

Abstract
Decreased expression of endothelial nitric oxide synthase (eNOS), a key mediator of perinatal transition, characterizes persistent pulmonary hypertension of the newborn (PPHN) in neonates and a fetal lamb model; the mechanisms are unclear. We investigated whether increased DNA CpG methylation at the eNOS promoter in estrogen response elements (EREs) and altered histone code together contribute to decreased eNOS expression in PPHN. We isolated pulmonary artery endothelial cells (PAEC) from fetal lambs with PPHN induced by prenatal ductus arteriosus constriction from 128 to 136 days gestation or gestation-matched twin controls. We measured right ventricular systolic pressure (RVSP) and Fulton index and determined eNOS expression in PAEC in control and PPHN lambs. We determined DNA CpG methylation by pyrosequencing and activity of ten eleven translocase demethylases (TET) by colorimetric assay. We quantified the occupancy of transcription factors, specificity protein 1 (Sp1), and estrogen receptors and density of four histone marks around Sp1 binding sites by chromatin immunoprecipitation (ChIP) assays. Fetal lambs with PPHN developed increased RVSP and Fulton index. Levels of eNOS mRNA and protein were decreased in PAEC from PPHN lambs. PPHN significantly increased the DNA CpG methylation in eNOS promoter and decreased TET activity in PAEC. PPHN decreased Sp1 occupancy and density of the active mark, lysine 12 acetylation of histone 4, and increased density of the repression mark, lysine 9 trimethylation of histone 3 around Sp1 binding sites in eNOS promoter. These results suggest that epigenetic modifications are primed to decrease Sp1 binding at the eNOS gene promoter in PPHN.
Keywords: DNA methylation, histone modifications, lung diseases, newborn
INTRODUCTION
Endothelial nitric oxide synthase (eNOS) is a critical mediator of perinatal pulmonary vasodilation, a physiologic event required for successful transition at birth (26, 44). Expression of eNOS increases toward term gestation in fetal lungs to prime the neonatal lung for this adaptation (36). Failure of this transition results in persistent pulmonary hypertension of the newborn (PPHN). PPHN affects 0.2% of all live births and is responsible for one-third of all neonatal mortality (22, 40). Human and animal studies have shown that eNOS expression is downregulated in PPHN human neonates and the fetal lamb model of PPHN (21, 37, 45). However, the mechanisms involved in the downregulation of eNOS gene expression in PPHN remain unknown.
Regulation of eNOS expression occurs at both transcriptional and translational levels. Transcription initiation of eNOS gene relies on the binding of RNA polymerase together with one or more transcription factor (TF) to its promoter. The proximal promoter of eNOS gene contains two binding sites for specificity protein 1 (Sp1) and four of estrogen response element (ERE) half sites (Fig. 1). Binding of Sp1 or estrogen receptors (ERs) to eNOS promoter facilitates eNOS gene transcription (50). Estrogens can upregulate eNOS in human endothelial cells by increasing eNOS promoter activity and enhancing the binding activity of the transcription factor Sp1 (17). Estrogen increases eNOS gene expression in fetal pulmonary artery endothelial cells through activation of ER at physiologic levels of estrogen (28). However, the ease of transcription factor binding to eNOS promoter depends upon accessibility, which is determined by epigenetic factors such as DNA methylation and histone modifications (4, 39).
Fig. 1.

Schematic representation of endothelial nitric oxide synthase (eNOS) proximal promoter region examined in this study. This promoter region contains two specificity protein 1 (Sp1) binding sites and four estrogen response element (ERE) half-sites. Vertical lines represent a total of 44 CpG sites examined with the locations relative to transcription start site in this region.
DNA methylation in the promoter region typically acts to repress gene transcription. Methylated CpG is now recognized as a gene-silencing signal (18, 51). DNA CpG methylation affects gene transcription by not only interfering with TF binding but also directly modulating chromatin structure via modifying the interaction between core histones and DNA (10). DNA methylation status is regulated in part by demethylation enzymes, ten-eleven translocation methylcytosine dioxygenases (TET). TET oxidize 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) (42). 5hmC and its further oxidized derivatives are subsequently replaced with an unmodified C by base-excision repair to achieve demethylation (38). Reduced 5mC oxidation due to decreased TET activity can increase DNA methylation (43).
Histone modifications, on the other hand, can act to activate or repress gene transcription. Acetylation of histones usually increases gene transcription, while methylation of histones can either increase or decrease transcription of genes depending on which amino acids in the histones are methylated (7). For example, trimethylated lysine 4 of histone H3 (H3K4me3) is associated with gene transcription activation (35) while trimethylated lysine 9 of histone H3 (H3K9me3) is related to gene transcription repression (6, 32). Recent evidence suggests that expression of eNOS is epigenetically regulated and controlled by a cell-specific histone code (11). Endothelial cells, but not a variety of nonendothelial cells, are particularly enriched in acetylated histone H3 lysine 9 (H3K9ac), H4K12ac, and H3K4me3 at the core promoter of eNOS gene. Histone modifications at eNOS promoter region has been demonstrated to be functionally relevant to eNOS expression (11).
Using a fetal lamb model of PPHN, we have previously demonstrated that PPHN significantly decreased eNOS expression in pulmonary artery endothelial cells (PAEC) (21, 23). However, whether this decrease in eNOS expression accompanies concurrent repressive epigenetic changes is so far unknown. In this study, we focused on the investigation of epigenetic changes induced by PPHN in eNOS proximal promoter, since this region contains multiple Sp1 binding sites and EREs, which are the key regulatory elements for eNOS transcription. We hypothesized that PPHN will increase DNA methylation and alter the histone code in the proximal promoter of eNOS gene primed to decrease the binding of TFs Sp1 and ERs at their corresponding binding sites. We further hypothesized that PPHN will decrease TET enzyme activity in PAEC concurrently. We investigated our hypothesis in PAEC isolated from PPHN and control fetal lambs, after they reached term gestation.
MATERIALS AND METHODS
Creation of PPHN lamb model.
