

Abstract
Chronic kidney disease (CKD) is a condition with significant morbidity and mortality that affects 15% of adults in the United States. One cause of CKD is acute kidney injury (AKI), which commonly occurs secondary to sepsis, ischemic events, and drug-induced nephrotoxicity. Unilateral ischemia-reperfusion injury (UIRI) without contralateral nephrectomy (CLN) and repeated low-dose cisplatin (RLDC) models of AKI to CKD demonstrate responses characteristic of the transition; however, previous studies have not effectively compared the pathogenesis. We demonstrate both models instigate renal dysfunction, inflammatory cytokine responses, and fibrosis. However, the models exhibit differences in urinary excretory function, inflammatory cell infiltration, and degree of fibrotic response. UIRI without CLN demonstrated worsening perfusion and function, measured with 99mTc-mercaptoacetyltriglycine-3 imaging, and physiologic compensation in the contralateral kidney. Furthermore, UIRI without CLN elicited a robust inflammatory response that was characterized by a prolonged polymorphonuclear cell and natural killer cell infiltrate and an early expansion of kidney resident macrophages, followed by T-cell infiltration. Symmetrical diminished function occurred in RLDC kidneys and progressively worsened until day 17 of the study. Surprisingly, RLDC mice demonstrated a decrease in inflammatory cell numbers relative to controls. However, RLDC kidneys expressed increased levels of kidney injury molecule-1 (KIM-1), high mobility group box-1 (HMGB1), and colony stimulating factor-1 (CSF-1), which likely recruits inflammatory cells in response to injury. These data emphasize how the divergent etiologies of AKI to CKD models affect the kidney microenvironment and outcomes. This study provides support for subtyping AKI by etiology in human studies, aiding in the elucidation of injury-specific pathophysiologic mechanisms of the AKI to CKD transition.
Keywords: cisplatin, chronic kidney disease, fibrosis, glomerular filtration rate, inflammation, ischemia-reperfusion
INTRODUCTION
Chronic kidney disease (CKD) affects an estimated 30 million people in the United States alone (8a) and is a significant cause of morbidity and mortality. Acute kidney injury (AKI) increases the risk of CKD. Better understanding of the AKI to CKD transition is necessary to intervene in this process. Several clinical scenarios, including cardiac surgery (57), rhabdomyolysis (8), contrast exposure (20), and sepsis (59) cause AKI. Furthermore, the risk of CKD is increased in the setting of common comorbidities such as diabetes (1, 42) and cardiovascular disease (9, 22, 29). In the clinic, CKD is often missed until disease is severe, as patients can demonstrate an insidious course with subclinical presentation until after significant structural kidney damage has occurred. Currently, a lack of information regarding the mechanisms of CKD and its transition from AKI has hampered the development of targeted interventions.
One factor contributing to this lack of effective intervention is that preclinical animal models of the AKI to CKD transition, such as 5/6 nephrectomy, low-dose cisplatin, aristolochic acid nephrotoxicity, ischemia-reperfusion (IR), and nitric oxide inhibition each have their own underlying mechanisms for progressive disease, cytoarchitectural features, and functional outcomes. Common elements of these models include persistent decreased kidney function, as measured by glomerular filtration rate (GFR), albuminuria, inflammatory cell infiltration, histopathologic signs of damage to the tubular epithelium, and interstitial fibrosis.
There are two widely used animal models of the AKI to CKD transition. The first, unilateral ischemia-reperfusion injury (UIRI) without contralateral nephrectomy (CLN), is a validated model of the AKI to CKD transition with extensive interstitial fibrosis (31, 58). Ischemia time and body temperature during surgery are variables that directly relate to injury severity. Leaving the contralateral kidney intact improves survival, even in the setting of severe injury, and provides a control organ to decrease systemic injury burden. The injured kidney develops inflammatory cell infiltration and interstitial fibrosis, consistent with a chronic disease phenotype. However, the presence of the uninjured organ raises questions about the function of the injured, fibrotic organ in terms of filtration, secretion, and excretion. Isolated left or right kidney function within this model represents an unknown variable that we address with this study.
The second model, repeated low-dose cisplatin administration (RLDC), is a clinically relevant model of repeated injury and development of subsequent CKD. Cisplatin is a chemotherapeutic drug used for the treatment of solid organ tumors and is known for its beneficial effects of crosslinking, damaging, and inhibiting DNA repair, thereby killing cancer cells (13). Arguably, the most deleterious effect of cisplatin is nephrotoxicity, which can manifest with renal salt washing, proteinuria, distal tubular acidosis, AKI, and even CKD (38, 48). In the clinic, patients undergo multiple low-dose infusions of cisplatin to circumvent nephrotoxicity; however, even with this measure in place, AKI still occurs in 20% of these patients (5, 30). To better understand cisplatin nephrotoxicity in the laboratory, a standard (20 mg/kg) dosing method of cisplatin administration in mice is used to model AKI (7). Although efficacious for understanding acute cisplatin nephrotoxicity, the dosing is not conducive to data acquisition more than 72 h after administration. To overcome this, we use a model of low-dose intraperitoneal injections of cisplatin, adapted from Sharp et al. (51, 52), which perpetuates persistent inflammation and renal interstitial fibrosis and models CKD development over time.
In these studies, we applied molecular, cellular, and systemic physiologic assays to better understand these models to facilitate their application in a clinically relevant manner. These models lead to similar outcomes: declining renal function, inflammation, and progressive fibrosis. They differ markedly in the pathophysiology and molecular mechanisms of the disease process. A detailed comparison of these two models, initiated by ischemic or cytotoxic injury, has enabled identification of pathways that are common and unique to each model.
MATERIALS AND METHODS
Reagents and general supplies were ordered from Fisher Scientific (Waltham, MA) or Sigma-Aldrich (St. Louis, MO), unless otherwise stated.
Animal models.
All animals used in these studies were on a C57/BL6 genetic background and bred and maintained at the University of Alabama at Birmingham. All animal experiments were approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care.
UIRI without CLN.
UIRI without CLN was induced in age-matched C57BL/6 male mice (10 wk of age), as described previously (31, 58). Briefly, mice were anesthetized with inhaled isoflurane, 1.5–2.0% induction, 1.0–1.5% maintenance. Mice were placed on a heating pad, and core body temperature was maintained at 36–37°C by rectal thermometer. Left UIRI was induced via flank incision by application an atraumatic vascular clamp (18055-05; Fine Science Tools) to the renal pedicle for 30 min. During ischemia, the kidney was covered with sterile gauze soaked with prewarmed saline. The clamp was removed, and reperfusion was confirmed visually. The kidney was placed back in anatomical position, and the incision was closed. For sham surgery, the left kidney was exposed, but the vessels were not clamped. Serum was collected via facial vein bleed on alternative sides at 7-day intervals for analysis.
RLDC administration.
For the RLDC model, age-matched C57BL/6 male mice (10 wk of age) were briefly anesthetized with isoflurane, and 9 mg/kg body wt cisplatin (1.0 mg/ml solution in sterile 0.9% saline) or vehicle (sterile 0.9% saline) were administered intraperitoneally once a week for 4 wk, adapted from Sharp et al. (51). Kidneys were harvested at 24 days after injury induction for analysis. Serum was collected 72 h after each injection.
Quantification of mRNA.
