

Abstract
Increased sodium appetite is a physiological response to sodium deficiency; however, it has also been implicated in disease conditions such as congestive heart failure, kidney failure, and salt-sensitive hypertension. The central nervous system is the major regulator of sodium appetite and intake behavior; however, the neural mechanisms underlying this behavior remain incompletely understood. Here, we investigated the involvement of the (pro)renin receptor (PRR), a component of the brain renin-angiotensin system, in the regulation of sodium intake in a neuron-specific PRR knockout (PRRKO) mouse model generated previously in our laboratory. Sodium intake following deoxycorticosterone (DOCA) stimulation was tested by assessing the preference of mice for 0.9% saline or regular water in single-animal metabolic cages. Blood pressure was monitored in conscious, freely moving mice by a telemetry system. We found that saline intake and total fluid intake were significantly reduced in PRRKO mice following DOCA treatment compared with that in wild-type (WT) mice, whereas regular water intake was similar between the genotypes. Sodium preference and total sodium intake were significantly reduced in PRRKO mice compared with WT mice. PRRKO mice also excreted less urine and urinary sodium compared with WT mice following DOCA treatment, whereas potassium excretion was similar between the two groups. Finally, we found that the sodium balance, calculated by subtracting urinary sodium excretion from sodium intake, was greater in WT mice than in PRRKO mice. Collectively, these findings suggest that the neuronal PRR plays a regulatory role in DOCA-induced sodium intake.
Keywords: central nervous system; deoxycorticosterone; (pro)renin receptor; sodium appetite, sodium intake
INTRODUCTION
Sodium appetite is a motivated behavioral state that drives animals or humans to seek foods or fluids containing sodium. Excess salt intake, caused by sodium appetite sensitization (28, 48), is an important risk factor for many types of cardiovascular disease, including congestive heart failure, kidney failure, and salt-sensitive hypertension (3, 27, 33). Salt restriction has been recommended for these medical conditions and has proven successful in clinical practice if strictly enforced (46). However, compliance among patients with these diseases is often low, at least in part because of paradoxical increases in sodium appetite (7, 38, 39, 52). The renin-angiotensin aldosterone system (RAAS) has been shown to play a vital role in the regulation of sodium appetite and intake, in particular, through aldosterone actions on 11β-hydroxysteroid dehydrogenase type 2-positive neurons in the nucleus of the solitary tract (NTS) (30, 41), and through angiotensin II (ANG II) actions on ANG II-sensing neurons in the subfornical organ (SFO) (37) in the central nervous system (CNS) (9, 17, 19, 22, 47).
The (pro)renin receptor (PRR) is a key component of the brain RAAS that mediates the majority of endogenous ANG II formation in the brain (1, 34, 35). However, whether the brain PRR plays a role in the regulation of sodium appetite or intake is not known. In this study, we examined the importance of the neuronal PRR in deoxycorticosterone (DOCA)-induced sodium intake using a neuron-specific PRR-knockout (PRRKO) mouse model generated previously in our laboratory (34). We report here that PRRKO in neurons decreased salt intake, total fluid intake, and sodium intake induced by DOCA in mice.
MATERIALS AND METHODS
Animals.
Neuron-specific PRRKO mice were generated in our laboratory by breeding PRR-floxed mice (31), provided by Dr. Atsuhiro Ichihara (Tokyo Women's Medical University), with transgenic mice expressing Cre-recombinase driven by the neuron-specific Nefh (neurofilament, heavy polypeptide) promoter (Nefh-Cre mice, stock #009102; The Jackson Laboratory, Bar Harbor, ME) (26). Hemizygous Nefh-Cre mice (FVB/N background) were backcrossed to wild-type (WT) C57Bl/6J mice for at least four generations according to a previously described breeding scheme (34). In brief, male Nefh-Cre+ mice were bred with female PRR-floxed mice. Cre-negative PRR-floxed littermates (C57Bl/6J background) containing WT PRR genes were used as controls. All mice were housed in a university animal facility under a 12 h light-dark cycle with ad libitum access to regular chow (0.1% sodium, catalog #2019; Envigo, Madison, WI) and water and were used for experiments at 16–18 wk old. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Nevada, Reno, and were performed in accordance with National Institutes of Health Guidelines for the care and use of experimental animals.
DOCA treatment and metabolic cage studies.
Before commencing the experimental protocol, we trained PRRKO and WT mice (n = 5/group) by placing them in metabolic cages (Hatteras Instruments, Cary, NC) for 24 h and then returning them to their home cages for at least two 24 h rest periods until baseline food and water intake was stable (~1 wk). Mice were then anesthetized by isoflurane inhalation, and a 50 mg pellet of DOCA was implanted as described previously (7). After recovery from anesthesia, animals were housed singly in standard forced-air cages. All animals were maintained on standard chow and provided ad libitum access to a 0.15 mol/l (0.9%) NaCl solution and tap water solution. Mice were placed singly in metabolic cages twice a week for 24 h each. Food, saline, and water allocated to each mouse were weighed at the initial time of placement and then recorded again after 24 h. We determined food intake by calculating differences in weights of food at the beginning and end of each 24 h period. Water and saline intake (via water dispensers) and changes in body weight were also recorded similarly. A schematic for this protocol is presented in Fig. 1A.
Fig. 1.

Food intake and body weight during metabolic cage studies. A: schematic showing the metabolic cage experimental protocol. B: daily food intake. C: body weight. D: food intake normalized to body weight. E: ANCOVA analysis of food intake, corrected for body weight. #P < 0.05 vs. day specified in the same genotype in individual graphs; two-way ANOVA (n = 5/group). BW, body weight; DOCA, deoxycorticosterone; PRRKO, (pro)renin receptor knockout; WT, wild type.
