

Abstract
Nonalcoholic fatty liver disease (NAFLD) is the most common form of chronic liver disease in the United States affecting 80–100 million Americans. NAFLD encompasses a spectrum of diseases ranging from excess liver fat (nonalcoholic fatty liver or NAFL), to necro-inflammation (nonalcoholic steatohepatitis or NASH), to fibrosis/ cirrhosis, and malignant transformation (hepatocellular carcinoma). Susceptibility to NAFLD is highly variable and it remains unclear why some patients with NAFLD exhibit NASH, whereas patients with known risk factors have NAFL only. The reasons for this variability can be a partially attributed to differences in genetic background. In the last decade, there have been multiple genome wide association studies, which have enriched our understanding of the genetic basis of NAFLD. The I148M PNPLA3 (patatin-like phospholipase domain-containing protein 3) variant has been identified as the major common genetic determinant of NAFLD. Variants with moderate effect size like TM6SF2, MBOAT7 and GCKR have also been shown to have a significant contribution. New research has uncovered major pathways leading to disease development and progression; therefore, multiple medications are being developed and tested for the treatment of advanced NAFLD. These agents target metabolic mechanisms as well as inflammation and fibrosis pathways. Several randomized clinical trials (RCTs) are evaluating the efficacy of these novel agents on histological improvement of disease severity and decreasing liver-related outcomes. FDA-approved medications for NASH and NASH-related fibrosis are expected by 2020.
Keywords: Nonalcoholic fatty liver disease (NAFLD), nonalcoholic steatohepatitis (NASH), treatment, polymorphism, clinical trial
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most prevalent etiology of abnormal liver enzyme tests in the United States (1). The term NAFLD ranges from the relatively benign nonalcoholic fatty liver (NAFL) to the aggressive nonalcoholic steatohepatitis (NASH) to fibrosis and eventually cirrhosis, in the absence of excessive alcohol consumption (2). NAFL is defined as the presence of at least 5% hepatic steatosis (HS) without evidence of hepatocellular injury or fibrosis. NASH is defined as the presence of HS with inflammation and hepatocyte injury such as ballooning, with or without liver fibrosis (3). By the year 2020, NASH is anticipated to be the most common indication for liver transplantation among adults in the USA (4).
In this article, we discuss the genetic and epigenetic factors of NAFLD progression, the roles of proteins encoded by the common risk variants in NAFLD pathogenesis, and the potential to translate that into the design of novel diagnostic and therapeutic approaches to improve patient stratification and management. Furthermore, we will examine medications that are currently undergoing phase III randomized clinical trials (RCTs) for the treatment of NASH as well as their mechanisms of action.
Risk factors and epidemiology
HS is commonly associated with obesity, type 2 diabetes mellitus (T2DM), and hyperlipidemia; all components of the metabolic syndrome. In terms of epidemiology, a recent meta-analysis of studies that involved more than 8.5 million persons from 22 countries showed that the global prevalence of NAFLD diagnosed by imaging was 25.24% with highest prevalence in the Middle East at 31.8% and South America at 30.5% and lowest in Africa at 13.5%. It also showed that more than 50% of patients with NAFLD were overweight or obese, 70% had dyslipidemia, and 23% had T2DM (5).
Gender has an important influence on the prevalence of NAFLD. Multiple manuscripts demonstrated higher predominance of NAFLD in men compared to women (6-8).
The genetic risk of fatty liver disease
Hereditary modifiers have a significant impact on the susceptibility to NAFLD and the risk for progression to advanced disease. Significant inter-individual variability has been observed. Some patients will have NAFL only, whereas others might develop NASH, fibrosis and eventually cirrhosis. Studies have identified at least four genetic variants in four distinct genes which are associated with the evolution and progression of NAFLD. Their proteins are involved in the regulation of hepatic lipid metabolism (Figure 1). However, only two genetic variants (PNPLA3 and TM6SF2) were replicated in different racial populations and geographic areas indicating broader clinical significance. Table 1 provides a summary of genetic polymorphisms that are associated with susceptibility to NAFLD.
Figure 1.