RNA was isolated from tissues using TRIzol (Invitrogen) and converted to cDNA (Qiagen). Quantitative real-time polymerase chain reaction (RT-PCR) was performed using SYBR Green Master Mix (Invitrogen). Reactions were performed in triplicate and monitored using melting curve analysis. Relative mRNA expression was quantified using comparative threshold cycle method and normalized to GAPDH mRNA as an internal control. Primers used to detect the specific genes included the following: inducible nitric oxide synthase (iNOS), fwd: 5′-CCAAGCCCTCACCTACTTCC-3′ and iNOS rev: 5′-CTCTGAGGGCTGACACAAGG-3′; tumor necrosis factor-α (TNF-α), fwd: 5′-ACGGCATGGATCTCAAAGAC-3 and TNF-α, rev: 5′-AGATAGCAAATCGGCTGACG-3′; colony stimulating factor-1 (CSF-1), fwd: 5′-CGGGCATCATCCTAGTCTTGCTGACTGT-3′ and CSF-1, rev: 5′-ATAGTGGCAGTATGTGGGGGGCATCCTC-3′; interleukin-6 (IL-6), fwd: 5′-CTGCAAGAGACTTCCATCCAG-3′ and IL-6, rev: 5′-AGTGGTATAGACAGGTCTGTTGG-3′; monocyte chemoattractant protein-1 (MCP-1), fwd: 5′-ACTCACCTGCTGCTACTCAT-3′; MCP-1, rev: 5′-CTACAGCTTCTTTGGGACA-3′; IL1-β fwd: 5′-TGGGCCTCAAAGGAAAGA-3′ and IL1-β, rev: 5′-GGTGCTGATGTACCAGTT-3′; high mobility group box-1 (HMGB1), fwd: 5′-TTGTGCAAACTTGCCGGGAGGA-3′ and HMGB,-1 rev: 5′-ACTTCTCCTTCAGCTTGGCAGC-3′; neutrophil gelatinase-associated lipocalin (NGAL), fwd: 5′-CTCAGAACTTGATCCCTGCC-3′ and NGAL, rev: 5′-TCCTTGAGGCCCAGAGACTT-3′; fibronectin (Fn) fwd: 5′-TCTGGGAAATGGAAAAGGGGAATGG-3′ and fibronectin, rev: 5′-CACTGAAGCAGGTTTCCTCGGTTGT-3′; α-smooth muscle actin (α-SMA), fwd: 5′-GACGCTGAAGTATCCGATAGAACAC-3′ and α-SMA, rev: 5′-CCACCATCTCCAGAGTCCAGCACAAT-3′; transforming growth factor-β (TGF-β), fwd: 5′-AGCGGACTACTATGCTAAAGAGGTCACCC-3′ and TGF-β, rev: 5′-CCAAGGTAACGCCAGGAATTGTTGCTATA-3′; collagen 1, fwd: 5′-GCAACAGTCGCTTCACCTACA-3′ and collagen I, rev: 5′-CAATGTCCAAGGGAGCCACAT-3′; and GAPDH, fwd: 5′-ATCATCCCTGCATCCACT-3′, GAPDH, rev: 5′-ATCCACGACGGACACATT-3′.
Western blot analysis.
Tissues were homogenized and protein was isolated, as described previously (7). Briefly, kidneys were lysed in lysis buffer (10 mM/l Tris·HCl, 5 mM/l EDTA, 150 mM/l NaCl, 10% Nonidet P-40, and 10% Triton-X) with protease (Sigma-Aldrich) and phosphatase (Biotool) inhibitors. Total protein was quantified with the bichinchoninic acid protein assay (ThermoFisher). Protein (75 ug) was resolved on 12% Tris-glycine SDS-PAGE and transferred to a PVDF membrane (EMB Millipore, Billerica, MA). Membranes were blocked with 5% nonfat dry milk in PBS-T for 1 h and then incubated with mouse anti-α-SMA (Sigma-Aldrich; 1:5,000), rabbit anti-Fn (Sigma-Aldrich; 1:10,000), goat anti-kidney injury molecule-1 (anti-KIM-1) antibody (R&D Systems; 1:800), and goat anti-NGAL (R&D; 1:5,000), followed by incubation with donkey anti-rabbit or donkey anti-mouse IgG-Alexa Fluor 680 (ThermoFisher) by the Odyssey CLx (Li-Cor, Lincoln, NE).
Renal function measurements.
Serum creatinine levels were measured by liquid chromatography-tandem mass spectrometry at the University of Alabama at Birmingham-University of California, San Diego O’Brien Center for Acute Kidney Injury Research bioanalytical core facility by previously described methods (56).
Transcutaneous measurement of GFR was measured in mice using FITC-labeled sinistrin. Briefly, mice were anesthetized with 1.5–2% isoflurane induction, 1–1.5% maintenance. A small patch on the flank of the mouse was shaved, and the transdermal GFR monitors were adhered to the skin using a double sided adhesive patch (MediBeacon). Devices were secured by wrapping the mouse in medical tape. FITC-sinistrin (15 mg/ml dissolved in 0.9% sterile saline) was administered intravenously (tail vein). Mice were placed back in their cages and GFR was monitored for 2 h, and then the devices were removed and data were analyzed using elimination kinetics curve of FITC-sinistrin clearance, as previously described (17, 49, 50).
99mTc-mercaptoacetyltriglycine-3 (MAG3) imaging was performed as previously described (45). Briefly, mice were anesthetized with isoflurane and injected intraperitoneally with 0.5 ml sterile saline before radioisotope administration. Mice were then positioned over an Anger gamma camera, equipped with a parallel hole collimator (420/550 Mobile Radioisotope Gamma camera; Technicare, Solon, OH). 99mTc-MAG3 was injected (1.0 mCi/25g body wt) intravenously via the tail vein. Dynamic acquisition (Numa Station; Numa, Amherst, NH) was performed at the time of injection at 15 s/frame for 30 min (120 frames, 1 frame/15 s). Percent injected dose, and subsequent parameters, were measured as previously described (45). Briefly, percent injected dose in respective kidneys was background-corrected intrarenal counts, relative to mean total body counts over the duration of the imaging session. Mice that did not achieve a rapid peak and subsequent stable but slightly declining total body count value were excluded as outliers due to poor intravenous injection of 99mTc-MAG3. Tails were excluded from total body counts. To determine the start-point for intrarenal quantitative measurements, image sequences were examined for first image where the kidney was clearly visible relative to background.
Histology.
Kidneys were cut transversely and fixed in 10% neutral buffered formalin for 24 h then embedded in paraffin. Tissues were stained with periodic acid-Schiff hematoxylin using standard protocols. Fibrosis was visualized and measured using picrosirius red (PSR). Sections were deparaffinized in xylene washes and rehydrated, incubated in PSR for 1 h, washed in acidified water, and dehydrated. Sections were mounted in a resinous medium. All images were acquired using either Nikon Eclipse TE2000-S or a Nikon SMZ800 microscope and background corrected and white balanced using Nikon NIS Elements software (Version 4.60). Percent area positive and intensity (integrated density) of PSR stain were measured using the “Triangle” autothreshold method in FIJI version 1.51n. Color, brightness, and contrast adjustments were made in an identical manner for matched sections.
Flow cytometry.