This study was approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin and conformed to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Persistent pulmonary hypertension was induced by prenatal ductus arteriosus constriction as previously described (1, 3, 21, 23). Time-dated pregnant ewes at 128 days of gestation (term, 145 days) were anesthetized with inhaled isoflurane (1–3%). The fetal lamb was partially exteriorized after by a midline laparotomy and hysterotomy incisions and a left lateral thoracotomy was done on the fetus to expose the ductus arteriosus, which was ligated with an umbilical tape passed around the vessel. The fetal chest was then closed, and the lamb was placed back in the uterus. Sham-operated and gestation-matched twin fetal lambs were used as controls. The ewe and the fetus were then allowed to recover for 8 days. Previous studies have shown that these lambs consistently develop severe PPHN with this intervention (21, 23, 25, 33). The fetal lamb was delivered by Cesarean section at 136 days gestation. Right ventricular systolic pressure (RVSP) was measured with a Millar pressure catheter (Mikro-Tip pressure catheter, size 3F; AD Instruments, Colorado Springs, CO) inserted into the right ventricle (RV) chamber and connected to appropriate amplifier and data acquisition system (AD Instruments). Heart and lungs were harvested immediately after euthanasia of the fetus and the ewe. Fulton index of the fetal heart was determined as the ratio of RV to LV + septum weights.
Cell culture.
The pulmonary arteries were dissected and isolated from the fetal lungs and PAEC were isolated from third-fifth generation pulmonary arteries by collagenase digestion, as we previously described (2, 3, 21). PAEC isolated from control (normal) and PPHN fetal lambs were cultured in DMEM with 20% fetal calf serum at 37°C in room air and 5% CO2. We used PAEC between passages 2 and 4 for our experiments. Endothelial cell identity was verified by staining for factor VIII antigen and by acetylated LDL uptake (2, 21).
Endothelial NOS expression.
PAEC from control and PPHN lambs were grown to near confluence in 100 mm flasks for RNA and protein extraction and in six-well plates for cyclic GMP (cGMP) measurement as an indicator of NOS catalytic function. RNA was extracted from PAEC with Trizol reagent (cat. #15596018; Ambion, Waltham, MA) and 500 ng of RNA was used as a template for cDNA synthesis reaction (cat. #11754, Superscript Vilo cDNA synthesis kit; Invitrogen, Waltham, MA). Real-time PCR to amplify mRNA for eNOS and GAPDH, used as internal control, was done using specific primers and SYBR Green master mix (cat. #A25741, Applied Biosystems, Waltham, MA). The primers for GAPDH were: forward: 5′-CTGGCCAAGGTCATCCAT-3′ and reverse: 5′-ACAGTCTTCTGGGTGGCAGT-3′. The primers for eNOS were: forward: 5′-TCTTCCACCAGGAGATGGTC-3′ and reverse: 5′-AGAGGC GTACAGGATGGTTG-3′. Levels of eNOS mRNA relative to GAPDH mRNA in control samples was normalized to 1, and the level of eNOS mRNA relative to GAPDH in PPHN samples was shown as fold change from control, per methods previously described (1, 34).
Cell lysates of PAEC for immunoblotting were prepared in modified RIPA buffer as we described previously (21). Protein content of the lysates was determined by bicinchoninic acid assay. Proteins in the sample were resolved by SDS-PAGE and separated proteins were transferred to nitrocellulose membranes and were blotted with specific monoclonal antibodies for eNOS (Invitrogen, cat. # 33-4600) and β-actin (cat. #A2228; Sigma-Aldrich, St. Louis, MO) overnight at 4°C. The membranes were blotted with horseradish peroxidase-conjugated anti-mouse IgG antibody (1:10,000; Bio-Rad, Hercules, CA) and exposed to CL-XPosure film (Thermo Fisher Scientific, Waltham, MA) after treatment with SuperSignal West Pico (Thermo Fisher Scientific). The signals were analyzed with ImageJ, and eNOS band density was normalized to the loading control, β-actin.
eNOS function.
PAEC in six-well plates were placed in serum-free medium overnight and washed with Hanks’ balanced salt solution (HBSS). NO bioavailability was assessed by accumulation of its downstream mediator, cGMP in the cells, detected with an enzyme linked immunosorbent assay (ELISA) method (cat. #ADI-900-164; Enzo Lifesciences, Farmingdale, NY) as we reported previously (23). Control and PPHN PAEC were exposed to HBSS with or without 10−5 M ATP for 15 min. The reaction was stopped by the addition of cell lysis buffer, and cGMP content of the cell lysate was measured by ELISA. The protein concentration in each well was measured to calculate the amount of cGMP in picoM per milligram of protein. The cGMP levels measured in unstimulated (basal) and ATP-stimulated cells were compared between control and PPHN cells as a measure of eNOS function.
DNA isolation and sodium bisulfite pyrosequencing.
PAEC genomic DNA was extracted from eight different control and PPHN lambs using DNeasy Blood & Tissue kit (cat. #69506; Qiagen, Valencia, CA) per the manufacturer’s protocol. Genomic DNA was then subjected to sodium bisulfite modification using EZ DNA Methylation-Lightning Kit (cat. #D5031; Zymo Research, Irvine, CA) per the manufacturer’s protocol to determine the site-specific CpG methylation. DNA methylation of the validation-set samples was determined through PCR amplification with biotinylated primers (Integrated DNA Technologies, Coralville, IA) using Pyromark Assay Design Software version 2.0 (Table 1) and Pyromark Q96 MD pyrosequencer (Qiagen) as previously described (14). Amplified products were confirmed with agarose gel electrophoresis. Methylation was quantified with the provided software (Qiagen). Ten primer sets (Table 1) were used to examine DNA CpG methylation status in the proximal promoter of eNOS gene. Promoter sequence we examined contains 44 CpG sites spanning from −1 to −1500 nucleotide position from transcription start site.
Table 1.
Primers for pyrosequencing
| Forward | Reverse | Sequencing |
|---|---|---|
| Set 1: 5′-GGGTTTTTGGGGATATAGGTTT | 5′Biosg/CCCTAAATCCCAACTTCCTACCCTTTTATA | 5′-GGTTTGGTATTAGGGTATTTTA |
| Set 2: 5′-TTGGGGGATATAAAAGGGTAGGAA | 5′Biosg/CCAAAACTCCACCATCTCCCCAATAA | 5′-ATATAAAAGGGTAGGAAGT |
| Set 3: 5′-GTGGGGGGTAGGTTAGTA | 5′Biosg/CCTTCTCCAACCCCTAAATAACTT | 5′-GGTAGGGATTGTTTATT |
| Set 4: 5′-AAGTTATTTAGGGGTTGGAGAAG | 5′Biosg/CCCATCATATCTTCCACTACTTTTC | 5′-GGGGTTGGAGAAGGG |
| Set 5: 5′-GATAGGGAGAGGTAGATAGGTAGATAAA | 5′Biosg/CTCACCCTCTTCCCTACAA | 5′-GTGTTGGTTGAGTTTTT |
| Set 6: 5′-TGTTTTAGTTTTGGTGGGGAGAA | 5′Biosg/CCTAAACTCCCAAAAACTTTACTAC | 5′-TTAGTGGAAGGGTGG |
| Set 7: 5′-GGGGTAGGTGAATTTAGAAGTAT | 5′Biosg/AAAAAAAACCATTCTCAATCATATACATTA | 5′-TTAGTGGAAGGGTGG |
| Set 8: 5′-TTTATAGGGAAGGGTTGTGG | 5′Biosg/TAAACCCCATTTCTAACTCCCATA | 5′-AGGGTTGTGGTTAGG |
| Set 9: 5′-AAGGTTTTAGGAAGGGTTAAAGTA | 5′Biosg/TCCAAATTCCAACTAACAACTATAACT | 5′-GTTTTGATTTTGAGATGGAT |
| Set 10: 5′-GGGTATGAGGGTGGTATAAGTAA | 5′Biosg/ATCTCAAAATCAAAACTAACCTCATACTT | 5′-AGGGTGGTATAAGTAAG |
Nuclear extraction and TET activity assay.