We calculated 24 h sodium intake by converting the total saline intake volume to mmol of sodium, as follows: NaCl mmol/24 h = mmol/58.4 × l/1,000 ml × ml/24 h. Sodium preference was calculated as the ratio of 24 h saline intake (ml) to total fluid intake (ml). A ratio of 0.5 indicates no preference for sodium or water. Cumulative bar graphs represent the sums of values recorded at each time point. Urine samples were collected during the 24 h period and stored in 2 ml tubes at −20°C for further measurements. Tissues were collected at the end of experiments for subsequent analyses.
Urine electrolyte and osmolality analyses.
We collected 24 h urine samples at baseline (day 0) and days 3, 7, 10, 14, 17, and 21. Urinary sodium and potassium concentrations and osmolality were measured by flame photometry (3× dilution factor). We calculated 24 h urinary sodium and potassium excretion according to the following equation: mmol/24 h = mmol/l × l/1,000 ml × ml/24 h. Sodium balance was calculated as the difference in 24 h sodium intake minus the 24 h urinary sodium excretion. Cumulative bar graphs represent the sums of values recorded at each time point. We analyzed 24 h average urinary osmolality, expressed in the original units of mmol/kg, at the end of the protocol with a VAPRO vapor pressure Osmometer (Wescor, Logan, UT).
Telemetry recordings.
Separate sets of PRRKO and WT mice (n = 6/group) were anesthetized by isoflurane inhalation and instrumented with a radio telemetric transmitter [TA11PA C-10; Data Science International (DSI), St. Paul, MN] into the carotid artery, as described previously (17, 53). After a 14-day recovery period, PRRKO and WT mice were then implanted with DOCA pellets and provided 0.9% saline and water ad libitum for 21 days, as described above. Blood pressure (BP) and heart rate (HR) were recording from 9 to 11 AM, both at baseline and at the end of the protocol, by a continuous recording method (six recordings per second at 500 Hz) and Ponemah 6.0 software (DSI). Data are presented as systolic BP, diastolic BP, mean arterial BP (MAP), pulse pressure, and HR. HR was automatically derived from the BP waveform with Panemah software. Spontaneous baroreflex sensitivity (SBRS) was assessed with Hemolab software ver. 20.7 (http://www.haraldstauss.com/HaraldStaussScientific/hemolab/).
Statistical analysis.
Data are expressed as means ± SE and were analyzed by paired or unpaired Student’s t-test or two-way analysis of variance (ANOVA) with Bonferroni post hoc tests for comparison of replicate means, as appropriate. Student’s t-tests and two-way ANOVAs were performed with Prism7 (GraphPad Software). Analysis of covariance (ANCOVA) was performed by Dr. Wei Yang at the School of Community Health Sciences, University of Nevada, Reno, using SAS 9.4. Differences were considered statistically significant at a P value < 0.05.
RESULTS
Neuron-specific PRRKO mice exhibit reduced sodium and total fluid intake following DOCA treatment.
To examine the role of the neuronal PRR in regulating salt intake, we subjected both PRRKO and WT mice to a two-bottle test in which both water and 0.9% saline were offered following implantation of a DOCA pellet; mice were monitored for 21 days in single-mouse metabolic cages, as illustrated in Fig. 1A. We also monitored food intake and body weight during the metabolic cage study. We found that food intake was similar between WT and PRRKO mice at baseline and throughout the protocol (Fig. 1, B and C). Although there was no difference in body weight between the two groups of mice, there was a very small, but significant, reduction in body weight in both WT and PRRKO mice (1.2–1.4 g; P < 0.05) compared with baseline weights (Fig. 1D), reflecting the stress associated with housing in metabolic cages. BP, HR, and SBRS were monitored in a separate set of mice housed in normal cages. At baseline, there was no difference in systolic BP, diastolic BP, MAP, pulse pressure, HR, or SBRS between WT and PRRKO mice (Fig. 2). DOCA treatment, with free choice of water or saline in drinking solution, induced a small, but significant, increase in BP (MAP) in both WT (108 ± 3.3 mmHg; P = 0.003) and PRRKO (111 ± 2.6 mmHg; P = 0.001) mice compared with baseline values (WT: 94 ± 2.5 mmHg; PRRKO: 96 ± 1.8 mmHg), whereas there was no difference between groups following 21 days of DOCA treatment. No alterations in pulse pressure, HR, or SBRS (Fig. 2, D–F) were observed in either group after DOCA treatment.
Fig. 2.

Blood pressure (BP), heart rate (HR), and spontaneous baroreflex sensitivity (SBRS) following DOCA treatment. A: systolic BP (sBP). B: diastolic BP (dBP). C: MAP. D: pulse pressure. E: HR. F: SBRS. Measurements were obtained at baseline and on day 21 of DOCA treatment. #P < 0.05 vs. baseline in the same genotype; two-way ANOVA (n = 6/group).
At baseline, mice preferred drinking regular water (daily intake: 3.2 ± 0.5 and 3.2 ± 0.2 ml for WT and PRRKO mice, respectively) to 0.9% saline (daily intake: 1.2 ± 0.3 and 0.9 ± 0.1 ml for WT and PRRKO mice, respectively) regardless of genotype (Fig. 3, A and C). We found that, following 21-day DOCA treatment, daily saline intake increased rapidly in both WT (saline: 27.7 ± 1.8 ml; water: 3.4 ± 0.6 ml) and PRRKO (saline: 15.9 ± 1.2 ml; water: 5.2 ± 0.3 ml) mice, but increases in water intake were small relative to baseline levels. As also shown by cumulative amounts of saline and water consumed (Fig. 3, B and D), WT mice drank significantly higher amounts of saline and lower amounts of water (saline: 376.9 ± 39.6 ml; water: 65.4 ± 4 ml) compared with PRRKO mice (saline: 238.7 ± 11.1 ml; water: 78.5 ± 2 ml). An analysis of daily saline and water intake by two-way ANOVA showed that saline intake was significantly lower in PRRKO mice (P = 0.0001; Fig. 3A), but water intake was similar (P = 0.229; Fig. 3C) compared with that in WT mice. Finally, the daily total volume of fluid consumed (water + saline) and cumulative fluid intake volume were lower in PRRKO mice compared with WT mice (P = 0.0001; Fig. 3E).