Mice were anesthetized with 1.5–2% isoflurane, and kidneys were perfused with cold PBS (20 ml) or until abdominal organs blanched, via left ventricle cardiac puncture. Kidneys were decapsulated and weighed. Flow cytometry was performed as previously described (15). Briefly, kidneys were minced and incubated with liberase DL (Roche Diagnostics) in DMEM medium with 10 mM HEPES for 30 min at 37°C shaking. Kidney cell suspensions were filtered through 40-μm nylon mesh and stained with fluorochrome-conjugated antibodies for 30 min on ice in the dark then analyzed on a Becton Dickinson LSRII analyzer. The following antibodies (with their respective clone numbers) were used for the myeloid panel: 7-AAD, MHCII-FITC (M5/114.15.2), Gr-1-APC (1A8), Ly6C-eF450 (HK1.4), CD11b-SuperBright600 (M1/70), CD45.2-BV650 (104), F4/80-APC-eF780 (BM8), and CD11c-BV785 (N418). The following antibodies (with their respective clone numbers) were used for the lymphoid panel: natural killer (NK)1.1-PE (PK136), CD3e-APC (145–2C11), CD8a-eF450 (53–6.7), CD4-SuperBright600 (RM4–5), and CD19-BV785 (6D5). Absolute numbers were measured using AccuCheck Counting Beads quantitative standard (Life Technologies) and normalized to tissue mass. Data were analyzed, and figures were generated using FlowJo software (Treestar Software). Cells were gated for viable singlets. Myeloid cells were gated negative for lymphoid lineage (CD3, CD19, and NK1.1). Lymphoid cells were gated negative for myeloid lineage [F4/80, Ly6G (RB6–8C5)].
Statistical analysis.
Data are presented as means ± SE. For comparisons between two groups, the unpaired two-tailed t-test was used. ANOVA and Tukey’s, Sidak’s, or Dunnett’s post hoc tests were used for comparisons between more than two groups and are specified in figure legends. P < 0.05 was considered significant. For MAG3 percent injected dose curves, two quantitative analyses were performed. First, to compare kidneys within individual animals, the difference of percent injected dose in paired kidneys was calculated for each time point over the duration of elimination of MAG3. The differences were fit to a mixed model with animal as a random effect and quadratic or linear time by treatment effects (if the quadratic terms were not significant) and tested for significant paired differences from zero. Second, a one-compartment elimination model was fit to the percent injected dose curves for individual kidneys. Area under the elimination curves were calculated and compared among groups using Two-way ANOVA and multiple comparisons by Tukey’s post hoc test.
RESULTS
Renal function in IR and low-dose cisplatin models of AKI to CKD.
Male C57BL/6 mice underwent 30 min UIRI without CLN or were treated with repeated intraperitoneal cisplatin at 9 mg/kg once a week for 4 wk (Fig. 1, A and B). We observed 100% survival (data not shown) and a modest rise in serum creatinine compared with noninjured controls in both models over the course of the study (Fig. 1C). To better understand the extent of injury, we used a more sensitive measure of kidney function: transcutaneous GFR measurement with FITC-sinistrin. With this method, mice that underwent UIRI without CLN also showed significant decline in GFR at day 7 (151.1 ± 11.8 μl/min) but stayed constant for the remainder of the time course. Cisplatin-treated mice experienced a significant decline in GFR by day 10, continuing to decrease to 53.5 ± 7.7% of baseline at 24 days after the first injection (Fig. 1D).
Fig. 1.

Renal function in unilateral ischemia-reperfusion injury without contralateral nephrectomy (UIRI without CLN) and repeated low-dose cisplatin models of acute kidney injury to chronic kidney disease. A and B: experimental design for UIRI without CLN (A) and cisplatin administration (9 mg/kg; B). Male C57BL/6 mice were used in all experiments. C: serum creatinine levels were measured and expressed as milligrams per deciliter as a time course from UIRI without CLN and cisplatin mice and their respective controls. D: glomerular filtration rate (GFR) was measured using transcutaneous FITC-sinistrin clearance over the course of the study in UIRI without CLN and cisplatin mice and their respective controls. Data represented as means ± SE; n = 3–8 per group. Statistical significance was determined by 2-way ANOVA followed by Sidak’s multiple comparisons test * P < 0.05; n = 3–8 per group.
99mTc-MAG3 imaging is a powerful tool for assessing split renal blood flow, tubular function, and kidney injury in patients and animal models. This technique also allows measurement of individual kidneys rather than global renal function measurement (45, 54). In this assay, radioisotope is injected intravenously, and a gamma camera captures images as the labeled MAG3 is taken up and then excreted by each kidney. Two methods were used to compare percent injected dose (%ID, renographic) curves in this study (Fig. 2). First, the difference in %ID in paired kidneys was calculated and fit to a quadratic or linear model. Percent ID in IRI kidneys was different compared with IR-contralateral at 7, 14, and 21 days after injury (Table 1). Notably, IRI kidneys attained a lower peak than IR contralateral, indicating poor perfusion or basolateral epithelial uptake (Fig. 2A). Unexpectedly, sham injury was different compared with sham contralateral at 14 days. Otherwise, as expected, sham-injury and sham-contralateral kidneys were not different (Table 1). IRI kidneys demonstrated a progressively decreasing renographic area under the curve (AUC) from 7 to 21 days after injury. IR-contralateral kidneys demonstrated increased AUC at 21 days compared with 14 days, providing direct functional evidence of physiologic compensation (Fig. 2C). At 7 days, IRI demonstrated increased AUC compared with sham injury, which is due to poor excretion (Fig. 2, A and C). Additionally, decrements in function were apparent from %ID curves for cisplatin kidneys relative to vehicle controls at days 17 and 24 (Fig. 2B). Similar peak %ID was achieved between groups (Fig. 3B), however, there was a trend toward decreased excretion at 17 and 24 days (Fig. 3D). Furthermore, AUC was increased for cisplatin kidneys at day 17 compared with day 10 (Fig. 2D). Appropriately, since cisplatin is administered systemically, no difference was observed between cisplatin-left and cisplatin-right kidneys (Table 1). Findings from renographic AUC and paired kidney differences indicated delayed or diminished uptake and/or poor excretion in AKI to CKD kidneys, although the shape of the curves in diseased kidneys in the two models appeared different. Further quantitative analysis follows for clinically relevant parameters.
Fig. 2.

99mTc-mercaptoacetyltriglycine-3 (MAG3) renal function percent injected dose curves at varying time points during acute kidney injury to chronic kidney disease. A and B: 99mTc-MAG3 imaging was performed to analyze renal function over the course of the study for unilateral ischemia-reperfusion injury (UIRI) without contralateral nephrectomy (CLN) (A) and repeated low-dose cisplatin (RLDC) (B). Background-corrected percent injected dose within individual kidneys are plotted as a function of time after IV administration of radioisotope. Varying time points after UIRI and cisplatin injury induction were tested. Representative still images, shown adjacent to graphs, were obtained from the first frame before signal saturation in the bladder (within 15–60 s of intravenous injection of 99mTc-MAG3). If present, curvilinear opacities represent mouse tails at the site of injection. C and D: area under the curve (AUC) measurements for elimination curves fit to a 1-compartment model for UIRI without CLN (C) and RLDC (D). Two-way ANOVA with Tukey’s posttest: *P < 0.05 within time points; #P < 0.05 across time points; n = 4–5 kidneys for IR; n = 3 kidneys for sham, quantifying each kidney independently; n = 6–14 kidneys for cisplatin (3–7 mice quantifying both kidneys); n = 6–8 kidneys for saline (3–4 mice quantifying both kidneys).
Table 1.
Analysis of MAG3 data for paired kidney function by mixed model
| Model/Group/Time Point | P |
|---|---|
| UIRI without CLNa | |
| IRc | |
| 7 days | 0.038 |
| 14 days | <0.0001 |
| 21 days | <0.0001 |
| Shamd | |
| 7 days | 0.397 |
| 14 days | <0.0001 |
| 21 days | 0.626 |
| RLDCb | |
| Cisplatine | |
| 10 days | 0.471 |
| 17 days | 0.265 |
| 24 days | 0.523 |
| Vehiclef | |
| 10 days | 0.517 |
| 17 days | 0.397 |
| 24 days | 0.464 |
CLN, contralateral nephrectomy; MAG3, mercaptoacetyltriglycine-3; RLDC, repeated low-dose cisplatin; UIRI, unilateral ischemia-reperfusion injury.