PAEC from four control and four PPHN lambs were harvested at 70% confluence. Nuclear extraction of the cells was performed as described by Luo et al. (27). The activity of TET family of 5mC dioxygenases was determined with the Epigenase 5mC Hydroxylase TET Activity/Inhibition Assay Kit (cat. # P-3086; Epigentek, Farmingdale, NY). The assay is based on the conversion of methylated substrate present in each well by TET enzyme present in the sample to hydroxymethylated product, which is detected by a specific antibody. The amount of hydroxymethylated product, which is proportional to TET activity in the sample, was measured colorimetrically by the absorbance in a microplate spectrophotometer at a wavelength of 450 nm. The individual value for TET activity in each sample was then derived from the absorbance of standards supplied with the kit and expressed as ng·min−1·mg−1 of protein in each sample. Each assay was done in triplicate and the average of three measurements was used for TET activity for each sample.
Chromatin isolation and chromatin immunoprecipitation assay.
PAEC chromatin was isolated from six different control and PPHN lambs with the tryChIP chromatin shearing reagent kit (#520154, Covaris) and focused-ultrasonicator M220 (Covaris). Chromatin immunoprecipitation (ChIP) was done with antibodies against ERα (sc-7207x; Santa Cruz Biotechnology, Santa Cruz, CA), ERβ (GTX23577; GeneTex, Irvine, CA), Sp1(sc-59x, Santa Cruz Biotechnology), H3K4me3 (cat. #39915; Active Motif, Carlsbad, CA), H3K9ac (cat. #39917, Active Motif), H3K9me3 (cat. #39765, Active Motif), and H4K12ac (cat. #39165, Active Motif) and were performed using EZ-Magna ChIP HT96 kit (cat. #17-10078; EMD Millipore, Temecula, CA). Real-time PCR was used to quantitate the amount of DNA from eNOS promoter regions of two Sp1 binding sites and three proximal ERE half-sites; 5% of input was used as a loading control (13, 16). The PCR primers are listed in Table 2. Two control experiments were performed simultaneously with our ChIP experiments. First, we performed a “mock” ChIP that included input but did not utilize antibody. Second, we performed a ChIP that utilized an anti-rabbit secondary antibody as negative control.
Table 2.
Primers for ChIP assays
| Forward | Reverse | Probe | |
|---|---|---|---|
| −209 Sp site | 5′-AGCTTCCTGCCCTTTTGTGTC | 5′-TTCCGGCCCCTATTTCCT | 5′-CCCAACTTGAGTCACAGG |
| −335 half ERE site | 5′-GCTCCACGATCTCCCCAGT | 5′-TCCAGTGACGAATGCTTCCC | 5′-TAGGTCAGCAGACAGACC |
| −879 half ERE site | 5′-GGCCTCATACTTCTGAATTCACCT | 5′-CCTTGTGTGCCCAGACAACC | 5′-TCCTGGGCTCCCAGAA |
| −1086 Sp site | 5′-CTACATGGGAGCCAGAAATGG | 5′-GCCCCTACCTCTCAGACATTCTC | 5′-CCATCTGCTTCCGCC |
| −1293 to −1327 (2 half EREs) | 5′-TCAGGAAGGGTCAAAGCATGA | 5′-GACTCTTGACCTCGAGGTCTCC | 5′-TCAGCCCTGACTTTG |
Statistical analysis.
All data are expressed as means ± SD. We used unpaired t-test for comparison of RVSP, Fulton index, eNOS mRNA and protein, and cGMP levels between control and PPHN lambs. We used Mann-Whitney test to determine statistical significance for data sets involving Tet activity and the occupancies of TFs, Sp1, and ERs, as well as histone marks in eNOS promoter. We used Fisher exact test to determine statistical significance for data sets of DNA CpG methylation in eNOS promoter. Significance level was set as P < 0.05.
RESULTS
Control and PPHN groups had equal distribution of male sex with three out of six control and four out of six PPHN lambs being male. No significant differences were noted in the hemodynamic or epigenetic alterations observed in PPHN lambs by sex.
RVSP and Fulton index are increased in PPHN lambs.
Lambs with PPHN had a significant increase in RVSP after 8 days of prenatal ductus arteriosus constriction, with a mean increase of 20 mmHg (Fig. 2, A and B; n = 4). These results are consistent with the increase in mean pulmonary artery pressure in PPHN lambs we reported previously (21). PPHN lambs also had significant increase in Fulton index, suggesting RV hypertrophy, consistent with pulmonary hypertension (Fig. 2C, n = 4).
Fig. 2.
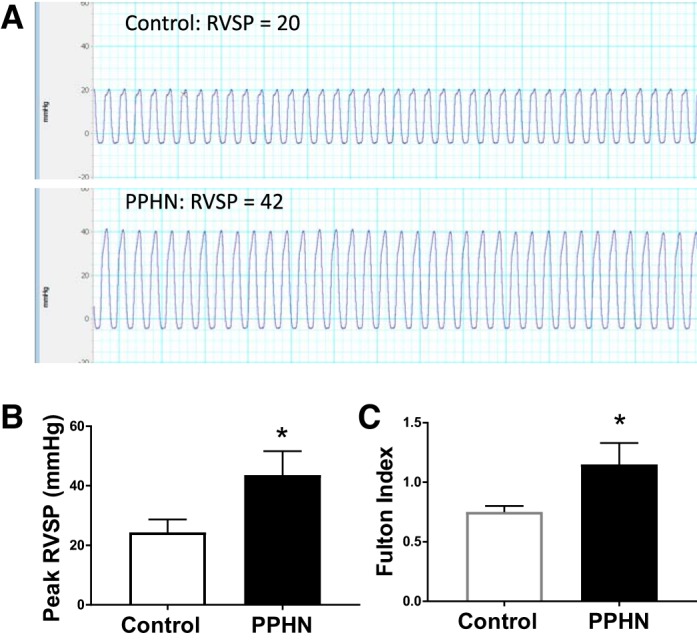
Representative right ventricular systolic pressures (RVSP) from one control and one persistent pulmonary hypertension of the newborn (PPHN) lamb (A) and summarized data from 4 control and PPHN lambs each (B) showing significant increase in RVSP in PPHN. Fulton index (RV/LV+septum weight) was also increased in PPHN (C, n = 4). *P < 0.05 from control by unpaired t-test.