Fig. 3.

Deletion of the neuronal PRR reduces saline intake and total fluid intake in DOCA-treated mice. Daily saline intake (A) and cumulative saline intake (B) at 21 days of DOCA treatment. Daily regular water intake (C) and cumulative regular water intake (D) at 21 days of DOCA treatment. Daily saline plus water intake (E) and cumulative saline plus water intake (F) at 21 days of DOCA treatment. *P < 0.05 vs. WT at the same time point; **P < 0.001 vs. WT at the end of treatment; #P < 0.05 vs. day 0 in mice with the same genotype; Student’s t-test or two-way ANOVA (n = 5/group).
PRR deletion in neurons reduces sodium preference in DOCA-treated mice.
To evaluate the role of the neuronal PRR in sodium preference, we calculated sodium appetite/preference, defined as the ratio of saline intake to total fluid intake. As shown in Fig. 4A, this ratio at baseline was less than 0.5 in both WT (0.26 ± 0.06) and PRRKO (0.19 ± 0.02) mice, indicating a preference for water over saline when provided ad libitum. The preference for saline over water in WT mice increased rapidly following DOCA treatment, reaching a plateau of 0.89 ± 0.02; sodium preference was also increased in PRRKO mice following the 21-day protocol (0.75 ± 0.02). Importantly, sodium preference was significantly lower in PRRKO mice compared with WT mice (P = 0.0138) at the end of the protocol. To determine the impact of PRRKO on total sodium intake following DOCA treatment, we calculated the total amount of sodium consumed (Fig. 4B). Compared with WT mice, PRRKO mice ingested a significantly smaller amount of sodium, determined as daily and cumulative sodium (WT: 58.0 ± 6.1 mmol; PRRKO: 36.8 ± 1.7 mmol) (P < 0.01; Fig. 4C). Taken together, these data suggest that the neuronal PRR plays an important role in sodium intake.
Fig. 4.
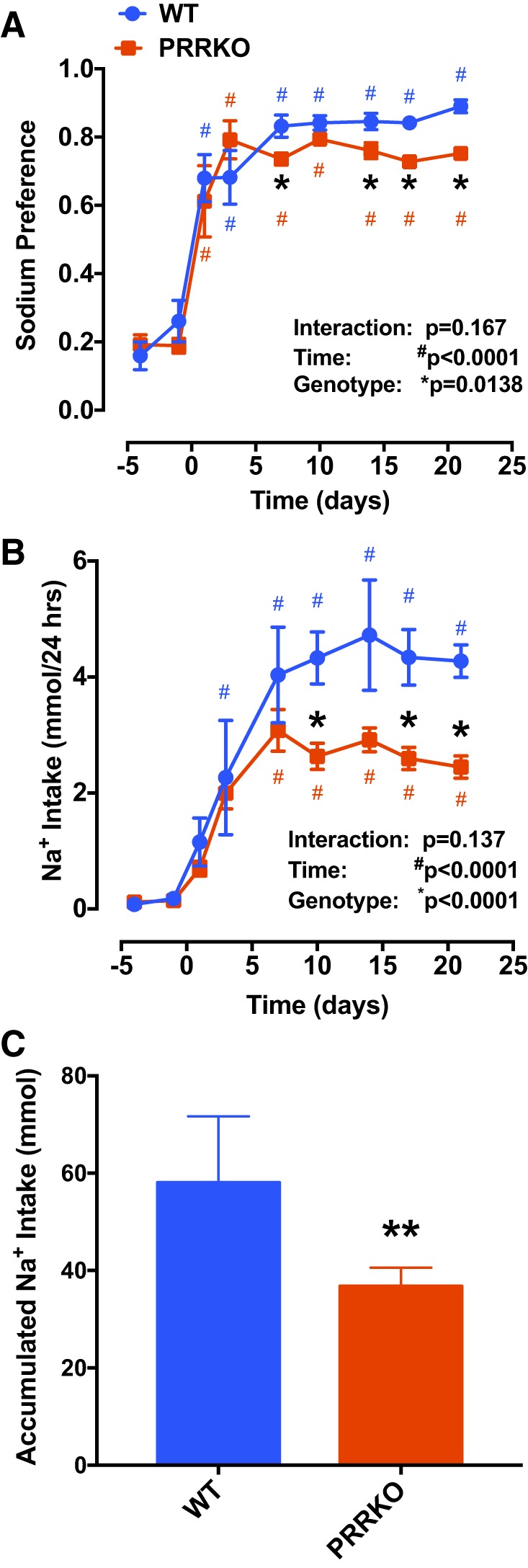
Reduction in sodium intake in neuron-specific PRRKO mice during DOCA treatment. A: sodium preference, calculated as the ratio of saline intake volume to total fluid intake volume. B: daily sodium intake. C: cumulative sodium intake. *P < 0.05 vs. WT at the same time point; **P < 0.001 vs. WT at the end of treatment; #P < 0.05 vs. day 0 in mice with the same genotype; Student’s t-test or two-way ANOVA (n = 5/group).
Neuronal PRR modulates urine excretion and urinary electrolytes.
Following DOCA treatment, daily urine excretion in WT mice gradually increased from baseline (1.6 ± 0.1 ml/day) to the end of the 21-day protocol (20 ± 1.1 ml/day). Neuronal PRRKO significantly reduced both daily (P < 0.0001) and cumulative (P = 0.04) urine output induced by DOCA stimulation (Fig. 5, A and B). Measurements of the osmolality of urine at the end of the study showed significantly higher osmolality in PRRKO mice (1,643 ± 44 osmol/kgH2O) compared with WT mice (1,414 ± 72 osmol/kgH2O; P = 0.03), possibly due to the diminished volume of urine excreted by PRRKO mice (Fig. 5C).
Fig. 5.