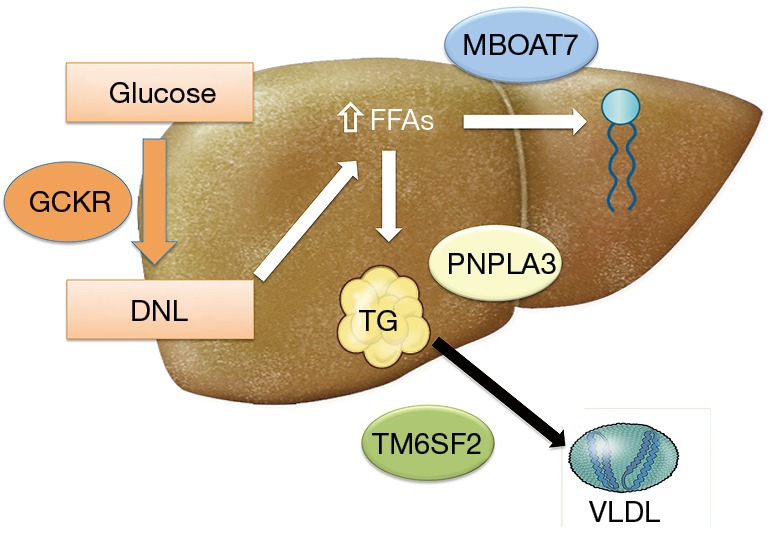
Different gene play a role in the development and progression of NAFLD. PNPLA3 gene product plays a role in lipid droplet remodeling; TM6SF2 product is involved in VLDL secretion leading to lipid accumulation in the liver while decreasing cardiovascular risk; GCKR gene is involved in de novo lipogenesis, and MBOAT7 is involved in remodeling of phosphatidylinositol. VLDL, very low-density lipoprotein.
Table 1. Genetic polymorphisms associated with susceptibility to NAFLD.
| Gene | Variant | Function | Phenotype | Impact | Effect |
|---|---|---|---|---|---|
| PNPLA3 | rs738409 | Lipid droplets remodeling | ↑NAFLD, ↑fibrosis | Loss-of-function | p.I148M |
| TM6SF2 | rs12137855 | VLDL secretion | ↑NAFLD, ↑fibrosis | Loss-of-function | p.E167k |
| GCKR | rs1260326 | Regulation of de novo lipogenesis | ↑NAFLD, ↑fibrosis | Loss-of-function | p.P446L |
| MBOAT7 | rs641738 | Remodeling of phosphatidylinositol | ↑NAFLD, ↑fibrosis | Loss-of-function | Protein change |
NAFLD, nonalcoholic fatty liver disease; VLDL, very low-density lipoprotein.
Genes associated with susceptibility to NAFLD
PNPLA3 gene
The PNPLA3 is the most replicated modifier NAFLD pathogenesis in different ethnicities (9). In this variant cytosine substitutes guanine which changes codon 148 from isoleucine to methionine (rs738409 C>G encoding for PNPLA3 I148M) (10). The wild type PNPLA3 has a hydrolysis function for triglycerides and retinyl esters. This enzymatic activity is lost with the substitution of the I148M which consequently leads to an accumulation of triglycerides and retinyl esters within hepatocytes (11). Studies have suggested that the severity of steatohepatitis and fibrosis related to the presence of rs738409 (I148M) variant (12). Pirazzi et al. (13) demonstrated in his study that PNPLA3 is highly prevalent in human hepatic stellate cells (HSCs). The wild-type PNPLA3 hydrolyzes retinyl palmitate into retinol and palmitic acid; however, the I148M variant reduces this enzymatic activity significantly. These findings indicate a possible association between HSCs, retinoid metabolism, and PNPLA3 in defining the tendency to hepatic fibrosis. Additional studies are required before this mechanism can be determined as relevant. Some researchers have also reported an association between the I148 variant and hepatocellular carcinoma in alcoholic liver disease (14) and chronic viral hepatitis (15).
Conflicting data has been reported in vitro studies. One study demonstrated the deletion of PNPLA3 did not lead to triglyceride accumulation in the hepatic cells (16). In other studies, expression of PNPLA3-I148M, but not wild type PNPLA3, caused steatosis (17). More recent study revealed that hepatic triglyceride content (HTGC) was associated with 40-fold increase in the PNPLA3-I148M which is consistent with a predominant loss of function effect of I148 polymorphism (18).
The association between NAFLD and PNPLA3 was identified in the first genome wide association study (GWAS) which evaluated North American population of diverse ethnicity and used non-invasive proton magnetic resonance spectroscopy (H-MRS) to measure the hepatic triglyceride content (HTGC) accumulation (10). This association was also validated in another GWAS that used CT to measure HS (19). Two Japanese histologically-based studies confirmed this association as well (20,21). A study conducted in European and Indian-Asian populations (22) and a cohort study of 61,089 individuals (23) revealed that PNPLA3 was associated with elevated serum ALT levels.
TM6SF2 gene
Within the hepatocytes, Transmembrane 6 superfamily member 2 (TM6SF2) plays a crucial role in the enrichment of triglycerides to apolipoproteins B 100 in the pathway of very low-density lipoprotein secretion. The variant is an adenine for guanine substitution in coding nucleotide 499, which subrogates glutamate at residue 167 with lysine. This substitution leads to a loss of activity and subsequently causes an elevated liver triglyceride content and a decreased serum lipoprotein level. Therefore, individuals who carry this variant have higher risk of NAFLD but lower risk for cardiovascular (CV) disease (24). Contrariwise, overexpression of TM6SF2 decreases the number and the size of lipid droplets and eventually lowers the liver cell steatosis (21,25). Hence, the major allele is linked to hyperlipidemia and increased CVD risk (21).