Differences fit using quadratic model;
differences fit using linear model;
IR-injury minus IR-contralateral;
sham-injury minus sham-contralateral;
cisplatin-left minus cisplatin-right;
vehicle-left minus vehicle-right.
Fig. 3.
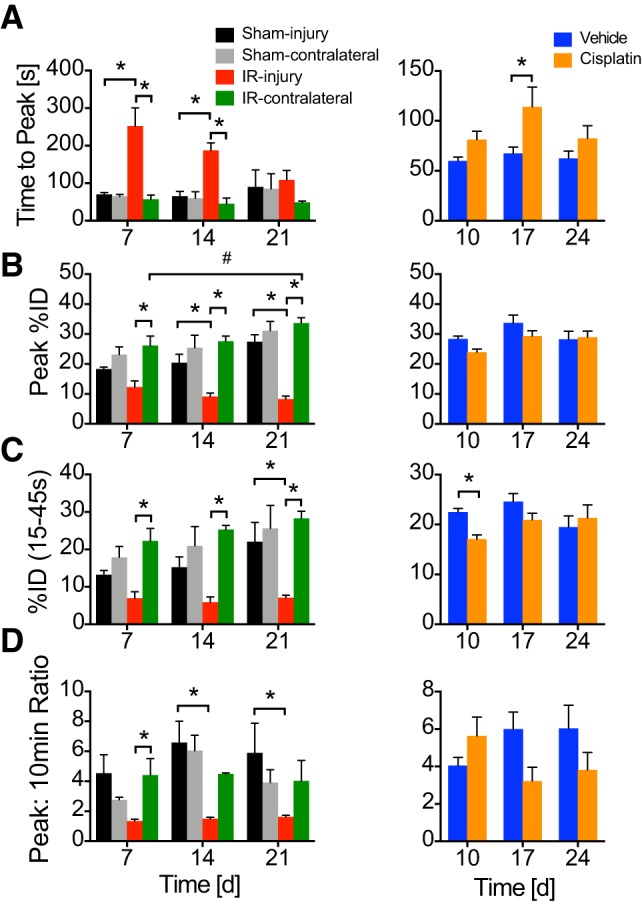
99mTc-mercaptoacetyltriglycine-3 physiologic parameters for kidney function related to perfusion and excretion. A–D: time to peak (A), peak percent injected dose (B), percent injected dose (%ID; C) during 15- to 45-s intervals after radioisotope injection, and peak:10-min ratio (D) were measured. Data represented as means ± SE. Two-way ANOVA with Tukey’s posttest: *P < 0.05 within time points; #P < 0.05 across time points. n = 4–5 for IR; n = 3 for sham; n = 6–14 for cisplatin (3–7 mice quantifying both kidneys); n = 6–8 for saline (3–4 mice quantifying both kidneys).
MAG3 imaging allows measurement of perfusion or basolateral epithelial organic anion uptake [peak %ID, time to peak, %ID 15–45 s (early %ID)] and urinary excretion (peak:10-min ratio) (Fig. 3). IRI kidneys demonstrated decreased perfusion and/or uptake across time points relative to IR-contralateral uninjured kidneys (Fig. 3, A, B, and C). The time to peak is notably delayed in IRI compared with IR-contralateral and sham-injury kidneys at days 7 and 14 (Fig. 3A). A statistically significant excretion defect was observed relative to IR-contralateral uninjured kidneys only at 7 days (Fig. 3D); however, the trend was maintained throughout the study. Effects from sham-injury surgery were observed in peak %ID, early %ID, and peak:10-min ratio, as was evident by a lack of statistically significant difference with IR-injury at early time points (Fig. 3, B–D). These effects appeared to resolve with significantly greater peak %ID, early %ID, and peak:10-min ratio in sham-injury compared with IR-injury at 21 days (Fig. 3, B–D). No differences were observed between sham-injury and sham-contralateral kidneys. Cisplatin mice maintain perfusion, uptake, and excretion, only demonstrating a delay in time to peak at 17 days in cisplatin vs. saline (Fig. 3A) and decrease in early %ID relative to saline at day 10 (Fig. 3C).
Leukocytic infiltration in AKI to CKD models.
To better understand how inflammation participates in nephrotoxic and IR AKI to CKD, we evaluated absolute numbers of intrarenal inflammatory cells by flow cytometry (Fig. 4A). Not surprisingly, CD45+ cells were increased in IR relative to sham control at days 7 and 14, resolving by day 21. However, in cisplatin kidneys, we found a decrease in CD45+ cells relative to vehicle (Fig. 4B).
Fig. 4.

Intrarenal leukocytic populations in unilateral ischemia-reperfusion injury without contralateral nephrectomy and repeated low-dose cisplatin. Flow cytometry from whole kidney single cell suspensions normalized to mass of tissue. A: gating scheme. B: CD45+ leukocytes and neutrophils [polymorphonuclear cell (PMNs)]. C: F4/80Hi resident macrophages and F4/80Low infiltrating macrophages and dendritic cells. D: CD4 and CD8 T cells. E: B cells and natural killer (NK) cells. Black dots: sham controls collapsed from 7, 14, and 21 days; red dots: IR; blue dots: vehicle; orange dots: cisplatin. MHCII, major histocompatibility complex class II. Data represented as means ± SE. *P < 0.05 by one-way ANOVA and Dunnett’s posttest, comparing each time point vs. sham or vehicle controls; n = 5–9 for IR; n = 10 for sham; n = 3 for cisplatin and vehicle groups.
For IR, further analysis revealed that myeloid lineage cells including polymorphonuclear cells, F4/80Hi tissue resident, and F4/80Low macrophages/dendritic cells predominate early, with significant increases relative to sham at day 7, and neutrophils remain elevated until day 14 (Fig. 4, B and C). Similarly, innate NK cells are increased relative to sham at days 7 and 14 (Fig. 4E). Adaptive CD4 and CD8 T cells were increased relative to sham in later phases at days 14 and 21 (Fig. 4, D), consistent with the principle that macrophages and T cells predominate in end organ fibrotic inflammatory responses. B cells showed no changes in the IR model (Fig. 4E).
After cisplatin dosing, neutrophils were decreased relative to vehicle control at day 10 but trended toward an increase at day 24 (Fig. 4B). Throughout the study, F4/80Hi resident macrophages and F4/80Low macrophages and dendritic cells were decreased in RLDC compared with vehicle controls (Fig. 4C). Notably, CD4 and CD8 T cells do not increase in later phases, even while the organ is actively experiencing interstitial fibrosis (Fig. 4D). Decreases were observed in B cells and NK cells in cisplatin mice at day 24 (Fig. 4E).
Proinflammatory cytokines, chemokines, and damage patterns in nephrotoxic and IR AKI to CKD.