The neuronal PRR modulates urine excretion and urinary electrolyte homeostasis. A: daily urine excretion. B: cumulative urine excretion. C: urine osmolality at the end of the protocol. D: daily urinary sodium excretion. E: cumulative urinary sodium excretion. F: urinary sodium concentration. G: daily urinary potassium excretion. H: cumulative urinary potassium excretion. I: urinary potassium concentration. *P < 0.05 vs. WT at the same time point; #P < 0.05 vs. day 0 in mice with the same genotype; Student’s t-test or two-way ANOVA (n = 5/group).
Daily urinary sodium excretion (Fig. 5D), calculated from the 24 h urine output and urinary sodium concentration, was significantly higher in WT compared with PRRKO mice during the 21 days of DOCA treatment (P = 0.002). Cumulative urinary sodium excretion (WT: 14.3 ± 1.5 mmol vs. PRRKO: 10.9 ± 0.8 mmol) trended lower in PRRKO mice but was not significantly different (P = 0.069; Fig. 5E). Interestingly, urinary sodium concentration was transiently elevated in both WT and PRRKO mice on day 3 of DOCA treatment (P < 0.0001; Fig. 5F), after which it returned to baseline levels (P > 0.05). However, a two-way ANOVA revealed a higher urinary sodium concentration in PRRKO mice compared with WT mice throughout the protocol (P = 0.011; Fig. 5F). Following DOCA implantation, urinary potassium excretion significantly increased in WT mice, especially at the end of the protocol, compared with that at baseline. However, overall urinary potassium excretion was similar between the genotypes (P = 0.108; Fig. 5, G and H). Urinary potassium concentration was markedly reduced in both groups of mice (Fig. 5I) during the first week (days 3 and 7) of treatment (P < 0.0001) compared with baseline (day 0) values and stabilized at this low level until the end of the experiment. There was a small, but significant, increase in urinary potassium concentration in PRRKO compared with WT mice. Daily and cumulative potassium excretion was similar between WT and PRRKO mice (Fig. 5H).
We further estimated the sodium balance by subtracting urinary sodium excretion from sodium intake (Fig. 6). Interestingly, following DOCA treatment, both WT and PRRKO mice tended to have an initial increase in positive sodium balance (P = 0.015), a phenomenon of mineralocorticoid “escape,” but this balance subsequently returned to normal. A two-way ANOVA revealed an overall lower positive sodium balance in PRRKO compared with WT mice during DOCA treatment (P = 0.005).
Fig. 6.
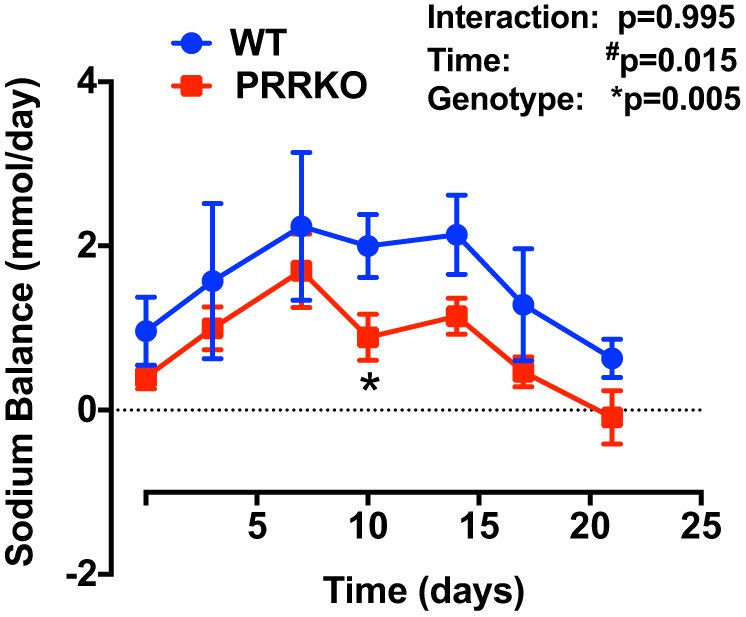
Effects of DOCA on sodium balance in WT and PRRKO mice. Sodium balance, calculated as urinary sodium excretion subtracted from sodium intake (n = 5/group). *P < 0.05 vs. WT at the same time point; #P < 0.05 vs. day 0 in mice with the same genotype; Student’s t-test or two-way ANOVA (n = 5/group).
DISCUSSION
The importance of the neuronal PRR in hypertension and other cardiovascular diseases has been demonstrated by a number of research groups (50, 54). Despite extensive evidence supporting this involvement (54), the mechanisms of action of the PRR remain incompletely understood. We previously reported that PRR deletion in neurons or blockade of the PRR by an antagonist attenuates the development of DOCA salt-induced hypertension (1, 34, 35). These previous studies used a model of DOCA salt-induced hypertension in which mice were given saline only in their drinking solution. Thus, we were unable to determine whether the PRR played a role in the regulation of sodium intake and preference if water is available. In the present study, we sought to investigate the role of the PRR in sodium intake in response to DOCA by giving mice free access to both saline and regular water. We found that neuronal PRR deletion attenuated DOCA-induced saline (0.9% sodium chloride) intake and modulated body fluid and electrolyte homeostasis.
In most experimental rodent models, a combination of the steroid DOCA and excess dietary salt is required to induce a series of pathological features, including elevated BP and the development of aortic aneurysms (12, 24, 36). In mice supplied saline as the sole drinking solution, DOCA induces a marked elevation in BP (~30 mmHg) and causes hypertension (7, 26). However, when offered both saline and water solutions ad libitum, as in the current study, WT mice treated with DOCA exhibited a modest, but significant, increase in BP (~15 mmHg) without reaching a hypertensive stage. These data suggest that mice with fully functional kidneys are able to balance their body fluid homeostasis and sodium excretion, as shown in Fig. 5. Sodium and total fluid volume intake were higher in WT mice treated with DOCA, and these mice also excreted more sodium by increasing their urine excretion volume. The net sodium balance was only slightly smaller in PRRKO mice compared with the WT mice as showed in Fig. 6. This observation possibly explains the absence of a difference in BP between PRRKO and WT mice in our model.