Expression and function of eNOS are decreased in PPHN.
PAEC obtained from lambs with PPHN had a significant decrease in eNOS protein levels, compared with control PAEC (Fig. 3A, n = 5). Levels of eNOS mRNA were also decreased by fivefold in PAEC from PPHN lambs compared with control PAEC (Fig. 3B, n = 5). Exposure of control PAEC to NOS agonist ATP (19, 20) resulted in a significant increase in cGMP levels (Fig. 3C, n = 4). In contrast, PAEC from PPHN lambs fail to show a change in cGMP levels with ATP stimulation, suggesting decreased NOS function (Fig. 3C, n = 4). These data suggest that eNOS expression and function are decreased in PPHN cells.
Fig. 3.

Representative immunoblots for endothelial NOS (eNOS) and β-actin in pulmonary artery endothelial cells (PAEC) from control and PPHN lambs and summarized data (A, n = 5) showing decrease in eNOS protein in PPHN. IOD, integrated optical density. The mRNA levels of eNOS were decreased in PPHN (B, n = 5). Levels of cGMP, an indicator of NOS function, were decreased in PPHN at basal level. Stimulation with NOS agonist ATP increased cGMP levels in control, but not PPHN cells (C, n = 4). *P < 0.05 from control by unpaired t-test.
PPHN significantly increased DNA CpG methylation in the proximal promoter region of eNOS gene at multiple sites especially around EREs in PAEC.
We have previously demonstrated that PPHN decreases eNOS expression in PAEC (21). The present study investigated the potential molecular mechanisms that lead to downregulation of eNOS expression in PAEC. PPHN significantly increased DNA CpG methylation at 19 of 44 CpG sites examined in eNOS proximal promoter region, especially around EREs compared with controls (Fig. 4). The percent of CpG methylation between control and PPHN groups at 44 CpG sites is listed in Table 3 (*P < 0.05, n = 8 in each group).
Fig. 4.

Percent of CpG methylation at 44 CpG sites in eNOS promoter in controls (white bars) and PPHN (black bars). n = 8, *P < 0.05 between control and PPHN samples by Fisher’s exact test.
Table 3.
% CpG methylation at 44 sites between control and PPHN
| Site | CG Position Relative to TSS | Control, mean ± SD, % | PPHN, mean ± SD, % | P Value |
|---|---|---|---|---|
| 1 | −1497 | 57.4 ± 13 | 44 ± 14.1 | 0.066 |
| 2 | −1462 | 32.3 ± 8.6 | 25.1 ± 8.7 | 0.347 |
| 3 | −1425 | 85.2 ± 3.6 | 88.3 ± 6.3 | 0.529 |
| 4 | −1418 | 79.4 ± 12.5 | 86.3 ± 9 | 0.187 |
| 5 | −1410 | 77.3 ± 13.2 | 80.9 ± 8.6 | 0.726 |
| 6 | −1397 | 75.4 ± 10 | 80.6 ± 12.5 | 0.397 |
| 7 | −1327 | 64.7 ± 11.2 | 82.5 ± 16.3* | 0.00583 |
| 8 | −1314 | 71.1 ± 13.5 | 74 ± 18.1 | 0.874 |
| 9 | −1293 | 31.7 ± 11.6 | 46.2 ± 12.7* | 0.042 |
| 10 | −1248 | 73.8 ± 14.7 | 81.6 ± 13.2 | 0.232 |
| 11 | −1240 | 76.3 ± 11.6 | 90 ± 10.4* | 0.021 |
| 12 | −1195 | 45.6 ± 10.9 | 75.5 ± 17.7* | 0.0001 |
| 13 | −1189 | 73 ± 7.6 | 76.1 ± 16.9 | 0.624 |
| 14 | −1086 | 73.1 ± 15.5 | 75.2 ± 10 | 0.746 |
| 15 | −1043 | 63.9 ± 22 | 72.9 ± 32.9 | 0.223 |
| 16 | −1020 | 68.8 ± 16.1 | 83.9 ± 13.4* | 0.019 |
| 17 | −1003 | 38 ± 27.3 | 67.8 ± 29.9* | 0.0001 |
| 18 | −960 | 0 | 0% | |
| 19 | −914 | 0 | 0% | |
| 20 | −826 | 33.1 ± 21.7 | 75.9 ± 29.5* | 0.0001 |
| 21 | −790 | 23.9 ± 14.2 | 43.1 ± 21.6* | 0.0041 |
| 22 | −778 | 23.9 ± 15.9 | 35.6 ± 25.2 | 0.121 |
| 23 | −769 | 19.1 ± 13.5 | 40.1 ± 29 | 0.0032 |
| 24 | −760 | 23.23 ± 10.6 | 36.1 ± 16.3 | 0.076 |
| 25 | −752 | 20.2 ± 13.2 | 30.3 ± 10.3 | 0.511 |
| 26 | −735 | 27.6 ± 15.5 | 46.4 ± 18.4 | 0.105 |
| 27 | −707 | 26.6 ± 15 | 57.1 ± 30.5* | 0.0001 |
| 28 | −649 | 19.2 ± 18.5 | 42.3 ± 19.6* | 0.0007 |
| 29 | −629 | 13.9 ± 8.8 | 39 ± 24.2* | 0.00001 |
| 30 | −607 | 14.4 ± 9.4 | 18.8 ± 15.4 | 0.851 |
| 31 | −588 | 18 ± 7.4 | 16.2 ± 9.9 | 0.851 |
| 32 | −533 | 15.6 ± 8 | 32.3 ± 11* | 0.0048 |
| 33 | −427 | 5.7 ± 5.3 | 25.5 ± 14.5* | 0.0072 |
| 34 | −410 | 0.4 ± 0.5 | 1.2 ± 2.3 | 1 |
| 35 | −406 | 5.8 ± 3.5 | 13.1 ± 6.5 | 0.146 |
| 36 | −389 | 0 | 0 | |
| 37 | −351 | 6.3 ± 1.2 | 16.8 ± 6.8* | 0.04 |
| 38 | −298 | 5.9 ± 2 | 34.6 ± 11.3* | 0.0001 |
| 39 | −285 | 5.3 ± 4 | 27.4 ± 20.9* | 0.0001 |
| 40 | −279 | 9.6 ± 6.1 | 52.4 ± 19.8* | 0.00001 |
| 41 | −255 | 10.1 ± 20.6 | 54.4 ± 11.9* | 0.00001 |
| 42 | −209 | 25.5 ± 9.2 | 35.7 ± 11.9 | 0.361 |
| 43 | −195 | 18.2 ± 7.9 | 25.6 ± 8.5 | 0.498 |
| 44 | −173 | 15.9 ± 4.3 | 22 ± 5.3 | 0.274 |
TSS, transcription start site; PPHN, persistent pulmonary hypertension of the newborn. Boldface refers to nucleotide positions where significant differences in % methylation were observed.