We next sought to investigate soluble mediators of inflammatory response in IR and cisplatin-mediated CKD. Consistent with the chronic proinflammatory state found in patients, we found that inflammatory markers IL-6, HMGB1, CSF-1, MCP-1, and TNF-α are elevated compared with sham and vehicle-treated mice in both models (Fig. 5, A–D and F). It is interesting to note that all tested inflammatory markers in UIRI mice were significantly elevated at 21 days compared with both sham controls and cisplatin-treated mice, while a modest induction of inflammatory markers occurred in cisplatin-treated mice at 24 days. The exception to this observation was with HMGB-1 and CSF-1, which were significantly elevated in cisplatin mice compared with vehicle controls (Fig. 5, B and C). We also investigated differences in renal NGAL and nitric oxide synthase-2 (NOS2) expression. In both models, NGAL was elevated compared with sham and vehicle controls (Fig. 5, G and I); however, NOS2 was only elevated after UIRI, not cisplatin (Fig. 5H). Proximal tubules are often the site of maximal injury in the kidney due to their energy-expensive responsibilities (10); therefore, we compared KIM-1 induction after UIRI and cisplatin administration. Although KIM-1 is induced in both injury models at end-point analysis, induction was greater after cisplatin administration (Fig. 5, I and J), consistent with the localized accumulation and S3 segment-specific nephrotoxicity of the drug (12, 14).
Fig. 5.

Proinflammatory cytokines and damage patterns are exacerbated in acute kidney injury to chronic kidney disease models. A–H: total RNA was isolated from whole kidney tissue at end points [ischemia-reperfusion (IR), 21 days; cisplatin (Cp), 24 days] and was analyzed for expression of interleukin-6 (IL-6; A), high mobility group box-1 (HMGB1; B), colony stimulating factor-1 (CSF-1; C), monocyte chemoattractant protein-1 (MCP-1; D), IL-1β (E), tumor necrosis factor-α (TNF-α; F), neutrophil gelatinase associated lipocalin (NGAL; G), and nitric oxide synthase-2 (NOS2; H) by real-time PCR. Data are normalized to GAPDH and expressed as fold change compared with sham or vehicle (veh) controls. Data are represented as means ± SE; n = 3–8 per group. Statistical significance was determined by one-way ANOVA and Tukey’s multiple comparisons test: *P < 0.05. I: protein lysates from whole kidney tissue were analyzed for kidney injury molecule-1 (KIM-1) and NGAL expression. Anti-GAPDH was used as a loading control. n = 2–5 per group. J and K: densitometric values were normalized to GAPDH and represented as arbitrary units (AU) for KIM-1 (J) and NGAL (K) using ImageJ. Statistical significance was determined by one-way ANOVA and Tukey’s multiple comparisons test: *P < 0.05; n = 2–5 per group.
Interstitial renal fibrosis in models of AKI to CKD.
One of the major hallmarks of CKD is the development of interstitial fibrosis (19); therefore, we assessed fibrotic remodeling in both models of AKI to CKD. Compared with UIRI without CLN, we found that cisplatin caused a modest induction of fibrosis in the RLDC model (Fig. 6, A–C). PSR staining demonstrated collagen deposition in the interstitial space with destruction of surrounding architecture in both models (Fig. 6, A–C). In both models, high-power PAS-hematoxylin-stained kidney sections demonstrated patchy periglomerular and perivascular fibrosis. Cyst-like spaces were observed in the corticomedullary region in UIRI without CLN (Fig. 6A). Healthy-appearing tubules were intermixed with affected areas, including anucleated tubular epithelial cells and injured tubules, demonstrating epithelial cell swelling (Fig. 6A). Although there was an elevation of Fn, TGF-β, and collagen I in the RLDC model, the expression of these markers in the UIRI model of CKD was much more pronounced (Fig. 7, A–C). Furthermore, a significant increase in α-SMA expression was observed after UIRI; however, α-SMA mRNA did not differ from vehicle controls after four doses of cisplatin (Fig. 7D). Similar to mRNA expression after UIRI, protein levels of Fn and α-SMA were increased relative to sham. Accordingly, mRNA and protein levels of Fn were increased twofold compared with vehicle controls after RLDC, while mRNA and protein levels of α-SMA between cisplatin treated and vehicle controls were not statistically different. Overall, fibrosis markers at the protein level were consistent with histopathologic observations (Fig. 7E), indicating that both models of injury demonstrated interstitial fibrosis but that UIRI changes were more severe given the experimental conditions.
Fig. 6.

Unilateral ischemia-reperfusion injury (UIRI) without contralateral nephrectomy (CLN) and repeated low-dose cisplatin (RLDC) demonstrate histopathologic signs of interstitial fibrosis at study end points. A: Picrosirius red (PSR) and periodic acid Schiff-hematoxylin (PASH) stained kidney transverse sections at low and high power. C, cyst-like space; arrow: perivascular and periglomerular fibrosis; arrowhead: anucleated epithelium; red arrowhead: cellular swelling. B: percent area above threshold for PSR. C: intensity of PSR stain encompassing positive staining areas above threshold. Statistical significance was determined by unpaired t-test: *P < 0.05; n = 2–8 per group.
Fig. 7.

Unilateral ischemia-reperfusion injury (UIRI) induces a more severe fibrotic response compared with cisplatin (Cp). Total RNA was isolated from whole kidney tissue at end points was analyzed for expression of fibronectin (Fn; A), transforming growth factor-β (TGF-β; B), collagen I (C), and α-smooth muscle actin (α-SMA; D) by real-time PCR. Data were normalized to GAPDH and expressed as fold change compared with sham or vehicle (veh) controls; n = 3–8 per group. E: protein lysates from whole kidney tissue were analyzed for Fn and α-SMA expression. Anti-GAPDH was used as a loading control; n = 2–5 per group. F and G: densitometric values were normalized to GAPDH and represented as arbitrary units (AU) for Fn (F) and α-SMA (G) using ImageJ. Statistical significance was determined by one-way ANOVA and Tukey’s multiple comparisons test: *P < 0.05; n = 2–5 per group.
DISCUSSION
In this study, we investigated two models of the AKI to CKD transition, UIRI without CLN and RLDC, with a focus on differences in pathophysiology. The relevance of this study stems from the facts that AKI: 1) causes morbidity and mortality in patients, and 2) is associated with the development of CKD (9, 34). Our data demonstrate a decline in renal function (serum creatinine and GFR), increased inflammatory response, and fibrotic remodeling in both of these models of the AKI to CKD transition; however, they manifest differently. First, IR allows for long-term study of inflammation and fibrosis after one incident of ischemic AKI. In contrast, RLDC uses multiple AKI insults that result in decreased kidney function, excretion, and development of CKD. Second, while UIRI without CLN produces a robust inflammatory phenotype, our data suggest that cisplatin administration suppresses inflammatory cell infiltration. Finally, while both models stimulate fibrosis, it is more pronounced in the UIRI model compared with cisplatin most likely due to the effects of cisplatin on inflammation.
Both UIRI without CLN and RLDC administration demonstrated only a modest rise in serum creatinine compared with their controls, likely due to the presence of the contralateral kidney and the low administered cisplatin doses, respectively. However, we observed a significant decline in GFR and renal structural damage in both models, indicating transdermal GFR is more sensitive than serum creatinine for measurement of kidney dysfunction in animal models of AKI to CKD.
MAG3 imaging is a robust technique for measuring renal dysfunction after injury, as it assesses renal perfusion, in combination with basolateral epithelial uptake, and excretion. UIRI without CLN was first established by Zager et al. (58) as a model of the AKI to CKD transition. In this model, the injured kidney atrophies and the contralateral kidney undergoes compensatory hypertrophy. Before this study, the extent to which the atrophic injured kidneys are able to maintain kidney physiologic function was not known; therefore, we applied the MAG3 assay, which allows measurement of function of individual kidneys. After UIRI without CLN, we observed a significant reduction in renal function within 7 days after injury, evidenced by increased time to peak and decreased peak %ID. By day 14, perfusion of the injured kidney decreases and reaches a minimum by day 21. This may result from vascular rarefaction, which has been observed in AKI to CKD models (3, 4, 11, 16, 27). Mice that underwent RLDC administration exhibited renal dysfunction as early as 10 days after induction. A notable feature of the RLDC model was that perfusion/uptake was maintained, as is indicated by a relative lack of differences in peak %ID, time to peak, early %ID, and peak:10-min ratio (Figs. 2 and 3).