It is well established that aldosterone and ANG II are two key factors that stimulate sodium appetite (20, 49). An increase in sodium appetite is a normal physiological response to sodium deprivation. This increase in sodium appetite stimulates aldosterone production in the adrenal cortex and thereby promotes sodium conservation by the kidney. Circulating ANG II plays an important role in the regulation of thirst, water-intake behavior, and maintenance of blood volume, with brain ANG II being a stronger stimulus for both sodium appetite and water intake (4, 6, 10, 51). Our study suggests that the brain PRR, a key component of the local brain ANG II-formation cascade, is part of the molecular signaling interacting with the mineralocorticoid receptor system that stimulates sodium intake, but additional molecular mechanisms and neural circuits remain to be determined. In addition to acting as a normal physiological response, increased sodium intake has been shown to be a pathological behavior induced by high levels of mineralocorticoid in animal models of salt-sensitive hypertension and heart failure (14, 21). Although the relative importance of sodium appetite in various disease states in humans remains unclear (23), salt restriction is recommended and has been shown to be effective in managing salt-sensitive hypertension and heart failure, given patient compliance (26, 27, 52).
The brain regions responsible for sodium appetite and sensing have been extensively studied (23). Previous lesion studies in the lamina terminalis, where the SFO is located, revealed important connections necessary for ANG II-induced sodium appetite, but not DOCA-induced sodium appetite (13, 18). However, a separate study showed that lesion of the SFO in rats results in elevated DOCA-induced sodium intake (40). More recent studies using cell-specific targeting and optogenetic approaches have shown that SFO neurons play an essential role in the regulation of sodium appetite (8, 23, 37). More importantly, signaling between ANG II-sensitive neurons in the SFO and aldosterone-sensitive neurons in the NTS in the ventral part of the bed nucleus of the stria terminalis synergize in the regulation of sodium intake (41). The present study showed that PRRKO reduced DOCA-induced sodium intake, a finding that does not completely agree with those lesion studies but is more in line with concepts developed in recent studies on the synergy of SFO and NTS. Since the SFO contains both type 1 angiotensin II receptor (AT1R)-positive excitatory neurons and GABAergic inhibitory neurons (37), which regulate sodium appetite and sodium intake, lesion of the SFO removes the entire structure and both types of neurons, possibly including interactions with or inputs from aldosterone-sensitive neurons. Discrepancies between previous lesion studies and the current study regarding DOCA-induced sodium appetite might attributable to the experimental approaches used.
A number of previous studies have shown that the brain RAS is involved in stimulation of sodium intake and that actions of RAS components, especially ANG II, in the SFO and lamina terminalis (2, 5, 25, 29, 44). We recently showed that the PRR is a functional component of the brain RAS that mediates ANG II formation and regulates other RAS components (1). In the current study, we found that deletion of the PRR reduces sodium intake, providing new evidence that the brain PRR, acting as part of the RAS, is important in sodium intake, possibly by reducing the effect of DOCA on the brain RAS, behavior, or both. It has been shown that RAS blockers suppress sodium appetite, an effect that is mainly attributable to blocking the central actions of ANG II (19, 23). For example, peripheral administration of an extremely large amount (e.g., 100 mg/kg) of the ACE inhibitor, captopril, or coadministration of an ACE inhibitor and AT1R blocker is required to reduce saline intake after a physiological sodium deficiency, whereas a much smaller amount is needed if directly administered into the brain (15, 16). We propose that the brain PRR contributes to DOCA-induced sodium intake through a mechanism that may be mediated by ANG II (54). The PRR also mediates an ANG-II-independent signaling cascade in the CNS (32, 50). It is not clear from the current study whether the ANG II-independent PRR signaling pathway is also involved, although this possibility cannot be excluded. Further studies are warranted to address this question.
In the original study by Richter (43), it was shown that adrenalectomized rats have an increased appetite for sodium-containing solutions, including sodium lactate, sodium phosphate, and sodium chloride, but not chlorides of iron, magnesium, calcium, or aluminum, among others. A more recent study by David et al. (11) showed that central ANG II increases appetite for sodium bicarbonate, but not sodium chloride. Both of these studies revealed an increased appetite for sodium, but not for other ions. In addition, Rice and Richter (42) showed that DOCA increases sodium chloride and water intake by normal rats, a finding similar to that reported in our current mouse study. However, a more recent study showed that preference for saline is not altered following DOCA treatment in mice with a CD-1 background (45). Since our study used C57Bl/6J mice and mice with a possible residual FVB/N background, differences in DOCA-induced sodium intake could be attributable to differences in mouse strains used. We conclude from our study that PRRKO affects DOCA-induced isotonic NaCl intake in mice. However, further work is required to demonstrate whether the effect is selective for solutions containing the mineral sodium (e.g., sodium bicarbonate, sodium chloride) or any other type of mineral solution (e.g., potassium chloride, calcium chloride, etc.).
In summary, the key findings of the current study are that neuronal deletion of the PRR results in a significant reduction in sodium intake and total fluid intake following DOCA treatment. We conclude that the brain PRR acts in conjunction with activation of the DOCA/mineralocorticoid receptor to play a role in regulating sodium intake.
Perspective
A better understanding of the mechanisms responsible for sodium appetite and intake behavior may make it possible to design PRR-targeted therapies that aid patients in reducing their hunger for salt. However, understanding the fundamental mechanisms of sodium appetite regulation by the brain RAS and dissecting the neural circuitry involved remain critically important scientific questions, quite apart from potential therapeutic applications. Our current study provides a glimpse into this topic. Considering the complexity of the neural circuits and systems involved, further investigations designed to map the brain PRR and angiotensinergic neural circuits in the regulation of sodium appetite will require integrated approaches. One of the significant findings of this study is that deletion of the neuronal PRR decreases DOCA-induced sodium intake. Going forward, potential pharmacological interventions targeting the brain PRR to decrease sodium appetite and intake in animal models of salt-sensitive hypertension or heart failure would be of considerable interest. Further investigations are warranted to broaden our understanding of the complex mechanisms that regulate salt appetite during physiological responses to a lack of sodium or in pathophysiological disease states.
