

Abstract
Numerous studies have demonstrated that Na+/H+ exchanger isoform 1 (NHE1) is elevated in myocardial diseases and its effect is detrimental. To better understand the involvement of NHE1, we have previously studied cardiac-specific NHE1 transgenic mice and shown that these mice develop cardiac hypertrophy, interstitial fibrosis, and cardiac dysfunction. The purpose of current study was to identify microRNAs and their mRNA targets involved in NHE1-mediated cardiac injury. An unbiased high-throughput sequencing study was performed on both microRNAs and mRNAs. RNA sequencing showed that differentially expressed genes were enriched in hypertrophic cardiomyopathy pathway by Kyoto Encyclopedia of Genes and Genomes annotation in NHE1 transgenic hearts. These genes were classified as contraction defects (e.g., Myl2, Myh6, Mybpc3, and Actb), impaired intracellular Ca2+ homeostasis (e.g., SERCA2a, Ryr2, Rcan1, and CaMKII delta), and signaling molecules for hypertrophic cardiomyopathy (e.g., Itga/b, IGF-1, Tgfb2/3, and Prkaa1/2). microRNA sequencing revealed that 15 microRNAs were differentially expressed (2-fold, P < 0.05). Six of them (miR-1, miR-208a-3p, miR-199a-5p, miR-21-5p, miR-146a-5p, and miR-30c-5p) were reported to be related to cardiac pathological functions. The integrative analysis of microRNA and RNA sequencing data identified several crucial microRNAs including miR-30c-5p, miR-199a-5p, miR-21-5p, and miR-34a-5p as well as 10 of their mRNA targets that may affect the heart via NFAT hypertrophy and cardiac hypertrophy signaling. Furthermore, important microRNAs and mRNA targets were validated by quantitative PCR. Our study comprehensively characterizes the expression patterns of microRNAs and mRNAs, establishes functional microRNA-mRNA pairs, elucidates the potential signaling pathways, and provides novel insights on the mechanisms underlying NHE1-medicated cardiac injury.
Keywords: cardiac hypertrophy, cardiovascular diseases, high-throughput sequencing, miRNA-mRNA interaction, NHE1
INTRODUCTION
Despite dramatic medical advances over the past years, cardiovascular diseases (CVD) remain the leading global cause of death. According to the American Heart Association Statistics - 2016 Update (62), CVD accounted for 31% of all global deaths in 2013. Nearly 801,000 people in the US died from heart disease, stroke, and other cardiovascular diseases in 2013. That’s about one of every three deaths in America. Therefore, continued research efforts are required to help improve the survival and quality of life of people suffering from CVD.
The mammalian Na+/H+ exchangers (NHEs) are a family of plasma membrane transport proteins that mediate an electroneutral 1:1 exchange of intracellular H+ for extracellular Na+ and, in so doing, regulate intracellular pH (pHi) and cell volume. To date, 10 NHE isoforms have been identified. NHE isoform 1 (NHE1) is ubiquitously expressed in the body and is the predominant NHE isoform in the sarcolemma of cardiomyocytes (18). Besides being a regulator of pHi and ionic homeostasis, NHE1 also acts as an anchor for actin filaments and a scaffold for an ensemble of signaling complexes. Thus NHE1 regulates a diverse range of cell behaviors, including migration, proliferation, growth, differentiation, cytoskeleton dynamics, and survival (44).
NHE1 has attracted significant attention in CVD field because 1) NHE1 protein expression and activity are elevated in various forms of cardiac injuries, including in hypertensive, hypertrophied, and diabetic myocardium (6, 15, 30, 33), in hearts subjected to ischemia (21), in isolated cardiomyocytes subjected to chronic acidosis (14), as well as in patients with end-stage heart failure (64), 2) this increase in NHE1 have been shown to mediate pathological changes and dysfunction in the heart (32, 34), 3) a plethora of experimental studies have demonstrated salutary effects of NHE inhibition in protecting the myocardium against ischemic and reperfusion injury as well as attenuating myocardial remodeling and heart failure (1, 9, 16, 18, 34), and 4) cardiac-specific overexpression of activated NHE1 itself is sufficient to induce cardiac hypertrophy, fibrosis, and dysfunction in two independent transgenic mouse lines (47, 63).