Although the TM6SF2 rs58542926 minor allele was demonstrated in multiple large cohorts for NAFLD phenotypes ranging from steatosis to fibrosis and cirrhosis (26,27), some studies have failed to prove that due to low minor allele frequency (28). In 2014, Kozlitina et al. identified a genetic variant in the TM6SF2, was associated with H-MRS quantified HTGC (24). Another study in 2,300 individuals validated the association between PNPLA3 and NAFLD, and identified a locus adjacent TM6SF2 (29).
MBOAT7 gene
Phospholipids have numerous structural and functional roles in cells and after they are formed, they undergo modifications by a remodeling pathway called the Lands cycle. Inside this cycle, the membrane-bound O-acyltransferase domain containing 7 (MBOAT7) plays an essential role in remodeling the phosphatidylinositol with arachidonic acid. The rs641738 C>T variant attached to the 3’ untranslated region of MBOAT7 is a commonly associated with reduced levels of phosphatidylinositol containing arachidonic acid within the hepatocytes and circulation through the down regulation of MBOAT7 at an mRNA and protein level (30). This MBOAT7 variant is associated with the risk of developing NAFLD among Caucasians.
GCKR gene
The GCKR gene product, the glucokinase regulatory protein (GCKRP), controls de novo lipogenesis by regulating the flux of glucose into hepatocytes. A common missense loss of function GCKR mutation (rs1260326) encoding for the P446L protein variant results in hepatic fat accumulation (31). Studies have demonstrated that the GCKRP L466 variant produces a protein that has reduced regulation by physiological concentrations of fructose 6 phosphate, therefore causing a persistent raise in GCKR activity. This increased activity in the liver will promote the glycolytic influx, consequently enhancing hepatic glucose metabolism and increasing the concentrations of malonyl-CoA (22), which inhibits carnitine-palmitoyltransferase blocking fatty acid oxidation (31) and increasing HTGC.
In the largest GWAS meta-analysis for NAFLD, Speliotes et al. analyzed four cohort studies (Age/Gene/Environment Susceptibility-Reykjavik Study, Old Order Amish Study, Family Heart Study, and Framingham Heart Study) in European and American ancestry. CT was used to measure steatosis. PNPLA3 and GCKR were identified and associated with increased HTGC (23).
Epigenetic factors associated with NAFLD
Epigenetic factors cause heritable changes in gene expression and phenotypic variation without directly altering the DNA code itself. These modifications might be caused by alterations in DNA methylation, remodeling of chromatin and RNA based mechanisms, such as non-coding RNAs. Their interaction with the hereditary factors defines an individual’s susceptibility towards NAFLD (32). For instance, animal studies demonstrated that exposure to high fat diet inside the uterus causes demethylation of hepatic DNA which eventually leads to NASH (33,34). DNA methylation interposes the effect of ageing on an individual’s susceptibility to NAFLD (35), and is also involved in the regulation of PNPLA3 gene expression (36). Interestingly, some epigenetic alterations could be reversible, as studies revealed partial reversibility of the methylation of genes associated insulin signalling and intermediate metabolism after bariatric surgeries (37).
Down regulation of miRNA-122, the most abundant hepatic miRNA, has been demonstrated in several studies (38). Conversely, the levels of miR-122 in the plasma are elevated in patients with NASH (39), which might represent a new approach to estimate and noninvasively monitor liver disease severity.
Interactions between gene variants and environmental factors
The complicated interaction between the genetic and environmental factors will influence the NAFLD susceptibility and progression. A recent study evaluated the influence of the major three genetic variants of NAFLD (PNPLA3, TM6SF2 and GCKR) in individuals stratified for adiposity. Interestingly, the study demonstrated that the effect size of the association between the variants and the risk of NAFLD and cirrhosis is proportional to the body mass index (40). Obesity might amplify the effects of the risk alleles by altering their expression. For example, PNPLA3 is a direct target of the insulin-regulated transcription factor sterol regulatory element binding protein-1c (SREBP-1c) and is regulated by fasting and re-feeding (41). GCKR expression is also increased by glucose and insulin (42). Therefore, the insulin resistance associated with obesity may increase expression of PNPLA3 and GCKR. However, TM6SF2 does not respond to food intake (43).