GRANTS
This work was supported, in part, by National Heart, Lung, and Blood Institute Grant R01HL-122770 and American Heart Association National Center Grant 17IRG33370128 to Y. Feng. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the granting agencies.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
F.T., W.L., and Y.F. conceived and designed research; F.T. and W.L. performed experiments; F.T., W.L., and Y.F. analyzed data; F.T., W.L., and Y.F. interpreted results of experiments; F.T., W.L., and Y.F. prepared figures; F.T. and Y.F. drafted manuscript; F.T., W.L., and Y.F. edited and revised manuscript; F.T., W.L., and Y.F. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Wei Yang at the School of Community Health Sciences, University of Nevada, Reno, for helping with ANCOVA and the expert inputs to the statistical analysis for this manuscript.
REFERENCES
- 1.Aoki H, Sadoshima J, Izumo S. Myosin light chain kinase mediates sarcomere organization during cardiac hypertrophy in vitro. Nat Med 6: 183–188, 2000. doi: 10.1038/72287. [DOI] [PubMed] [Google Scholar]
- 2.Avrith DB, Fitzsimons JT. Increased sodium appetite in the rat induced by intracranial administration of components of the renin-angiotensin system. J Physiol 301: 349–364, 1980. doi: 10.1113/jphysiol.1980.sp013210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blaustein MP, Leenen FHH, Chen L, Golovina VA, Hamlyn JM, Pallone TL, Van Huysse JW, Zhang J, Wier WG. How NaCl raises blood pressure: a new paradigm for the pathogenesis of salt-dependent hypertension. Am J Physiol Heart Circ Physiol 302: H1031–H1049, 2012. doi: 10.1152/ajpheart.00899.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buggy J, Fink GD, Johnson AK, Brody MJ. Prevention of the development of renal hypertension by anteroventral third ventricular tissue lesions. Circ Res 40: I110–I117, 1977. [PubMed] [Google Scholar]
- 5.Buggy J, Fisher AE. Evidence for a dual central role for angiotensin in water and sodium intake. Nature 250: 733–735, 1974. doi: 10.1038/250733a0. [DOI] [PubMed] [Google Scholar]
- 6.Buggy J, Fisher AE, Hoffman WE, Johnson AL, Phillips MI. Ventricular obstruction: effect on drinking induced by intracranial injection of angiotensin. Science 190: 72–74, 1975. doi: 10.1126/science.1166302. [DOI] [PubMed] [Google Scholar]
- 7.Butler J, Papadimitriou L, Georgiopoulou V, Skopicki H, Dunbar S, Kalogeropoulos A. Comparing Sodium Intake Strategies in Heart Failure: Rationale and Design of the PROHIBIT Sodium (PRevent adverse Outcomes in Heart faIlure By limITing Sodium) Study. Circ Heart Fail 8: 636–645, 2015. doi: 10.1161/CIRCHEARTFAILURE.114.001700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cerqueira DR, Ferreira HS, Moiteiro ALBB, Fregoneze JB. Effects of interleukin-1 beta injections into the subfornical organ and median preoptic nucleus on sodium appetite, blood pressure and body temperature of sodium-depleted rats. Physiol Behav 163: 149–160, 2016. doi: 10.1016/j.physbeh.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 9.Coghlan JP, Considine PJ, Denton DA, Fei DT, Leksell LG, McKinley MJ, Muller AF, Tarjan E, Weisinger RS, Bradshaw RA. Sodium appetite in sheep induced by cerebral ventricular infusion of angiotensin: comparison with sodium deficiency. Science 214: 195–197, 1981. doi: 10.1126/science.6169149. [DOI] [PubMed] [Google Scholar]
- 10.Daniels D. Frontiers in neuroscience diverse roles of angiotensin receptor intracellular signaling pathways in the control of water and salt intake. In: Neurobiology of Body Fluid Homeostasis: Transduction and Integration, edited by De Luca LA Jr, Menani JV, Johnson AK. Boca Raton, FL: CRC, 2014. [PubMed] [Google Scholar]
- 11.David RB, Menani JV, De Luca LA Jr. Central angiotensin II induces sodium bicarbonate intake in the rat. Appetite 51: 82–89, 2008. doi: 10.1016/j.appet.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 12.de Gracia MC, Osuna A, O’Valle F, del Moral RG, Wangensteen R, del Rio CG, Vargas F. Deoxycorticosterone suppresses the effects of losartan in nitric oxide-deficient hypertensive rats. J Am Soc Nephrol 11: 1995–2000, 2000. [DOI] [PubMed] [Google Scholar]
- 13.De Luca LA Jr, Galaverna O, Schulkin J, Yao S-Z, Epstein AN. The anteroventral wall of the third ventricle and the angiotensinergic component of need-induced sodium intake in the rat. Brain Res Bull 28: 73–87, 1992. doi: 10.1016/0361-9230(92)90233-N. [DOI] [PubMed] [Google Scholar]
- 14.DiNicolantonio R, Hutchinson JS, Mendelsohn FA. Exaggerated salt appetite of spontaneously hypertensive rats is decreased by central angiotensin-converting enzyme blockade. Nature 298: 846–848, 1982. doi: 10.1038/298846a0. [DOI] [PubMed] [Google Scholar]
- 15.Elfont RM, Epstein AN, Fitzsimons JT. Involvement of the renin-angiotensin system in captopril-induced sodium appetite in the rat. J Physiol 354: 11–27, 1984. doi: 10.1113/jphysiol.1984.sp015359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evered MD, Robinson MM. Effects of captopril on salt appetite in sodium-replete rats and rats treated with desoxycorticosterone acetate (DOCA). J Pharmacol Exp Ther 225: 416–421, 1983. [PubMed] [Google Scholar]