P < 0.05 from control by Fisher exact test.
PPHN significantly decreased DNA demethylation enzyme Tet activity in PAEC.
PPHN significantly decreased TET activity in PAEC to 25 ± 2.7 ng·min−1·mg−1 protein compared with the controls 81 ± 6.3 ng·min−1·mg−1 protein (Fig. 5) (*P < 0.001, n = 4 in each group).
Fig. 5.
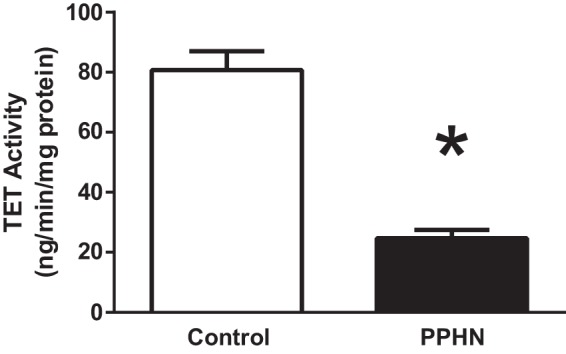
Ten eleven translocase demethylase (Tet) enzyme activity measured by colorimetric assay in controls and PPHN. n = 4 samples each, *P < 0.05 from control by t-test.
PPHN significantly decreased Sp1 occupancy in the proximal promoter of eNOS gene in PAEC.
PPHN significantly decreased Sp1 occupancy at all five sites examined in the proximal promoter of eNOS in PAEC compared with controls (Fig. 6) (*P < 0.05, n = 6 in each group). However, PPHN did not affect the occupancy of ERα or ERβ in the proximal promoter of eNOS gene in PAEC (data not shown).
Fig. 6.

Chromatin immunoprecipitation (ChIP) analysis for the occupancy of Sp1, around 2 sp1 binding sites, and 3 ERE half-sites in eNOS promoter in controls (white bars) and PPHN (black bars). Black arrows on the schematic representation of eNOS promoter are the locations of primer sets designed. Data presented as 2(-∆CT) relative to 5% input. n = 6, *P < 0.05 from control.
PPHN altered the histone code in the proximal promoter of eNOS gene in PAEC.
PPHN significantly decreased H4K12ac density in the proximal promoter of eNOS gene at both Sp1 binding sites and two distal EREs in PAEC compared with controls (Fig. 7). Concurrently, PPHN significantly increased the density of the repression mark H3K9me3 around the distal Sp1 binding site (*P < 0.05, n = 6) in PAEC compared with the controls (Fig. 8) and appears to increase it at the proximal site, although the difference was not significant for this site (P = 0.06). PPHN did not change the density of H3K4me3 or H3K9ac in the proximal promoter of eNOS (data not shown).
Fig. 7.

ChIP analysis for the occupancy of H4K12ac around 2 sp1 binding sites and 3 ERE half-sites in eNOS promoter in controls (white bars) and PPHN (black bars). Black arrows on the schematic representation of eNOS promoter are the locations of primer sets designed. Data presented as 2(-∆CT) relative to 5% input. n = 6, *P < 0.05 from control.
Fig. 8.

ChIP analysis for the occupancy of H3K9me3 around 2 sp1 binding sites and 3 ERE half-sites in eNOS promoter in controls (white bars) and PPHN (black bars). Black arrows on the schematic representation of eNOS promoter are the locations of primer sets designed. Data presented as 2(-∆CT) relative to 5% input. n = 6, *P < 0.05 from control.
DISCUSSION
Our novel findings in this study can be grouped into four categories. In the proximal promoter of eNOS gene, PPHN has significantly 1) increased DNA CpG methylation, 2) decreased Sp1 occupancy, 3) altered histone code primed to decrease Sp1 binding in PAEC, and 4) also significantly decreased TET enzyme activity in PAEC. Additionally, we observed that eNOS expression and function are decreased in PPHN lambs, as we reported previously (21, 23).
DNA methylation in the promoter of certain genes is associated with transcriptional silencing (18, 49). Our finding of increased DNA CpG methylation in the eNOS promoter in PPHN PAEC is consistent with our current and previous finding that PPHN decreases eNOS expression in PAEC from our fetal lamb model (21). Previous studies have shown that DNA is hypomethylated in the eNOS promoter in vascular endothelial cells, which express eNOS exclusively, while DNA is hypermethylated in the eNOS promoter in nonendothelial cell types that do not express the native eNOS transcript (9). However, DNA methylation of eNOS gene promoter showed a trend for decrease in a relation to increased eNOS expression in pulmonary vascular endothelial cells in a rat model of pulmonary hypertension induced by hypoxia and indomethacin (46). These data suggest that alterations in DNA methylation in eNOS gene promoter and eNOS expression are specific to the cell type and mechanism of pulmonary hypertension.
DNA methylation status is regulated, in part, by demethylation enzyme TET. Decreased TET activity induced by hypoxia has been shown to induce hypermethylation at gene promoters in vitro (43). Moreover, TET2 expression and activity have been shown to be negatively correlated with the long noncoding RNA MEG3 expression and DNA methylation in the promoter of MEG3 in acute myeloid leukemia patients (47). Thus, our finding of decreased TET activity is consistent with decreased DNA demethylation, and this alteration may contribute to DNA hypermethylation seen in eNOS promoter in PPHN PAEC.