PNPLA3, TM6SF2 and MBOAT7 were identified as genetic variants for alcoholic liver cirrhosis (44). Adiposity was also found to amplify the effect of alcohol consumption on liver disease (45) by its actions on PNPLA3 (46), which indicates that NAFLD and alcoholic liver disease have similar mechanisms of disease development, which are activated by obesity and insulin resistance on the one hand, and excessive alcohol consumption on the other.
Clinical implications
The identified NAFLD risk loci might be helpful in developing risk algorithms to improve patient stratification and managements. Up until now, none of these variants by itself has successfully translated into a clinical tool that merits diagnostic application. However, the development of a polygenic score of the cumulative effects of the genetic loci in conjunction with clinical and laboratory factors can offer new risk prediction models to screen high-risk individuals (47). Hopefully, in the near future, we will be able to identify new genetic loci, add more biomarkers such as epigenetic and immunological factors that will increase the tool’s precision.
Treatment
Current therapy
Currently, NAFLD is treated with life style modification such as weight loss and exercise as there has not been an approved pharmacologic therapy yet.
In a prospective study about the effects of weight loss on NAFLD, 90% of patients had resolution of NASH and 45% of patients had fibrosis retrogression when they achieved weight loss ≥10%. However, only 30% of subjects were able to lose ≥5% of their weight and only 10% were able to lose ≥10% (48). Other current treatments for NAFLD focus on treating any associated metabolic comorbidities (3). Vitamin E and pioglitazone are recommended for patients with NASH, although there are concerns regarding side effects (49), and treatment should be limited to patients with biopsy-proven NASH.
Taking into consideration the role of intestinal microbiota in the pathogenesis of NAFLD, probiotics, antibiotics and prebiotics might play a therapeutic role in the NAFLD via modulating the gut microbiota (50) but more evidence is needed before making any recommendations. Genetic screening for polymorphisms that identify individuals at high risk for NASH and advanced fibrosis might further improve allocation to the appropriate therapeutic agent (51).
Upcoming therapeutic targets
With increased understanding of the pathogenesis of NAFLD, numerous therapeutic agents were developed. Medications that inhibit recruitment of inflammatory cells/block inflammatory signaling, reduce oxidative or cellular stress, modulate bile acid signaling, and improve insulin sensitivity in preclinical models are being tested in patients with NASH. Elafibranor, obeticholic acid, cenicriviroc, and selonsertib are four medications that are undergoing phase III RCTs. Table 2 provides a concise summary about the clinical development of these medications.
Table 2. New agents in phase III RCTs for the treatment of NASH and NASH fibrosis.
| Medication | Mechanism | Effective dose (mg/day) | Phase II efficacy data | Phase III RCT | Planned interim analysis duration (weeks) | |
|---|---|---|---|---|---|---|
| Resolution of NASH | Decrease in fibrosis stage | |||||
| Obeticholic acid | FXR agonist | 10–25 | No | Yes | REGENERATE (NCT02548351) | 72 |
| Elafibranor | PPARα/δ agonist | 120 | Yes | No | RESOLVE-IT (NCT02704403) | 72 |
| Cenicriviroc | CCR2/CCR5 antagonist | 150 | No | Yes | AURORA (NCT03028740) | 52 |
| Selonsertib | ASK1 inhibitor | 6 and 18 | No | Yes | STELLAR 3 (NCT03053050), STELLAR-4 (NCT03053063) | 48 |
NASH, nonalcoholic steatohepatitis; RCT, randomized clinical trial; FXR, farnesoid X receptors; PPAR, peroxisome proliferator-activated receptor; CCR, C-C chemokine receptor.
Peroxisome proliferator-activated receptor (PPAR) agonist
PPARs are a family of ligand-activated transcription factors that regulate some metabolic processes, such as lipid and glucose homeostasis (52,53). PPARα is expressed in the liver and other metabolically active tissues (52); its activity lowers lipid levels, and leads the expression of genes that regulate fatty acid β-oxidation, lipid transport, and the hormone fibroblast growth factor (FGF)-21 (53). PPARδ is highly expressed in hepatocytes, it participates in fatty acid oxidation, decreases hepatic glucose production, and improves insulin sensitivity (52,53).
Elafibranor functions as a dual PPARα/δ agonist, thus it improves glucose metabolism and insulin sensitivity and decreases inflammation. A phase IIb RCT (NCT01694849, GOLDEN-505), studied the effects of elafibranor (120, 80 mg/day, or placebo) for 52 weeks. Its primary endpoint was the reversal of NASH without worsening fibrosis (54). By using a modified definition of NASH resolution that emphasized the disappearance of hepatocyte ballooning, NASH was resolved in 19% of patients in the elafibranor high dose group compared with 9% in the placebo group (P=0.045) (54).