Interstitial nephritis is associated with CKD and functional decline in patients (39, 41, 44, 46). Many studies have linked inflammation with tissue damage and eventual dysfunction (23). The damage that is caused by initial AKI is controlled by a balance between adaptive and maladaptive renal repair mechanisms that depends on macrophage lineage, transcriptional profile, and phenotype (18, 24–26, 32, 33, 36, 61). The presence of chronic inflammation drives further damage and subsequent fibrosis; therefore, it is important to understand the inflammatory milieu in different modes of injury (ischemic, cytotoxic, septic, etc.), as the recruitment time, length of stay, and cell populations may differ. In this study, we examined myeloid and lymphoid cell populations during disease progression. In UIRI without CLN, we observed an increase in resident and infiltrating macrophages, neutrophils, and NK cells 7 days after IR, which resolved by day 21. Absolute numbers of T cells and B cells increased into the chronic phase, and we suggest they were recruited to participate in a chronic inflammatory and fibrotic response consistent with previously published data (37). The possibility of a cognate antigen receptor-mediated adaptive immune response in this model is intriguing. The presence of exposed autoantigens secondary to cellular injury and a breakdown of tolerance may participate in chronic inflammation in AKI to CKD. Likewise, we found a more robust increased expression of inflammatory markers in the kidneys at 21 days. Notably, we observed significant upregulation of NGAL and NOS2 expression, consistent with severity of injury and the oxidative stress of IR injury.
Previous studies with higher doses of cisplatin show robust inflammation and immune cell infiltration at early time points in AKI (2, 6, 35, 60). In contrast, with the repeated low doses of cisplatin, we observed a decrease in inflammatory cell absolute numbers in the cisplatin model of AKI to CKD. Absolute numbers were normalized to tissue mass; therefore, decreased absolute numbers could be explained if cisplatin kidneys increased in mass. However, we did not observe significant changes in mass of kidneys between vehicle and cisplatin groups (data not shown). We hypothesize that these repeated doses of cisplatin cause apoptotic cell death of renal tubular cells, as well as depletion of resident and infiltrating immune cells at later time points. In fact, cisplatin in combination with cyclophosphamide, etoposide, and/or radiation is associated with severe leukopenia in humans (15, 21, 55) and has been administered in combination with colony stimulating factor to counter the myelosuppression (40, 47). Furthermore, it has been associated with leukopenia in mice (28, 53). At the protein and RNA levels, we did not observe a change in most inflammatory markers at day 24. However, we did observe significant increases in HMGB-1 and CSF-1 mRNA expression compared with vehicle controls. While both models exhibited increased renal NGAL expression at end-point analysis, RLDC-treated mice had more robust KIM-1 expression, a marker for proximal tubule-specific injury, compared with UIRI mice. Similar to trends in inflammatory marker expression and cell infiltration, UIRI caused more substantial fibrotic remodeling compared with that of RLDC. A robust fibrotic response may not be observed after four doses of cisplatin due to the suppression of inflammatory cell infiltration. Sharp et al. (52) recently reported that fibrosis was increased 6 mo after RLDC administration, consistent with our hypothesis. It is possible that the lack of inflammatory infiltrates, which include macrophages, causes gradual accumulation of cellular debris, and when cisplatin dosing ceases, structural damage may be exacerbated.
In summary, we have compared IR and nephrotoxic models of AKI to CKD, with specific emphasis on the unique pathophysiologic processes involved. The specific causes of disease influenced the character and timing of renal function decline, inflammation, and fibrosis. Therefore, we conclude kidney injury resulting from nephrotoxicity and IR proceeds through unique physiologic, cellular, and molecular pathways.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grants R01-DK-59600 (to A. Agarwal and J. George), 1T32-DK-116672-01 (to L. M. Black), and F31-DK-115169-01 (to J. M. Lever) and the core resource of the UAB-University of California, San Diego O’Brien Center (NIDDK Grant P30-DK-079337 to A. Agarwal). The UAB Comprehensive Cancer Center’s Preclinical Imaging Shared Facility was supported by NIH grant P30-CA-013148 (to K. R. Zinn).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.M.B., J.M.L., J.G., and A.A. conceived and designed research; L.M.B., J.M.L., A.M.T., B.C., Z.Y., S.E., Y.J., and R.B. performed experiments; L.M.B., J.M.L., A.M.T., G.C., and J.G. analyzed data; L.M.B., J.M.L., B.C., and G.C. interpreted results of experiments; L.M.B. and J.M.L. prepared figures; L.M.B., J.M.L., and J.G. drafted manuscript; L.M.B., J.M.L., A.M.T., B.C., Z.Y., S.E., Y.J., G.C., R.B., J.G., and A.A. edited and revised manuscript; L.M.B., J.M.L., A.M.T., B.C., Z.Y., S.E., Y.J., G.C., R.B., J.G., and A.A. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the University of Alabama at Birmingham (UAB) Comprehensive Cancer Center’s Preclinical Imaging Shared Facility and Sharon Samuel for assistance with MAG3 imaging. We also thank the UAB-University of California, San Diego O’Brien Center Bioanalytical Core for serum creatinine measurements. We appreciate the work of the UAB Comparative Pathology Core for assistance with histology and the technical assistance of Farah Abou Daya, Hannah Eckenrode, and Kayla McCullough.