- 17.Feng Y, Yue X, Xia H, Bindom SM, Hickman PJ, Filipeanu CM, Wu G, Lazartigues E. Angiotensin-converting enzyme 2 overexpression in the subfornical organ prevents the angiotensin II-mediated pressor and drinking responses and is associated with angiotensin II type 1 receptor downregulation. Circ Res 102: 729–736, 2008. doi: 10.1161/CIRCRESAHA.107.169110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fitts DA. Effects of lesions of the ventral ventral median preoptic nucleus or subfornical organ on drinking and salt appetite after deoxycorticosterone acetate or yohimbine. Behav Neurosci 105: 721–726, 1991. doi: 10.1037/0735-7044.105.5.721. [DOI] [PubMed] [Google Scholar]
- 19.Fitzsimons JT. Angiotensin, thirst, and sodium appetite. Physiol Rev 78: 583–686, 1998. doi: 10.1152/physrev.1998.78.3.583. [DOI] [PubMed] [Google Scholar]
- 20.Fluharty SJ, Epstein AN. Sodium appetite elicited by intracerebroventricular infusion of angiotensin II in the rat: II. Synergistic interaction with systemic mineralocorticoids. Behav Neurosci 97: 746–758, 1983. doi: 10.1037/0735-7044.97.5.746. [DOI] [PubMed] [Google Scholar]
- 21.Francis J, Weiss RM, Wei SG, Johnson AK, Beltz TG, Zimmerman K, Felder RB. Central mineralocorticoid receptor blockade improves volume regulation and reduces sympathetic drive in heart failure. Am J Physiol Heart Circ Physiol 281: H2241–H2251, 2001. doi: 10.1152/ajpheart.2001.281.5.H2241. [DOI] [PubMed] [Google Scholar]
- 22.Geerling JC, Kawata M, Loewy AD. Aldosterone-sensitive neurons in the rat central nervous system. J Comp Neurol 494: 515–527, 2006. doi: 10.1002/cne.20808. [DOI] [PubMed] [Google Scholar]
- 23.Geerling JC, Loewy AD. Central regulation of sodium appetite. Exp Physiol 93: 177–209, 2008. doi: 10.1113/expphysiol.2007.039891. [DOI] [PubMed] [Google Scholar]
- 24.Goodwin FJ, Knowlton AI, Laragh JH. Absence of renin suppression by deoxycorticosterone acetate in rats. Am J Physiol 216: 1476–1480, 1969. [DOI] [PubMed] [Google Scholar]
- 25.Grafe LA, Takacs AE, Yee DK, Flanagan-Cato LM. The role of the hypothalamic paraventricular nucleus and the organum vasculosum lateral terminalis in the control of sodium appetite in male rats. J Neurosci 34: 9249–9260, 2014. doi: 10.1523/JNEUROSCI.3979-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ha SK. Dietary salt intake and hypertension. Electrolyte Blood Press 12: 7–18, 2014. doi: 10.5049/EBP.2014.12.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hummel SL, Konerman MC. Dietary Sodium Restriction in Heart Failure: A Recommendation Worth its Salt? JACC Heart Fail 4: 36–38, 2016. doi: 10.1016/j.jchf.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 28.Hurley SW, Johnson AK. The biopsychology of salt hunger and sodium deficiency. Pflugers Arch 467: 445–456, 2015. doi: 10.1007/s00424-014-1676-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hurley SW, Zhang Z, Beltz TG, Xue B, Johnson AK. Sensitization of sodium appetite: evidence for sustained molecular changes in the lamina terminalis. Am J Physiol Regul Integr Comp Physiol 307: R1405–R1412, 2014. doi: 10.1152/ajpregu.00210.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jarvie BC, Palmiter RD. HSD2 neurons in the hindbrain drive sodium appetite. Nat Neurosci 20: 167–169, 2017. doi: 10.1038/nn.4451. [DOI] [PubMed] [Google Scholar]
- 31.Kinouchi K, Ichihara A, Sano M, Sun-Wada G-H, Wada Y, Kurauchi-Mito A, Bokuda K, Narita T, Oshima Y, Sakoda M, Tamai Y, Sato H, Fukuda K, Itoh H. The (pro)renin receptor/ATP6AP2 is essential for vacuolar H+-ATPase assembly in murine cardiomyocytes. Circ Res 107: 30–34, 2010. doi: 10.1161/CIRCRESAHA.110.224667. [DOI] [PubMed] [Google Scholar]
- 32.Krop M, Lu X, Danser AH, Meima ME. The (pro)renin receptor. A decade of research: what have we learned? Pflugers Arch 465: 87–97, 2013. doi: 10.1007/s00424-012-1105-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuehn BM. How Low Is Too Low With Salt in Heart Failure? Randomized Studies Needed to Resolve Concern. Circulation 136: 597–598, 2017. doi: 10.1161/CIRCULATIONAHA.117.030211. [DOI] [PubMed] [Google Scholar]
- 34.Li W, Peng H, Mehaffey EP, Kimball CD, Grobe JL, van Gool JM, Sullivan MN, Earley S, Danser AH, Ichihara A, Feng Y. Neuron-specific (pro)renin receptor knockout prevents the development of salt-sensitive hypertension. Hypertension 63: 316–323, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li W, Sullivan MN, Zhang S, Worker CJ, Xiong Z, Speth RC, Feng Y. Intracerebroventricular infusion of the (Pro)renin receptor antagonist PRO20 attenuates deoxycorticosterone acetate-salt-induced hypertension. Hypertension 65: 352–361, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu S, Gong MC, Guo Z. A New Mouse Model for Introduction of Aortic Aneurysm by Implantation of Deoxycorticosterone Acetate Pellets or Aldosterone Infusion in the Presence of High Salt. Methods Mol Biol 1614: 155–163, 2017. doi: 10.1007/978-1-4939-7030-8_12. [DOI] [PubMed] [Google Scholar]