A phase III RCT (NCT02704403, RESOLVE-IT) is recruiting patients to assess the effects of elafibranor (120 mg/day or placebo) for 72 weeks on liver histology. Patients will also be followed for long-term outcomes, including cirrhosis, mortality, and other liver-related clinical events, over approximately 4 years. Additional novel PPAR agonists that are being tested for NAFLD include saroglitazar and lanifibranor.
Farnesoid X receptor agonist
Farnesoid X receptors (FXR) is a nuclear receptor that is highly expressed in the liver and small intestine (55). Bile acids (BAs) are the natural ligand of FXR (55) and together, they regulate lipid/glucose homeostasis, promote insulin sensitivity, and potentially modulate liver fibrosis (56).
Obeticholic acid (OCA, 6-ethylchenodeoxycholic acid) is a synthetic bile acid and an activator of the Farnesoid X receptors (57). In a phase IIb RCT (NCT01265498, FLINT), OCA (25 mg/day or placebo) for 72 weeks was evaluated for the treatment of patients with histological evidence of NASH based upon a liver biopsy and a histological NAFLD activity score (NAS) score of 4 or greater. The primary outcome was a decrease in NAS by 2 points without worsening fibrosis (57).
A larger number of patients in the OCA group had a histological improvement compared to those in the placebo group (45% vs. 21%, P=0.0002); additionally, 35% of the OCA group has achieved improvement in fibrosis compared to 19% of the placebo (P=0.004). Despite the histological improvement and improved fibrosis, there was no difference in the resolution of NASH between the two groups (22% in the OCA group vs. 13% in the placebo group, P=0.08) (57). Although the FLINT study showed that OCA is effective, long-term safety and tolerability need to be assessed.
A phase III RCT (NCT02548351, REGENERATE) is currently recruiting patients with biopsy-proven NASH to assess the effectiveness of OCA (10, 25 mg/day, or placebo) for 72 weeks. Long-term effects, including histological progression and liver-related morbidity and mortality, will also be evaluated through a 7-year period. Several other FXR agonists are being studied for the treatment of NASH including GS-9674, which is currently in a phase II RCT (NCT02854605).
CC chemokine receptor type 2/5 antagoni
The inflammatory cascade is activated after the hepatocyte injury. It starts when the Kupffer cells (KCs) recruit additional macrophages and monocytes to the site of injury, which then produce pro-inflammatory cytokines and chemokines. This leads to additional involvement of inflammatory cells as well as the activation of hepatic stellate cells (HSCs) (58). KCs, monocytes, and HSCs all express C-C chemokine receptor types 2 (CCR2) and CCR5 which enhance the inflammatory response in hepatic injury (58) and eventually leads to hepatic fibrogenesis.
Cenicriviroc (CVC) functions as a dual antagonist of CCR2 and CCR5. It demonstrated abilities to decrease fibrosis in preclinical models. A phase IIb RCT (NCT02217475, CENTAUR) evaluated the effectiveness of CVC (150 mg/day or placebo) for the treatment of NASH with liver fibrosis. The primary endpoint was histological improvement of NAS without worsening fibrosis. The study included 289 patients with NASH, liver fibrosis, and diabetes mellitus/metabolic syndrome (38). There was no significant improvement in terms of NASH resolution after receiving CVC for twelve months. However, liver fibrosis improved (least one stage) in 20% of patients in the CVC arm as compared to 10% in the placebo arm (P=0.02) (38).
These findings justify the rationale to directly target inflammatory mechanisms of NASH separate from the underlying metabolic liver disease. A phase III RCT is recruiting patients with NASH to evaluate the effectiveness of CVC (150 mg/day or placebo) on liver fibrosis (NCT03028740, AURORA). Histological endpoints will be assessed at 12 months, while long-term outcomes will be evaluated over a 5-year period to determine the incidence of cirrhosis, liver-related mortality and morbidity.
Apoptosis signal-regulating kinase 1 (ASK1) inhibitor
ASK1 is a member of the mitogen-activated protein kinase (MAP3k) family that has a significant role in stress responses, including cell death, differentiation, and production of inflammatory cytokines (59). Hepatocyte injury and apoptosis are both driven by tumor necrosis factor (TNF)-α signaling. The TNF-α, oxidative stress, and endoplasmic reticulum stress all activate ASK1, which can then lead to hepatic inflammation, hepatocyte apoptosis, and fibrosis (53).
Selonsertib (SEL, GS-4997) is an oral, selective inhibitor of ASK1. A phase II RCT (NCT02466516) evaluated the effects of SEL (6 or 18 mg/day) in patients with NASH and fibrosis (60). Patients received SEL showed more fibrosis improvement than the placebo arm with up to 43% of the subjects in the 18 mg/day SEL arm showing improvement by at least 1 fibrosis stage after 24 weeks of treatment (60). The findings warrant additional studies to evaluate the long-term effects of SEL on NASH fibrosis.