Our findings of decreased Sp1 occupancy in the eNOS proximal promoter in PPHN PAEC suggest that Sp1 binding facilitates eNOS gene transcription. Endothelial NOS expression has been reported as an indicator of Sp1 transactivation activity (48). Functional analysis of human eNOS promoter has demonstrated that Sp1 is necessary for eNOS basal transcription in human endothelial cells (50). Moreover, blocking NF-κB activation in mice deficient in NF-κB p50 gene (NF-κB-KO) suppressed LPS-inducible Sp1-degrading enzyme (LISPDE) activity, prevented Sp1 protein degradation, and reversed the downregulation of Sp1 DNA binding activity and eNOS expression. Pretreatment of LPS-challenged WT mice with a selective LISPDE inhibitor increased nuclear Sp1 protein content, restored Sp1 DNA binding activity, and reversed eNOS protein downregulation in lungs (48). Although PPHN did not change CpG methylation status at Sp1 binding site to directly affect Sp1 binding in our study, methylation of adjacent CpG sites can affect Sp1 binding and activity in the promoter indirectly by recruiting histone deacetylases through methyl-DNA-binding proteins (15, 31). We speculate that PPHN decreases the Sp1 occupancy in eNOS promoter by increasing DNA CpG methylation and subsequently recruiting histone deacetylase. These events further lead to decreased densities of acetylated histones in the eNOS promoter and contribute to downregulation of eNOS expression in PPHN PAEC.
Specific histone modifications at eNOS promoter in this study were associated with decreased eNOS expression in PPHN PAEC. Histone modifications H3K4me3, H3K9ac, and H4K12ac are found to be particularly enriched at eNOS proximal promoter in endothelial cells (11). Inhibition of histone deacetylase activity by trichostatin A increased acetylation of histones H3 and H4 at the eNOS proximal promoter in nonexpressing cell types and led to increased steady-state eNOS mRNA transcript levels (11). Furthermore, histone acetylation including H3K9ac and H4K12ac are decreased at eNOS proximal promoter in human umbilical vein endothelial cells exposed to hypoxia (12). In contrast, increased H3K9me3 at the promoter has been shown to repress the expression of 3β-HSD in Leydig cells exposed to arsenic (5). Consistent with this observation, decreased H3K9me3 levels at the promoters of IL-6 is associated with increase IL-6 expression in cardiomyocyte cells exposed to high glucose (49). In our study, we found PPHN selectively decreased the density of activation mark H4K12ac in eNOS promoter and increased the density of repression mark H3K9me3 around Sp1 binding sites in PAEC. We speculate that these specific histone modifications, along with increased DNA CpG methylation in eNOS proximal promoter, lead to decreased Sp1 binding and contributes to eNOS gene repression in PPHN PAEC.
Activation of eNOS gene is mediated in part by estrogen via interaction of ERs through both genomic and nongenomic mechanisms (24). Estrogen upregulates eNOS gene expression in fetal PAEC through the activation of PAEC ERs (28). Nuclear run-on assays indicated that the increase in eNOS mRNA is the result of an estrogen-induced enhancement of eNOS gene transcription, which may be due to estrogen-induced enhanced binding of transcription factor Sp1 to eNOS promoter in human endothelial EA.hy 926 cells (17). Further work in cultured endothelial cells indicated that eNOS downregulation by TNF-α, which is due to enhanced eNOS mRNA degradation, is prevented by 17β-estradiol (41), indicating that there is transcriptional regulation of eNOS expression by estradiol through mechanisms that do not involve classical ERE-mediated processes (8). In our study, PPHN did not change the occupancy of either ERα or ERβ in the eNOS promoter, suggesting that downregulated eNOS expression in PPHN may involve other transcriptional mechanisms. However, estrogens have nongenomic effects involving the binding of estradiol to the membrane ER and the activation of phosphatidylinositol 3-kinase/protein kinase B (Akt) pathway, resulting in eNOS phosphorylation and increased eNOS activity (29, 30). Future studies are needed to explore whether alterations in this nongenomic mechanism for ERs are involved in the downregulation of eNOS expression in PPHN PAEC.
The limitation of this study is that we focused on the epigenetic changes of eNOS gene as a mechanism for downregulation of eNOS expression in PPHN. Whether decreased mRNA stability and increased proteasomal degradation contribute to decreased eNOS protein levels in PPHN requires additional studies. Further investigations examining the effect of epigenetic modification enzymes such as DNA methyltransferases or histone acetyltransferase and deacetylases on downregulation of eNOS gene in PPHN will be an important next step in establishing cause and effect relationships between the observed epigenetic modifications and decreased expression in PPHN.
In conclusion, PPHN decreased Sp1 occupancy in the eNOS promoter in PAEC. These changes occur within the context of epigenetic modifications primed to decrease Sp1 binding at Sp1 binding sites of eNOS gene. These results together build upon our previous finding of decreased eNOS expression in PPHN. We speculate that epigenetic modifications of the eNOS gene promoter may contribute to the pathogenesis of PPHN.
GRANTS
Supported by National Heart, Lung, and Blood Institute Grant R01HL-057268, funding from the Children’s Research Institute, and the Muma Endowed Chair in Neonatology (G. G. Konduri).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
X.K., H.J., X.J., T.M., and Y.-W.H. performed experiments; X.K., Y.-W.H., and G.G.K. analyzed data; X.K., R.H.L., and G.G.K. interpreted results of experiments; X.K. and G.G.K. prepared figures; X.K. drafted manuscript; R.H.L. and G.G.K. edited and revised manuscript; R.H.L. and G.G.K. approved final version of manuscript; G.G.K. conceived and designed research.