The purpose of the current study was to investigate the molecular basis of NHE1-mediated cardiac injury. In the past decade, emerging evidence has indicated that microRNAs (miRNAs) are powerful and dynamic modifiers of CVD (11, 12, 22, 37, 61). miRNAs are small noncoding RNAs that regulate gene expression through inhibiting mRNA translation or promoting mRNA degradation. miRNAs play essential roles in mediating many biological processes as well as the pathogenesis of human diseases such as CVD, cancer, and neurological disorders. Thus, the expression of mRNAs and miRNAs was profiled through an unbiased deep-sequencing study. With the aid of bioinformatics tools, miRNA-mRNA regulatory networks were explored. We identified several critical miRNA-mRNA pairs and elucidated potential signaling pathways. The knowledge obtained help us better understand the roles of miRNA-mRNA in cardiac pathology mediated by NHE1.
MATERIALS AND METHODS
NHE1 transgenic mice.
Transgenic mice that expressed constitutively activated NHE1 in the mouse myocardium have been described earlier (2, 10, 28). The NHE1 construct has a mutation in the cytoplasmic regulatory calmodulin binding domain of the protein (Lys641, Arg643, Arg645, and Arg647 were mutated to Glu residues). The mutation causes activation of NHE1 via an alkaline shift in NHE1 pH dependency (3, 10, 28). The expression of the transgene is under control of the cardiac-specific alpha myosin heavy chain promoter. Female mice at the age of 7 mo old were used for all experiments and age-matched nontransgenic littermates were used as controls. All animal protocols (#S05534) were approved by the Institutional Animal Care and Use Committee at University of California San Diego.
Histology.
Hearts were fixed in 10% buffered formalin overnight, embedded in paraffin, serially sectioned into 5 µm slices, and stained with hematoxylin and eosin (H&E) (63). Cross-sectional area (CSA) was measured by computer-assisted image analysis software (ImageJ, NIH Image) (53). Visual fields were accepted for quantification if the nuclei were visible and cell membranes were intact. Ten fields were randomly selected from two sections per heart. Dimensions of ~500 cells per heart were measured. Five hearts were examined for each group (wild-type control vs. NHE1 transgenic). Interstitial fibrosis (IF) was visualized and determined with picro-sirius red staining. Ten fields were randomly selected from two sections per heart. Three hearts were examined for each group (wild-type control vs. NHE1 transgenic). The maximum fibrosis observed for any section was calculated as the area occupied by red-stained connective tissue divided by the areas occupied by connective tissue plus cardiac myocytes × 100. Intramural vessels, perivascular collagen, endocardium, and trabeculae were excluded (63). All the measurements were done by two blinded investigators. Data are presented as means ± SE. Student's t-test was employed, and P < 0.05 was considered statistically significant.
Small and total RNA isolation.
Small RNA (including miRNA) and total RNA were isolated using mirVana miRNA Isolation Kit (AM1560; Life Technologies, Grand Island, NY) according to manufacturer’s instructions. RNA yield was measured using a NanoDrop 1000 (Thermo Fisher Scientific, Wilmington, DE). The RNA quality was examined by Agilent Bioanalyzer with an RNA 6000 Nano Kit (Agilent Technologies, Santa Clara, CA). RNA samples with an RNA integrity number >8 were used for sequencing.
Library construction for the sequencing.