Two phase III studies recently completed recruiting patients with NASH and bridging fibrosis (NCT03053050, STELLAR-3) and compensated cirrhosis (NCT03053063, STELLAR-4) to assess the effects of SEL (6, 18 mg/day, or placebo). Both studies will evaluate patients for histological improvement at week 48 and monitor for long-term outcomes through week 240.
The future of NAFLD management
We anticipate that the clinical approach of NAFLD will be similar to that of type 2 diabetes. Genetic testing and noninvasive methods will help identify the severity of NAFLD without the need for liver biopsy. Patients will be divided into 3 categories and will be managed accordingly: (I) patients with NAFL, the relatively benign form of the disease, will be managed similar to those with pre-diabetes with a focus on lifestyle modifications/weight loss through diet and exercise; (II) patients with NASH and fibrosis, the aggressive form of the disease, will be managed similar to those with established type 2 diabetes with pharmacologic treatment with different classes of medications regardless of weight loss; (III) patients with NASH cirrhosis, the final stage of disease progression, will be managed similar to those with diabetes complications such as chronic kidney disease where the focus will be on preventing/treating complications with liver transplantation as a last resort. This approach to NAFLD management is summarized in Figure 2.
Figure 2.

The future of NAFLD management. NAFLD patients will be divided into different disease stages and managed in a similar fashion to the current management of individuals within the type 2 diabetes spectrum. OCA, obeticholic acid; CVC, cenicriviroc; SEL, selonsertib; CAD, coronary artery disease; CKD, chronic kidney disease; HCC, hepatocellular carcinoma; EV, esophageal varices; NAFLD, nonalcoholic fatty liver disease;
Conclusions
NAFLD is a complicated disease in which genetic variations and environmental factors interact to define disease phenotype and progression. It is associated with adiposity, T2DM, hyperlipidemia, and the metabolic syndrome. Gene-adiposity interaction has a major role in the development and progression of NAFLD. The identified NAFLD risk loci might be helpful in developing algorithms to improve patient stratification and management. However, the clinical implications of the genetic and epigenetic discoveries are still developing and GWAS performed for NAFLD have been small in terms of participant number. Future studies should involve larger number patients at risk (obesity and type 2 diabetes) might be more successful in identifying new genetic variant.
Additional research is necessary to help develop successful treatment for NASH. Currently, there is no FDA-approved treatment for NASH. The recommendation for lifestyle intervention, which includes weight loss, diet, and exercise, has found positive, but short-lived results. Dozens of medications are being tested in preclinical and clinical trials and are focused on different mechanisms of action including metabolic, anti-inflammatory, and anti-fibrotics targets. However, additional studies are necessary to identify the most effective treatment for NASH in different patient populations as it is becoming more prevalent in the world.
Acknowledgements
None.
Footnotes
Conflicts of Interest: N Alkhouri is on the speaker bureau for Intercept Pharmaceuticals and Gilead Sciences and received research funding from Intercept Pharmaceuticals, Allergan, and Gilead Sciences. The other authors have no conflicts of interest to declare.
References
- 1.Clark JM, Brancati FL, Diehl AM. The prevalence and etiology of elevated aminotransferase levels in the United States. Am J Gastroenterol 2003;98:960-7. 10.1111/j.1572-0241.2003.07486.x [DOI] [PubMed] [Google Scholar]
- 2.Anstee QM, McPherson S, Day CP. How big a problem is non-alcoholic fatty liver disease? BMJ 2011;343:d3897. 10.1136/bmj.d3897 [DOI] [PubMed] [Google Scholar]
- 3.Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328-57. 10.1002/hep.29367 [DOI] [PubMed] [Google Scholar]
- 4.Charlton MR, Burns JM, Pedersen RA, et al. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology 2011;141:1249-53. 10.1053/j.gastro.2011.06.061 [DOI] [PubMed] [Google Scholar]
- 5.Younossi ZM, Koenig AB, Abdelatif D, et al. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73-84. 10.1002/hep.28431 [DOI] [PubMed] [Google Scholar]
- 6.Chen ZW, Chen LY, Dai HL, et al. Relationship between alanine aminotransferase levels and metabolic syndrome in nonalcoholic fatty liver disease. J Zhejiang Univ Sci B 2008;9:616-22. 10.1631/jzus.B0720016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amarapurkar D, Kamani P, Patel N, et al. Prevalence of non-alcoholic fatty liver disease: population based study. Ann Hepatol 2007;6:161-3. [PubMed] [Google Scholar]
- 8.Browning JD, Szczepaniak LS, Dobbins R, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 2004;40:1387-95. 10.1002/hep.20466 [DOI] [PubMed] [Google Scholar]
