy for many human pathologies. However, there are few, nontoxic compounds that selectively induce MB (27). This presents an opportunity to target 5-HT receptors for the treatment of human acute organ injuries and cardiovascular diseases because the 5-HT1F receptor and other 5-HT1 and 5-HT2 receptors are present in human renal tissue. Although classical functional roles of serotonin and serotonin receptors in the cardiovascular system include renal vascular dilation, vasoconstriction, and platelet aggregation (17), future studies will be needed to understand the involvement of 5-HT receptors in MB and homeostasis in the kidney, as well as other organs involved in cardiovascular health.
In summary, these results provide the first evidence that the 5-HT1F receptor regulates mitochondrial homeostasis in renal cortical tissue under physiological conditions. Our data also demonstrate that the 5-HT1F receptor plays a protective role in I/R-induced AKI and this protection is mediated through mitochondrial homeostasis and biogenesis. This study sets the stage for the use of 5-HT receptors for the treatment of acute organ injuries that are characterized by mitochondrial dysfunction.
GRANTS
This study was funded by National Institutes of Health National Institute of General Medical Sciences: R01-GM-084147 (to R. Schnellmann), the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs no. BX000851 (to R. Schnellmann), the National Institutes of Health Ruth L. Kirschstein National Research Services Award no. T32-DK-083262 (to W. Gibbs), and Southwest Environmental Health Sciences Center grant no. P30 ES006694.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
W.S.G., C.C.B., and R.G.S. conceived and designed research; W.S.G., J.B.C., and M.M. performed experiments; W.S.G., J.B.C., M.M., and J.M. analyzed data; W.S.G. interpreted results of experiments; W.S.G. prepared figures; W.S.G. drafted manuscript; W.S.G., C.C.B., and R.G.S. edited and revised manuscript; W.S.G., J.B.C., M.M., C.C.B., J.M., and R.G.S. approved final version of manuscript.
REFERENCES
- 1.Andreux PA, Houtkooper RH, Auwerx J. Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov 12: 465–483, 2013. doi: 10.1038/nrd4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arany Z, Wagner BK, Ma Y, Chinsomboon J, Laznik D, Spiegelman BM. Gene expression-based screening identifies microtubule inhibitors as inducers of PGC-1α and oxidative phosphorylation. Proc Natl Acad Sci USA 105: 4721–4726, 2008. doi: 10.1073/pnas.0800979105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444: 337–342, 2006. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol 61: 599–610, 2013. doi: 10.1016/j.jacc.2012.08.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bazovkina DV, Kondaurova EM, Naumenko VS, Ponimaskin E. Genotype-dependent difference in 5-HT2C receptor-induced hypolocomotion: comparison with 5-HT2A receptor functional activity. Neural Plast 2015: 846589, 2015. doi: 10.1155/2015/846589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cameron RB, Beeson CC, Schnellmann RG. Development of therapeutics that induce mitochondrial biogenesis for the treatment of acute and chronic degenerative diseases. J Med Chem 59: 10411–10434, 2016. doi: 10.1021/acs.jmedchem.6b00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng Z, Ristow M. Mitochondria and metabolic homeostasis. Antioxid Redox Signal 19: 240–242, 2013. doi: 10.1089/ars.2013.5255. [DOI] [PubMed] [Google Scholar]
- 8.Clifton PG, Lee MD, Somerville EM, Kennett GA, Dourish CT. 5-HT1B receptor knockout mice show a compensatory reduction in 5-HT2C receptor function. Eur J Neurosci 17: 185–190, 2003. doi: 10.1046/j.1460-9568.2003.02437.x. [DOI] [PubMed] [Google Scholar]
- 9.Cooper MP. Interplay of mitochondrial biogenesis and oxidative stress in heart failure. Circulation 127: 1932–1934, 2013. doi: 10.1161/CIRCULATIONAHA.113.003177. [DOI] [PubMed] [Google Scholar]