REFERENCES
- 1.Afolayan AJ, Eis A, Alexander M, Michalkiewicz T, Teng RJ, Lakshminrusimha S, Konduri GG. Decreased endothelial nitric oxide synthase expression and function contribute to impaired mitochondrial biogenesis and oxidative stress in fetal lambs with persistent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 310: L40–L49, 2016. doi: 10.1152/ajplung.00392.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Afolayan AJ, Eis A, Teng RJ, Bakhutashvili I, Kaul S, Davis JM, Konduri GG. Decreases in manganese superoxide dismutase expression and activity contribute to oxidative stress in persistent pulmonary hypertension of the newborn. Am J Physiol Lung Cell Mol Physiol 303: L870–L879, 2012. doi: 10.1152/ajplung.00098.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Afolayan AJ, Teng RJ, Eis A, Rana U, Broniowska KA, Corbett JA, Pritchard K, Konduri GG. Inducible HSP70 regulates superoxide dismutase-2 and mitochondrial oxidative stress in the endothelial cells from developing lungs. Am J Physiol Lung Cell Mol Physiol 306: L351–L360, 2014. doi: 10.1152/ajplung.00264.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agalioti T, Chen G, Thanos D. Deciphering the transcriptional histone acetylation code for a human gene. Cell 111: 381–392, 2002. doi: 10.1016/S0092-8674(02)01077-2. [DOI] [PubMed] [Google Scholar]
- 5.Alamdar A, Xi G, Huang Q, Tian M, Eqani SAMAS, Shen H. Arsenic activates the expression of 3β-HSD in mouse Leydig cells through repression of histone H3K9 methylation. Toxicol Appl Pharmacol 326: 7–14, 2017. doi: 10.1016/j.taap.2017.04.012. [DOI] [PubMed] [Google Scholar]
- 6.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410: 120–124, 2001. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ III, Gingeras TR, Schreiber SL, Lander ES. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120: 169–181, 2005. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Chambliss KL, Shaul PW. Estrogen modulation of endothelial nitric oxide synthase. Endocr Rev 23: 665–686, 2002. doi: 10.1210/er.2001-0045. [DOI] [PubMed] [Google Scholar]
- 9.Chan Y, Fish JE, D’Abreo C, Lin S, Robb GB, Teichert AM, Karantzoulis-Fegaras F, Keightley A, Steer BM, Marsden PA. The cell-specific expression of endothelial nitric-oxide synthase: a role for DNA methylation. J Biol Chem 279: 35087–35100, 2004. doi: 10.1074/jbc.M405063200. [DOI] [PubMed] [Google Scholar]
- 10.Davey C, Pennings S, Allan J. CpG methylation remodels chromatin structure in vitro. J Mol Biol 267: 276–288, 1997. doi: 10.1006/jmbi.1997.0899. [DOI] [PubMed] [Google Scholar]
- 11.Fish JE, Matouk CC, Rachlis A, Lin S, Tai SC, D’Abreo C, Marsden PA. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J Biol Chem 280: 24824–24838, 2005. doi: 10.1074/jbc.M502115200. [DOI] [PubMed] [Google Scholar]
- 12.Fish JE, Yan MS, Matouk CC, St Bernard R, Ho JJ, Gavryushova A, Srivastava D, Marsden PA. Hypoxic repression of endothelial nitric-oxide synthase transcription is coupled with eviction of promoter histones. J Biol Chem 285: 810–826, 2010. doi: 10.1074/jbc.M109.067868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu Q, Yu X, Callaway CW, Lane RH, McKnight RA. Epigenetics: intrauterine growth retardation (IUGR) modifies the histone code along the rat hepatic IGF-1 gene. FASEB J 23: 2438–2449, 2009. doi: 10.1096/fj.08-124768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang YW, Kuo CT, Chen JH, Goodfellow PJ, Huang TH, Rader JS, Uyar DS. Hypermethylation of miR-203 in endometrial carcinomas. Gynecol Oncol 133: 340–345, 2014. doi: 10.1016/j.ygyno.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 19: 187–191, 1998. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 16.Ke X, Schober ME, McKnight RA, O’Grady S, Caprau D, Yu X, Callaway CW, Lane RH. Intrauterine growth retardation affects expression and epigenetic characteristics of the rat hippocampal glucocorticoid receptor gene. Physiol Genomics 42: 177–189, 2010. doi: 10.1152/physiolgenomics.00201.2009. [DOI] [PubMed] [Google Scholar]
- 17.Kleinert H, Wallerath T, Euchenhofer C, Ihrig-Biedert I, Li H, Förstermann U. Estrogens increase transcription of the human endothelial NO synthase gene: analysis of the transcription factors involved. Hypertension 31: 582–588, 1998. doi: 10.1161/01.HYP.31.2.582. [DOI] [PubMed] [Google Scholar]
- 18.Klose R, Bird A. Molecular biology. MeCP2 repression goes nonglobal. Science 302: 793–795, 2003. doi: 10.1126/science.1091762. [DOI] [PubMed] [Google Scholar]
- 19.Konduri GG, Mattei J. Role of oxidative phosphorylation and ATP release in mediating birth-related pulmonary vasodilation in fetal lambs. Am J Physiol Heart Circ Physiol 283: H1600–H1608, 2002. doi: 10.1152/ajpheart.00245.2002. [DOI] [PubMed] [Google Scholar]
- 20.Konduri GG, Mital S. Adenosine and ATP cause nitric oxide-dependent pulmonary vasodilation in fetal lambs. Biol Neonate 78: 220–229, 2000. doi: 10.1159/000014274. [DOI] [PubMed] [Google Scholar]
- 21.Konduri GG, Ou J, Shi Y, Pritchard KA Jr. Decreased association of HSP90 impairs endothelial nitric oxide synthase in fetal lambs with persistent pulmonary hypertension. Am J Physiol Heart Circ Physiol 285: H204–H211, 2003. doi: 10.1152/ajpheart.00837.2002. [DOI] [PubMed] [Google Scholar]
- 22.Konduri GG, Solimano A, Sokol GM, Singer J, Ehrenkranz RA, Singhal N, Wright LL, Van Meurs K, Stork E, Kirpalani H, Peliowski A; Neonatal Inhaled Nitric Oxide Study Group . A randomized trial of early versus standard inhaled nitric oxide therapy in term and near-term newborn infants with hypoxic respiratory failure. Pediatrics 113: 559–564, 2004. doi: 10.1542/peds.113.3.559. [DOI] [PubMed] [Google Scholar]
- 23.Konduri GG, Bakhutashvili I, Eis A, Pritchard KA. Oxidant stress from uncoupled nitric oxide synthase impairs vasodilation in fetal lambs with persistent pulmonary hypertension. Am J Physiol Heart Circ Physiol 292: H1812–H1820, 2007. doi: 10.1152/ajpheart.00425.2006. [DOI] [PubMed] [Google Scholar]
- 24.Kypreos KE, Zafirovic S, Petropoulou PI, Bjelogrlic P, Resanovic I, Traish A, Isenovic ER. Regulation of endothelial nitric oxide synthase and high-density lipoprotein quality by estradiol in cardiovascular pathology. J Cardiovasc Pharmacol Ther 19: 256–268, 2014. doi: 10.1177/1074248413513499. [DOI] [PubMed] [Google Scholar]