- 9.Singal AG, Manjunath H, Yopp AC, et al. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: a meta-analysis. Am J Gastroenterol 2014;109:325-34. 10.1038/ajg.2013.476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461-5. 10.1038/ng.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pingitore P, Pirazzi C, Mancina RM, et al. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim Biophys Acta 2014;1841:574-80. 10.1016/j.bbalip.2013.12.006 [DOI] [PubMed] [Google Scholar]
- 12.Sookoian S, Castano GO, Burgueno AL, et al. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J Lipid Res 2009;50:2111-6. 10.1194/jlr.P900013-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pirazzi C, Valenti L, Motta BM, et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum Mol Genet 2014;23:4077-85. 10.1093/hmg/ddu121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guyot E, Sutton A, Rufat P, et al. PNPLA3 rs738409, hepatocellular carcinoma occurrence and risk model prediction in patients with cirrhosis. J Hepatol 2013;58:312-8. 10.1016/j.jhep.2012.09.036 [DOI] [PubMed] [Google Scholar]
- 15.Valenti L, Rumi M, Galmozzi E, et al. Patatin-like phospholipase domain-containing 3 I148M polymorphism, steatosis, and liver damage in chronic hepatitis C. Hepatology 2011;53:791-9. 10.1002/hep.24123 [DOI] [PubMed] [Google Scholar]
- 16.Chen W, Chang B, Li L, et al. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 2010;52:1134-42. 10.1002/hep.23812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He S, McPhaul C, Li JZ, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem 2010;285:6706-15. 10.1074/jbc.M109.064501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smagris E, BasuRay S, Li J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015;61:108-18. 10.1002/hep.27242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feitosa MF, Wojczynski MK, North KE, et al. The ERLIN1-CHUK-CWF19L1 gene cluster influences liver fat deposition and hepatic inflammation in the NHLBI Family Heart Study. Atherosclerosis 2013;228:175-80. 10.1016/j.atherosclerosis.2013.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawaguchi T, Sumida Y, Umemura A, et al. Genetic polymorphisms of the human PNPLA3 gene are strongly associated with severity of non-alcoholic fatty liver disease in Japanese. PLoS One 2012;7:e38322. 10.1371/journal.pone.0038322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holmen OL, Zhang H, Fan Y, et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat Genet 2014;46:345-51. 10.1038/ng.2926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donnelly KL, Smith CI, Schwarzenberg SJ, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115:1343-51. 10.1172/JCI23621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Speliotes EK, Yerges-Armstrong LM, Wu J, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 2011;7:e1001324. 10.1371/journal.pgen.1001324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kozlitina J, Smagris E, Stender S, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352-6. 10.1038/ng.2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mahdessian H, Taxiarchis A, Popov S, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci U S A 2014;111:8913-8. 10.1073/pnas.1323785111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu YL, Reeves HL, Burt AD, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun 2014;5:4309. 10.1038/ncomms5309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dongiovanni P, Petta S, Maglio C, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015;61:506-14. 10.1002/hep.27490 [DOI] [PubMed] [Google Scholar]
- 28.Wong VW, Wong GL, Tse CH, et al. Prevalence of the TM6SF2 variant and non-alcoholic fatty liver disease in Chinese. J Hepatol 2014;61:708-9. 10.1016/j.jhep.2014.04.047 [DOI] [PubMed] [Google Scholar]
- 29.DiStefano JK, Kingsley C, Craig Wood G, et al. Genome-wide analysis of hepatic lipid content in extreme obesity. Acta Diabetol 2015;52:373-82. 10.1007/s00592-014-0654-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mancina RM, Dongiovanni P, Petta S, et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016;150:1219-30.e6. 10.1053/j.gastro.2016.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beer NL, Tribble ND, McCulloch LJ, et al. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet 2009;18:4081-8. 10.1093/hmg/ddp357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy SK, Yang H, Moylan CA, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology 2013;145:1076-87. 10.1053/j.gastro.2013.07.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bruce KD, Cagampang FR, Argenton M, et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 2009;50:1796-808. 10.1002/hep.23205 [DOI] [PubMed] [Google Scholar]