- 10.Funk JA, Schnellmann RG. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am J Physiol Renal Physiol 302: F853–F864, 2012. doi: 10.1152/ajprenal.00035.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garrett SM, Whitaker RM, Beeson CC, Schnellmann RG. Agonism of the 5-hydroxytryptamine 1F receptor promotes mitochondrial biogenesis and recovery from acute kidney injury. J Pharmacol Exp Ther 350: 257–264, 2014. doi: 10.1124/jpet.114.214700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibbs WS, Garrett SM, Beeson CC, Schnellmann RG. Identification of dual mechanisms mediating 5-hydroxytryptamine receptor 1F induced mitochondrial biogenesis. Am J Physiol Renal Physiol 314: F260–F268, 2018. doi: 10.1152/ajprenal.00324.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Golpich M, Amini E, Mohamed Z, Azman Ali R, Mohamed Ibrahim N, Ahmadiani A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: pathogenesis and treatment. CNS Neurosci Ther 23: 5–22, 2017. doi: 10.1111/cns.12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harmon JL, Wills LP, McOmish CE, Demireva EY, Gingrich JA, Beeson CC, Schnellmann RG. 5-HT2 receptor regulation of mitochondrial genes: unexpected pharmacological effects of agonists and antagonists. J Pharmacol Exp Ther 357: 1–9, 2016. doi: 10.1124/jpet.115.228395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jesinkey SR, Funk JA, Stallons LJ, Wills LP, Megyesi JK, Beeson CC, Schnellmann RG. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J Am Soc Nephrol 25: 1157–1162, 2014. doi: 10.1681/ASN.2013090952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalogeris T, Bao Y, Korthuis RJ. Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol 2: 702–714, 2014. doi: 10.1016/j.redox.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaumann AJ, Levy FO. 5-hydroxytryptamine receptors in the human cardiovascular system. Pharmacol Ther 111: 674–706, 2006. doi: 10.1016/j.pharmthera.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 18.Naumenko VS, Popova NK, Lacivita E, Leopoldo M, Ponimaskin EG. Interplay between serotonin 5-HT1A and 5-HT7 receptors in depressive disorders. CNS Neurosci Ther 20: 582–590, 2014. doi: 10.1111/cns.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nebigil CG, Jaffré F, Messaddeq N, Hickel P, Monassier L, Launay JM, Maroteaux L. Overexpression of the serotonin 5-HT2B receptor in heart leads to abnormal mitochondrial function and cardiac hypertrophy. Circulation 107: 3223–3229, 2003. doi: 10.1161/01.CIR.0000074224.57016.01. [DOI] [PubMed] [Google Scholar]
- 20.Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521: 525–528, 2015. doi: 10.1038/nature14300. [DOI] [PubMed] [Google Scholar]
- 21.Rosca MG, Hoppel CL. Mitochondrial dysfunction in heart failure. Heart Fail Rev 18: 607–622, 2013. doi: 10.1007/s10741-012-9340-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruiz-Andres O, Suarez-Alvarez B, Sánchez-Ramos C, Monsalve M, Sanchez-Niño MD, Ruiz-Ortega M, Egido J, Ortiz A, Sanz AB. The inflammatory cytokine TWEAK decreases PGC-1α expression and mitochondrial function in acute kidney injury. Kidney Int 89: 399–410, 2016. doi: 10.1038/ki.2015.332. [DOI] [PubMed] [Google Scholar]
- 23.Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 1813: 1269–1278, 2011. doi: 10.1016/j.bbamcr.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stallons LJ, Funk JA, Schnellmann RG. Mitochondrial homeostasis in acute organ failure. Curr Pathobiol Rep 1: 169–177, 2013. doi: 10.1007/s40139-013-0023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, Clish CB, Stillman IE, Karumanchi SA, Rhee EP, Parikh SM. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531: 528–532, 2016. doi: 10.1038/nature17184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weinberg JM. Mitochondrial biogenesis in kidney disease. J Am Soc Nephrol 22: 431–436, 2011. doi: 10.1681/ASN.2010060643. [DOI] [PubMed] [Google Scholar]
- 27.Whitaker RM, Corum D, Beeson CC, Schnellmann RG. Mitochondrial biogenesis as a pharmacological target: a new approach to acute and chronic diseases. Annu Rev Pharmacol Toxicol 56: 229–249, 2016. doi: 10.1146/annurev-pharmtox-010715-103155. [DOI] [PubMed] [Google Scholar]
- 28.Whitaker RM, Wills LP, Stallons LJ, Schnellmann RG. cGMP-selective phosphodiesterase inhibitors stimulate mitochondrial biogenesis and promote recovery from acute kidney injury. J Pharmacol Exp Ther 347: 626–634, 2013. doi: 10.1124/jpet.113.208017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 337: 1062–1065, 2012. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]