- 37.Matsuda T, Hiyama TY, Niimura F, Matsusaka T, Fukamizu A, Kobayashi K, Kobayashi K, Noda M. Distinct neural mechanisms for the control of thirst and salt appetite in the subfornical organ. Nat Neurosci 20: 230–241, 2017. doi: 10.1038/nn.4463. [DOI] [PubMed] [Google Scholar]
- 38.Ohta Y, Kimura Y, Kitaoka C, Sakata T, Abe I, Kawano Y. Blood pressure control status and relationship between salt intake and lifestyle including diet in hypertensive outpatients treated at a general hospital. Clin Exp Hypertens 39: 29–33, 2017. doi: 10.1080/10641963.2016.1200605. [DOI] [PubMed] [Google Scholar]
- 39.Ohta Y, Tsuchihashi T, Ueno M, Kajioka T, Onaka U, Tominaga M, Eto K. Relationship between the awareness of salt restriction and the actual salt intake in hypertensive patients. Hypertens Res 27: 243–246, 2004. doi: 10.1291/hypres.27.243. [DOI] [PubMed] [Google Scholar]
- 40.Osborn JW, Jacob F, Hendel M, Collister JP, Clark L, Guzman PA. Effect of subfornical organ lesion on the development of mineralocorticoid-salt hypertension. Brain Res 1109: 74–82, 2006. doi: 10.1016/j.brainres.2006.06.073. [DOI] [PubMed] [Google Scholar]
- 41.Resch JM, Fenselau H, Madara JC, Wu C, Campbell JN, Lyubetskaya A, Dawes BA, Tsai LT, Li MM, Livneh Y, Ke Q, Kang PM, Fejes-Tóth G, Náray-Fejes-Tóth A, Geerling JC, Lowell BB. Aldosterone-Sensing Neurons in the NTS Exhibit State-Dependent Pacemaker Activity and Drive Sodium Appetite via Synergy with Angiotensin II Signaling. Neuron 96: 190–206.e7, 2017. doi: 10.1016/j.neuron.2017.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rice KK, Richter CP. Increased sodium chloride and water intake of normal rats treated with desoxycorticosterone acetate. Endocrinology 33: 106–115, 1943. doi: 10.1210/endo-33-2-106. [DOI] [Google Scholar]
- 43.Richter CP. Increased salt appetite in adrenalectomized rats. Am J Physiol 115: 155–161, 1936. doi: 10.1152/ajplegacy.1936.115.1.155. [DOI] [Google Scholar]
- 44.Roncari CF, Barbosa RM, Vendramini RC, De Luca LA Jr, Menani JV, Colombari E, Colombari DSA. Enhanced angiotensin II induced sodium appetite in renovascular hypertensive rats. Peptides 101: 82–88, 2018. doi: 10.1016/j.peptides.2017.12.025. [DOI] [PubMed] [Google Scholar]
- 45.Rowland NE, Fregly MJ. Characteristics of thirst and sodium appetite in mice (Mus musculus). Behav Neurosci 102: 969–974, 1988. doi: 10.1037/0735-7044.102.6.969. [DOI] [PubMed] [Google Scholar]
- 46.Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, Obarzanek E, Conlin PR, Miller ER III, Simons-Morton DG, Karanja N, Lin PH, Aickin M, Most-Windhauser MM, Moore TJ, Proschan MA, Cutler JA; DASH-Sodium Collaborative Research Group . Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. N Engl J Med 344: 3–10, 2001. doi: 10.1056/NEJM200101043440101. [DOI] [PubMed] [Google Scholar]
- 47.Sakai RR, Chow SY, Epstein AN. Peripheral angiotensin II is not the cause of sodium appetite in the rat. Appetite 15: 161–170, 1990. doi: 10.1016/0195-6663(90)90017-3. [DOI] [PubMed] [Google Scholar]
- 48.Sakai RR, Fine WB, Epstein AN, Frankmann SP. Salt appetite is enhanced by one prior episode of sodium depletion in the rat. Behav Neurosci 101: 724–731, 1987. doi: 10.1037/0735-7044.101.5.724. [DOI] [PubMed] [Google Scholar]
- 49.Sakai RR, Nicolaidis S, Epstein AN. Salt appetite is suppressed by interference with angiotensin II and aldosterone. Am J Physiol Regul Integr Comp Physiol 251: R762–R768, 1986. doi: 10.1152/ajpregu.1986.251.4.R762. [DOI] [PubMed] [Google Scholar]
- 50.Sun Y, Danser AHJ, Lu X. (Pro)renin receptor as a therapeutic target for the treatment of cardiovascular diseases? Pharmacol Res 125, Pt A: 48–56, 2017. doi: 10.1016/j.phrs.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 51.Weisinger RS, Blair-West JR, Burns P, Denton DA, Tarjan E. Role of brain angiotensin in thirst and sodium appetite of rats. Peptides 18: 977–984, 1997. doi: 10.1016/S0196-9781(97)00077-6. [DOI] [PubMed] [Google Scholar]
- 52.Wessler JD, Hummel SL, Maurer MS. Dietary interventions for heart failure in older adults: re-emergence of the hedonic shift. Prog Cardiovasc Dis 57: 160–167, 2014. doi: 10.1016/j.pcad.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia H, Feng Y, Obr TD, Hickman PJ, Lazartigues E. Angiotensin II type 1 receptor-mediated reduction of angiotensin-converting enzyme 2 activity in the brain impairs baroreflex function in hypertensive mice. Hypertension 53: 210–216, 2009. doi: 10.1161/HYPERTENSIONAHA.108.123844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu Q, Jensen DD, Peng H, Feng Y. The critical role of the central nervous system (pro)renin receptor in regulating systemic blood pressure. Pharmacol Ther 164: 126–134, 2016. doi: 10.1016/j.pharmthera.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]