- 34.Dudley KJ, Sloboda DM, Connor KL, et al. Offspring of mothers fed a high fat diet display hepatic cell cycle inhibition and associated changes in gene expression and DNA methylation. PLoS One 2011;6:e21662. 10.1371/journal.pone.0021662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horvath S, Erhart W, Brosch M, et al. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci U S A 2014;111:15538-43. 10.1073/pnas.1412759111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitamoto T, Kitamoto A, Ogawa Y, et al. Targeted-bisulfite sequence analysis of the methylation of CpG islands in genes encoding PNPLA3, SAMM50, and PARVB of patients with non-alcoholic fatty liver disease. J Hepatol 2015;63:494-502. 10.1016/j.jhep.2015.02.049 [DOI] [PubMed] [Google Scholar]
- 37.Ahrens M, Ammerpohl O, von Schonfels W, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab 2013;18:296-302. 10.1016/j.cmet.2013.07.004 [DOI] [PubMed] [Google Scholar]
- 38.Friedman SL, Ratziu V, Harrison SA, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018;67:1754-67. 10.1002/hep.29477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gerhard GS, DiStefano JK. Micro RNAs in the development of non-alcoholic fatty liver disease. World J Hepatol 2015;7:226-34. 10.4254/wjh.v7.i2.226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stender S, Kozlitina J, Nordestgaard BG, et al. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet 2017;49:842-7. 10.1038/ng.3855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang Y, He S, Li JZ, et al. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci U S A 2010;107:7892-7. 10.1073/pnas.1003585107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arden C, Petrie JL, Tudhope SJ, et al. Elevated glucose represses liver glucokinase and induces its regulatory protein to safeguard hepatic phosphate homeostasis. Diabetes 2011;60:3110-20. 10.2337/db11-0061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smagris E, Gilyard S, BasuRay S, et al. Inactivation of Tm6sf2, a Gene Defective in Fatty Liver Disease, Impairs Lipidation but Not Secretion of Very Low Density Lipoproteins. J Biol Chem 2016;291:10659-76. 10.1074/jbc.M116.719955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buch S, Stickel F, Trepo E, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet 2015;47:1443-8. 10.1038/ng.3417 [DOI] [PubMed] [Google Scholar]
- 45.Hart CL, Morrison DS, Batty GD, et al. Effect of body mass index and alcohol consumption on liver disease: analysis of data from two prospective cohort studies. BMJ 2010;340:c1240. 10.1136/bmj.c1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tian C, Stokowski RP, Kershenobich D, et al. Variant in PNPLA3 is associated with alcoholic liver disease. Nat Genet 2010;42:21-3. 10.1038/ng.488 [DOI] [PubMed] [Google Scholar]
- 47.Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J Hepatol 2018;68:268-79. 10.1016/j.jhep.2017.09.003 [DOI] [PubMed] [Google Scholar]
- 48.Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, et al. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015;149:367-78.e5; quiz e14-5. [DOI] [PubMed]
- 49.Townsend SA, Newsome PN. Review article: new treatments in non-alcoholic fatty liver disease. Aliment Pharmacol Ther 2017;46:494-507. 10.1111/apt.14210 [DOI] [PubMed] [Google Scholar]
- 50.Machado MV, Cortez-Pinto H. Diet, Microbiota, Obesity, and NAFLD: A Dangerous Quartet. Int J Mol Sci 2016;17:481. 10.3390/ijms17040481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anstee QM, Seth D, Day CP. Genetic Factors That Affect Risk of Alcoholic and Nonalcoholic Fatty Liver Disease. Gastroenterology 2016;150:1728-44.e7. 10.1053/j.gastro.2016.01.037 [DOI] [PubMed] [Google Scholar]
- 52.Grygiel-Górniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications – a review. Nutr J 2014;13:17. 10.1186/1475-2891-13-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rotman Y, Sanyal AJ. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut 2017;66:180-90. 10.1136/gutjnl-2016-312431 [DOI] [PubMed] [Google Scholar]
- 54.Ratziu V, Harrison SA, Francque S, et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016;150:1147-59.e5. 10.1053/j.gastro.2016.01.038 [DOI] [PubMed] [Google Scholar]
- 55.Ali AH, Carey EJ, Lindor KD. Recent advances in the development of farnesoid X receptor agonists. Ann Transl Med 2015;3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carr RM, Reid AE. FXR agonists as therapeutic agents for non-alcoholic fatty liver disease. Curr Atheroscler Rep 2015;17:500. 10.1007/s11883-015-0500-2 [DOI] [PubMed] [Google Scholar]
- 57.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 2015;385:956-65. 10.1016/S0140-6736(14)61933-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lefebvre E, Moyle G, Reshef R, et al. Antifibrotic Effects of the Dual CCR2/CCR5 Antagonist Cenicriviroc in Animal Models of Liver and Kidney Fibrosis. PLoS One 2016;11:e0158156. 10.1371/journal.pone.0158156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hayakawa R, Hayakawa T, Takeda K, et al. Therapeutic targets in the ASK1-dependent stress signaling pathways. Proc Jpn Acad Ser B Phys Biol Sci 2012;88:434-53. 10.2183/pjab.88.434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Loomba R, Lawitz E, Mantry PS, et al. The ASK1 Inhibitor Selonsertib in Patients with Nonalcoholic Steatohepatitis: A Randomized, Phase 2 Trial. Hepatology 2017. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]