- 25.Lakshminrusimha S, Russell JA, Steinhorn RH, Ryan RM, Gugino SF, Morin FC III, Swartz DD, Kumar VH. Pulmonary arterial contractility in neonatal lambs increases with 100% oxygen resuscitation. Pediatr Res 59: 137–141, 2006. doi: 10.1203/01.pdr.0000191136.69142.8c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lévy M, Maurey C, Dinh-Xuan AT, Vouhé P, Israël-Biet D. Developmental expression of vasoactive and growth factors in human lung. Role in pulmonary vascular resistance adaptation at birth. Pediatr Res 57: 21R–25R, 2005. doi: 10.1203/01.PDR.0000159575.58834.8D. [DOI] [PubMed] [Google Scholar]
- 27.Luo Y, Hara T, Ishido Y, Yoshihara A, Oda K, Makino M, Ishii N, Hiroi N, Suzuki K. Rapid preparation of high-purity nuclear proteins from a small number of cultured cells for use in electrophoretic mobility shift assays. BMC Immunol 15: 586, 2014. doi: 10.1186/s12865-014-0062-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacRitchie AN, Jun SS, Chen Z, German Z, Yuhanna IS, Sherman TS, Shaul PW. Estrogen upregulates endothelial nitric oxide synthase gene expression in fetal pulmonary artery endothelium. Circ Res 81: 355–362, 1997. doi: 10.1161/01.RES.81.3.355. [DOI] [PubMed] [Google Scholar]
- 29.McNeill AM, Kim N, Duckles SP, Krause DN, Kontos HA. Chronic estrogen treatment increases levels of endothelial nitric oxide synthase protein in rat cerebral microvessels. Stroke 30: 2186–2190, 1999. doi: 10.1161/01.STR.30.10.2186. [DOI] [PubMed] [Google Scholar]
- 30.Moriarty K, Kim KH, Bender JR. Minireview: estrogen receptor-mediated rapid signaling. Endocrinology 147: 5557–5563, 2006. doi: 10.1210/en.2006-0729. [DOI] [PubMed] [Google Scholar]
- 31.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393: 386–389, 1998. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 32.Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O’Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, Kouzarides T. Rb targets histone H3 methylation and HP1 to promoters. Nature 412: 561–565, 2001. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- 33.Papamatheakis DG, Chundu M, Blood AB, Wilson SM. Prenatal programming of pulmonary hypertension induced by chronic hypoxia or ductal ligation in sheep. Pulm Circ 3: 757–780, 2013. doi: 10.1086/674767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45, 2001. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Active genes are tri-methylated at K4 of histone H3. Nature 419: 407–411, 2002. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 36.Shaul PW, Afshar S, Gibson LL, Sherman TS, Kerecman JD, Grubb PH, Yoder BA, McCurnin DC. Developmental changes in nitric oxide synthase isoform expression and nitric oxide production in fetal baboon lung. Am J Physiol Lung Cell Mol Physiol 283: L1192–L1199, 2002. doi: 10.1152/ajplung.00112.2002. [DOI] [PubMed] [Google Scholar]
- 37.Shaul PW, Yuhanna IS, German Z, Chen Z, Steinhorn RH, Morin FC III. Pulmonary endothelial NO synthase gene expression is decreased in fetal lambs with pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 272: L1005–L1012, 1997. doi: 10.1152/ajplung.1997.272.5.L1005. [DOI] [PubMed] [Google Scholar]
- 38.Shen L, Wu H, Diep D, Yamaguchi S, D’Alessio AC, Fung HL, Zhang K, Zhang Y. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell 153: 692–706, 2013. doi: 10.1016/j.cell.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119: 941–953, 2004. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 40.Steinhorn RH. Neonatal pulmonary hypertension. Pediatr Crit Care Med 11, Suppl: S79–S84, 2010. doi: 10.1097/PCC.0b013e3181c76cdc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sumi D, Hayashi T, Jayachandran M, Iguchi A. Estrogen prevents destabilization of endothelial nitric oxide synthase mRNA induced by tumor necrosis factor alpha through estrogen receptor mediated system. Life Sci 69: 1651–1660, 2001. doi: 10.1016/S0024-3205(01)01251-6. [DOI] [PubMed] [Google Scholar]
- 42.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324: 930–935, 2009. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thienpont B, Steinbacher J, Zhao H, D’Anna F, Kuchnio A, Ploumakis A, Ghesquière B, Van Dyck L, Boeckx B, Schoonjans L, Hermans E, Amant F, Kristensen VN, Peng Koh K, Mazzone M, Coleman M, Carell T, Carmeliet P, Lambrechts D. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 537: 63–68, 2016. doi: 10.1038/nature19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tiktinsky MH, Morin FC III. Increasing oxygen tension dilates fetal pulmonary circulation via endothelium-derived relaxing factor. Am J Physiol Heart Circ Physiol 265: H376–H380, 1993. doi: 10.1152/ajpheart.1993.265.1.H376. [DOI] [PubMed] [Google Scholar]
- 45.Villanueva ME, Zaher FM, Svinarich DM, Konduri GG. Decreased gene expression of endothelial nitric oxide synthase in newborns with persistent pulmonary hypertension. Pediatr Res 44: 338–343, 1998. doi: 10.1203/00006450-199809000-00012. [DOI] [PubMed] [Google Scholar]
- 46.Xu XF, Ma XL, Shen Z, Wu XL, Cheng F, Du LZ. Epigenetic regulation of the endothelial nitric oxide synthase gene in persistent pulmonary hypertension of the newborn rat. J Hypertens 28: 2227–2235, 2010. doi: 10.1097/HJH.0b013e32833e08f1. [DOI] [PubMed] [Google Scholar]
- 47.Yao H, Duan M, Lin L, Wu C, Fu X, Wang H, Guo L, Chen W, Huang L, Liu D, Rao R, Wang S, Ding Y. TET2 and MEG3 promoter methylation is associated with acute myeloid leukemia in a Hainan population. Oncotarget 8: 18337–18347, 2017. doi: 10.18632/oncotarget.15440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ye X, Liu H, Gong YS, Liu SF. LPS Down-Regulates Specificity Protein 1 Activity by Activating NF-κB Pathway in Endotoxemic Mice. PLoS One 10: e0130317, 2015. doi: 10.1371/journal.pone.0130317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu XY, Geng YJ, Liang JL, Zhang S, Lei HP, Zhong SL, Lin QX, Shan ZX, Lin SG, Li Y. High levels of glucose induce “metabolic memory” in cardiomyocyte via epigenetic histone H3 lysine 9 methylation. Mol Biol Rep 39: 8891–8898, 2012. doi: 10.1007/s11033-012-1756-z. [DOI] [PubMed] [Google Scholar]
- 50.Zhang R, Min W, Sessa WC. Functional analysis of the human endothelial nitric oxide synthase promoter. Sp1 and GATA factors are necessary for basal transcription in endothelial cells. J Biol Chem 270: 15320–15326, 1995. doi: 10.1074/jbc.270.25.15320. [DOI] [PubMed] [Google Scholar]
- 51.Zhu WG, Srinivasan K, Dai Z, Duan W, Druhan LJ, Ding H, Yee L, Villalona-Calero MA, Plass C, Otterson GA. Methylation of adjacent CpG sites affects Sp1/Sp3 binding and activity in the p21(Cip1) promoter. Mol Cell Biol 23: 4056–4065, 2003. doi: 10.1128/MCB.23.12.4056-4065.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]