REFERENCES
- 1.Afkarian M, Katz R, Bansal N, Correa A, Kestenbaum B, Himmelfarb J, de Boer IH, Young B. Diabetes, kidney disease, and cardiovascular outcomes in the Jackson Heart Study. Clin J Am Soc Nephrol 11: 1384–1391, 2016. doi: 10.2215/CJN.13111215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amirshahrokhi K, Khalili AR. Thalidomide ameliorates cisplatin-induced nephrotoxicity by inhibiting renal inflammation in an experimental model. Inflammation 38: 476–484, 2015. doi: 10.1007/s10753-014-9953-7. [DOI] [PubMed] [Google Scholar]
- 3.Basile DP, Collett JA, Yoder MC. Endothelial colony-forming cells and pro-angiogenic cells: clarifying definitions and their potential role in mitigating acute kidney injury. Acta Physiol (Oxf) 222: e12914, 2018. doi: 10.1111/apha.12914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basile DP, Friedrich JL, Spahic J, Knipe N, Mang H, Leonard EC, Changizi-Ashtiyani S, Bacallao RL, Molitoris BA, Sutton TA. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol 300: F721–F733, 2011. doi: 10.1152/ajprenal.00546.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennis Y, Savry A, Rocca M, Gauthier-Villano L, Pisano P, Pourroy B. Cisplatin dose adjustment in patients with renal impairment, which recommendations should we follow? Int J Clin Pharm 36: 420–429, 2014. doi: 10.1007/s11096-013-9912-7. [DOI] [PubMed] [Google Scholar]
- 6.Boddu R, Fan C, Rangarajan S, Sunil B, Bolisetty S, Curtis LM. Unique sex- and age-dependent effects in protective pathways in acute kidney injury. Am J Physiol Renal Physiol 313: F740–F755, 2017. doi: 10.1152/ajprenal.00049.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolisetty S, Traylor A, Joseph R, Zarjou A, Agarwal A. Proximal tubule-targeted heme oxygenase-1 in cisplatin-induced acute kidney injury. Am J Physiol Renal Physiol 310: F385–F394, 2016. doi: 10.1152/ajprenal.00335.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bywaters EG, Beall D. Crush injuries with impairment of renal function. BMJ 1: 427–432, 1941. doi: 10.1136/bmj.1.4185.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8a.Centers for Disease Control and Prevention National Chronic Kidney Disease Fact Sheet (Online) Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention; https://www.cdc.gov/kidneydisease/pdf/kidney_factsheet.pdf, 2017. [April 10, 2018]. [Google Scholar]
- 9.Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med 371: 58–66, 2014. doi: 10.1056/NEJMra1214243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chevalier RL. The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. Am J Physiol Renal Physiol 311: F145–F161, 2016. doi: 10.1152/ajprenal.00164.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clements ME, Chaber CJ, Ledbetter SR, Zuk A. Increased cellular senescence and vascular rarefaction exacerbate the progression of kidney fibrosis in aged mice following transient ischemic injury. PLoS One 8: e70464, 2013. doi: 10.1371/journal.pone.0070464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cristofori P, Zanetti E, Fregona D, Piaia A, Trevisan A. Renal proximal tubule segment-specific nephrotoxicity: an overview on biomarkers and histopathology. Toxicol Pathol 35: 270–275, 2007. doi: 10.1080/01926230601187430. [DOI] [PubMed] [Google Scholar]
- 13.Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 740: 364–378, 2014. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobyan DC, Levi J, Jacobs C, Kosek J, Weiner MW. Mechanism of cis-platinum nephrotoxicity: II. Morphologic observations. J Pharmacol Exp Ther 213: 551–556, 1980. [PubMed] [Google Scholar]
- 15.Egger SJ, Willson ML, Morgan J, Walker HS, Carrick S, Ghersi D, Wilcken N. Platinum-containing regimens for metastatic breast cancer. Cochrane Database Syst Rev 6: CD003374, 2017. doi: 10.1002/14651858.CD003374.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ehling J, Bábíčková J, Gremse F, Klinkhammer BM, Baetke S, Knuechel R, Kiessling F, Floege J, Lammers T, Boor P. Quantitative micro-computed tomography imaging of vascular dysfunction in progressive kidney diseases. J Am Soc Nephrol 27: 520–532, 2016. doi: 10.1681/ASN.2015020204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellery SJ, Cai X, Walker DD, Dickinson H, Kett MM. Transcutaneous measurement of glomerular filtration rate in small rodents: through the skin for the win? Nephrology (Carlton) 20: 117–123, 2015. doi: 10.1111/nep.12363. [DOI] [PubMed] [Google Scholar]
- 18.George JF, Lever JM, Agarwal A. Mononuclear phagocyte subpopulations in the mouse kidney. Am J Physiol Renal Physiol 312: F640–F646, 2017. doi: 10.1152/ajprenal.00369.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gewin LS. Renal fibrosis: primacy of the proximal tubule. Matrix Biol 68–69: 248–262, 2018. doi: 10.1016/j.matbio.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodman AI, Olszanecki R, Yang LM, Quan S, Li M, Omura S, Stec DE, Abraham NG. Heme oxygenase-1 protects against radiocontrast-induced acute kidney injury by regulating anti-apoptotic proteins. Kidney Int 72: 945–953, 2007. doi: 10.1038/sj.ki.5002447. [DOI] [PubMed] [Google Scholar]
- 21.Graziano SL, Lee K, Propert KJ, Tinsley R, Hayes DM, Green M, Comis RL. Phase II trial of etoposide and cisplatin for refractory small cell lung cancer: a Cancer and Leukemia Group B Study. Med Pediatr Oncol 18: 22–26, 1990. doi: 10.1002/mpo.2950180105. [DOI] [PubMed] [Google Scholar]
- 22.Hill NR, Fatoba ST, Oke JL, Hirst JA, O’Callaghan CA, Lasserson DS, Hobbs FD. Global prevalence of chronic kidney disease - a systematic review and meta-analysis. PLoS One 11: e0158765, 2016. doi: 10.1371/journal.pone.0158765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hodgkins KS, Schnaper HW. Tubulointerstitial injury and the progression of chronic kidney disease. Pediatr Nephrol 27: 901–909, 2012. doi: 10.1007/s00467-011-1992-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huen SC, Cantley LG. Macrophages in renal injury and repair. Annu Rev Physiol 79: 449–469, 2017. doi: 10.1146/annurev-physiol-022516-034219. [DOI] [PubMed] [Google Scholar]
- 25.Huen SC, Huynh L, Marlier A, Lee Y, Moeckel GW, Cantley LG. GM-CSF promotes macrophage alternative activation after renal ischemia/reperfusion injury. J Am Soc Nephrol 26: 1334–1345, 2015. doi: 10.1681/ASN.2014060612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hull TD, Kamal AI, Boddu R, Bolisetty S, Guo L, Tisher CC, Rangarajan S, Chen B, Curtis LM, George JF, Agarwal A. Heme oxygenase-1 regulates myeloid cell trafficking in AKI. J Am Soc Nephrol 26: 2139–2151, 2015. doi: 10.1681/ASN.2014080770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Humphreys BD. Mechanisms of renal fibrosis. Annu Rev Physiol 80: 309–326, 2018. doi: 10.1146/annurev-physiol-022516-034227. [DOI] [PubMed] [Google Scholar]
- 28.Kaku S, Ushioda N, Ishii H, Murakami T, Takahashi K, Nakai Y, Shimoya K, Nakamura T. Timing of cisplatin administration for chemoradiotherapy in transgenic mice bearing lens tumors. Oncol Rep 32: 16–22, 2014. doi: 10.3892/or.2014.3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanda T, Wakino S, Hayashi K, Plutzky J. Cardiovascular disease, chronic kidney disease, and type 2 diabetes mellitus: proceeding with caution at a dangerous intersection. J Am Soc Nephrol 19: 4–7, 2008. doi: 10.1681/ASN.2007111182. [DOI] [PubMed] [Google Scholar]
- 30.Latcha S, Jaimes EA, Patil S, Glezerman IG, Mehta S, Flombaum CD. Long-term renal outcomes after cisplatin treatment. Clin J Am Soc Nephrol 11: 1173–1179, 2016. doi: 10.2215/CJN.08070715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Clef N, Verhulst A, D’Haese PC, Vervaet BA. Unilateral renal ischemia-reperfusion as a robust model for acute to chronic kidney injury in mice. PLoS One 11: e0152153, 2016. doi: 10.1371/journal.pone.0152153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi B-S, Ruhrberg C, Cantley LG. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 22: 317–326, 2011. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lever JM, Yang Z, Boddu R, Adedoyin OO, Guo L, Joseph R, Traylor AM, Agarwal A, George JF. Parabiosis reveals leukocyte dynamics in the kidney. Lab Invest 98: 391–402, 2018. doi: 10.1038/labinvest.2017.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewington AJP, Cerdá J, Mehta RL. Raising awareness of acute kidney injury: a global perspective of a silent killer. Kidney Int 84: 457–467, 2013. doi: 10.1038/ki.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J, Gui Y, Ren J, Liu X, Feng Y, Zeng Z, He W, Yang J, Dai C. Metformin protects against cisplatin-induced tubular cell apoptosis and acute kidney injury via AMPKα-regulated autophagy induction. Sci Rep 6: 23975, 2016. doi: 10.1038/srep23975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li L, Huang L, Sung SS, Vergis AL, Rosin DL, Rose CE Jr, Lobo PI, Okusa MD. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int 74: 1526–1537, 2008. doi: 10.1038/ki.2008.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu J, Kumar S, Dolzhenko E, Alvarado GF, Guo J, Lu C, Chen Y, Li M, Dessing MC, Parvez RK, Cippà PE, Krautzberger AM, Saribekyan G, Smith AD, McMahon AP. Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight 2: e94716, 2017. doi: 10.1172/jci.insight.94716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of cisplatin nephrotoxicity. Toxins (Basel) 2: 2490–2518, 2010. doi: 10.3390/toxins2112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muriithi AK, Leung N, Valeri AM, Cornell LD, Sethi S, Fidler ME, Nasr SH. Clinical characteristics, causes and outcomes of acute interstitial nephritis in the elderly. Kidney Int 87: 458–464, 2015. doi: 10.1038/ki.2014.294. [DOI] [PubMed] [Google Scholar]
- 40.Neidhart JA, Mangalik A, Stidley CA, Tebich SL, Sarmiento LE, Pfile JE, Oette DH, Oldham FB. Dosing regimen of granulocyte-macrophage colony-stimulating factor to support dose-intensive chemotherapy. J Clin Oncol 10: 1460–1469, 1992. doi: 10.1200/JCO.1992.10.9.1460. [DOI] [PubMed] [Google Scholar]
- 41.Neilson EG. Pathogenesis and therapy of interstitial nephritis. Kidney Int 35: 1257–1270, 1989. doi: 10.1038/ki.1989.118. [DOI] [PubMed] [Google Scholar]
- 42.Patschan D, Müller GA. Acute kidney injury in diabetes mellitus. Int J Nephrol 2016: 6232909, 2016. doi: 10.1155/2016/6232909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Racusen LC, Solez K. Nephrotoxic tubular and interstitial lesions: morphology and classification. Toxicol Pathol 14: 45–57, 1986. doi: 10.1177/019262338601400106. [DOI] [PubMed] [Google Scholar]
- 45.Roberts J, Chen B, Curtis LM, Agarwal A, Sanders PW, Zinn KR. Detection of early changes in renal function using 99mTc-MAG3 imaging in a murine model of ischemia-reperfusion injury. Am J Physiol Renal Physiol 293: F1408–F1412, 2007. doi: 10.1152/ajprenal.00083.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodríguez-Iturbe B, García García G. The role of tubulointerstitial inflammation in the progression of chronic renal failure. Nephron Clin Pract 116: c81–c88, 2010. doi: 10.1159/000314656. [DOI] [PubMed] [Google Scholar]
- 47.Saito Y, Mori K, Tominaga K, Yokoi K, Miyazawa N. Phase II study of cisplatin as a 5-day continuous infusion with vindesine plus recombinant human granulocyte-colony-stimulating factor in the treatment of advanced non-small-cell lung cancer. Cancer Chemother Pharmacol 31: 81–84, 1992. doi: 10.1007/BF00685091. [DOI] [PubMed] [Google Scholar]
- 48.Sancho-Martínez SM, Prieto-García L, Prieto M, López-Novoa JM, López-Hernández FJ. Subcellular targets of cisplatin cytotoxicity: an integrated view. Pharmacol Ther 136: 35–55, 2012. doi: 10.1016/j.pharmthera.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 49.Schock-Kusch D, Xie Q, Shulhevich Y, Hesser J, Stsepankou D, Sadick M, Koenig S, Hoecklin F, Pill J, Gretz N. Transcutaneous assessment of renal function in conscious rats with a device for measuring FITC-sinistrin disappearance curves. Kidney Int 79: 1254–1258, 2011. doi: 10.1038/ki.2011.31. [DOI] [PubMed] [Google Scholar]
- 50.Schreiber A, Shulhevich Y, Geraci S, Hesser J, Stsepankou D, Neudecker S, Koenig S, Heinrich R, Hoecklin F, Pill J, Friedemann J, Schweda F, Gretz N, Schock-Kusch D. Transcutaneous measurement of renal function in conscious mice. Am J Physiol Renal Physiol 303: F783–F788, 2012. doi: 10.1152/ajprenal.00279.2012. [DOI] [PubMed] [Google Scholar]
- 51.Sharp CN, Doll MA, Dupre TV, Shah PP, Subathra M, Siow D, Arteel GE, Megyesi J, Beverly LJ, Siskind LJ. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am J Physiol Renal Physiol 310: F560–F568, 2016. doi: 10.1152/ajprenal.00512.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharp CN, Doll MA, Megyesi J, Oropilla GB, Beverly LJ, Siskind LJ. Sub-clinical kidney injury induced by repeated cisplatin administration results in progressive chronic kidney disease. Am J Physiol Renal Physiol 315: F161–F172, 2018. doi: 10.1152/ajprenal.00636.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Son JY, Shin JW, Wang JH, Park HJ, Kim HG, Raghavendran HR, Son CG. Chemotherapy-induced myelotoxicity and incidence of lung metastasis in an animal model. Hum Exp Toxicol 30: 649–655, 2011. doi: 10.1177/0960327110377521. [DOI] [PubMed] [Google Scholar]
- 54.Szabo Z, Alachkar N, Xia J, Mathews WB, Rabb H. Molecular imaging of the kidneys. Semin Nucl Med 41: 20–28, 2011. doi: 10.1053/j.semnuclmed.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szturz P, Wouters K, Kiyota N, Tahara M, Prabhash K, Noronha V, Castro A, Licitra L, Adelstein D, Vermorken JB. Weekly low-dose versus three-weekly high-dose cisplatin for concurrent chemoradiation in locoregionally advanced non-nasopharyngeal head and neck cancer: A systematic review and meta-analysis of aggregate data. Oncologist 22: 1056–1066, 2017. doi: 10.1634/theoncologist.2017-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takahashi N, Boysen G, Li F, Li Y, Swenberg JA. Tandem mass spectrometry measurements of creatinine in mouse plasma and urine for determining glomerular filtration rate. Kidney Int 71: 266–271, 2007. doi: 10.1038/sj.ki.5002033. [DOI] [PubMed] [Google Scholar]
- 57.Wang Y, Bellomo R. Cardiac surgery-associated acute kidney injury: risk factors, pathophysiology and treatment. Nat Rev Nephrol 13: 697–711, 2017. doi: 10.1038/nrneph.2017.119. [DOI] [PubMed] [Google Scholar]
- 58.Zager RA, Johnson ACM, Becker K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and “end-stage” kidney disease. Am J Physiol Renal Physiol 301: F1334–F1345, 2011. doi: 10.1152/ajprenal.00431.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zarjou A, Agarwal A. Sepsis and acute kidney injury. J Am Soc Nephrol 22: 999–1006, 2011. doi: 10.1681/ASN.2010050484. [DOI] [PubMed] [Google Scholar]
- 60.Zhang B, Ramesh G, Uematsu S, Akira S, Reeves WB. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J Am Soc Nephrol 19: 923–932, 2008. doi: 10.1681/ASN.2007090982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang MZ, Yao B, Yang S, Jiang L, Wang S, Fan X, Yin H, Wong K, Miyazawa T, Chen J, Chang I, Singh A, Harris RC. CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest 122: 4519–4532, 2012. doi: 10.1172/JCI60363. [DOI] [PMC free article] [PubMed] [Google Scholar]