Total RNA (10 μg) was treated with RiboMinus Transcriptome Isolation Kit (Life Technologies, Grand Island, NY). Using 200 ng of rRNA depleted RNA, we constructed mRNA-Seq libraries with ScriptSeq mRNA-Seq Library Preparation Kit (Epicentre, Madison, WI) according to manufacturer’s instructions. Small RNA libraries were constructed with ScriptMiner Small RNA-Seq library prep kit (Epicentre) according to manufacturer’s instructions. We used 100 ng of small RNA for each library. Libraries were checked on a Bioanalyzer with Agilent DNA High Sensitivity Kit (Agilent Technologies) and quantified with quantitative PCR using a KAPA Library Quantification Kit for Illumina Sequencing (KAPA Biosystems, Wilmington, MA). RNA sequencing (RNA-Seq) was done with 10 pM of total RNA libraries, paired end read at 75 cycles each, and miRNA sequencing was done with 10 pM of small RNA libraries, single read at 36 cycles, both using the Illumina Genome Analyzer IIx (Illumina, San Diego, CA).
Transcriptome profiling with RNA-Seq.
Libraries were prepared with three replicates from both control and NHE1 transgenic left ventricle tissue and sequenced using a 75 cycle paired end run on an Illumina platform. The resulting reads were aligned with TopHat2 (36) with default settings for Mus musculus using the mm9 genome index from Illumina iGenome. The aligned reads were processed with Cufflinks (57) to produce a list of differentially expressed genes. The resulting gene was filtered to remove gene where q > 0.05 and the calculated fold change was <1.5×. The data set of sequence analyses can be traced by accession number SRP118869 in the National Center for Biotechnology Information Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra).
miRNA profiling with miRNA-Seq.
Libraries prepared from left ventricle samples of control and NHE1 transgenic mice were sequenced on an Illumina platform using a 36 cycle single end run. Two different methods for determining miRNA expression levels were used on the resulting reads. First, miRNAKey (52) was used to produce a report of both mature and hairpin miRNAs that were differentially expressed in NHE1 transgenic hearts relative to controls. Additionally, miRanalyzer (23) was used to perform the same task to compare the sensitivity of the different methods employed by both pieces of software. The data set of sequence analyses can be traced by accession number SRP118869 in the National Center for Biotechnology Information SRA (https://www.ncbi.nlm.nih.gov/sra).
Functional categorization of differentially expressed mRNAs and miRNAs.
With the aid of The Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.7 program (https://david.abcc.ncifcrf.gov/) (26), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotation (31) was performed on the differentially expressed genes to facilitate biological interpretation of gene function in a network context. Fisher exact test with Benjamini-Hochberg (B-H) correction was used to select significant pathways. The threshold for significance was P < 0.05. Ingenuity Pathways Analysis program (Ingenuity Systems, Redwood City, CA; https://www.ingenuity.com) was chosen to classify differentially expressed miRNAs in terms of cardiac pathologies. The B-H multiple-testing corrected P value was used. The score for each function was shown as –log10 [B-H P value]. The significance threshold was set to a score of 1.3 (i.e., P ≤ 0.05).
qRT-PCR.
Seven differentially expressed miRNAs randomly selected from miRNA-Seq data and seven differentially expressed mRNA targets of microRNA (miR)-30c were validated by qPCR (63). For miRNAs, the standard TaqMan MicroRNA Assays, which employ target-specific stem-loop reverse transcription primers for 3′ extended templates, were carried out according to the manufacturer's instructions (Applied Biosystems, Carlsbad, CA). For mRNAs, the primer sets for specific genes were designed using Primer3 program (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) and synthesized by Invitrogen (Invitrogen, Carlsbad, CA). The fold change in expression level for each specific miRNA and mRNA was calculated by the 2–ΔΔCt method (39), after normalization to snoRNA202 for miRNA and GAPDH for mRNA (loading control). The final result represents the mean fold change of three individual transgenic samples over controls. Student's t-test was performed and statistical significance was set to P < 0.05.
Integration of miRNA and RNA-Seq data.
Ingenuity Upstream Regulator Analysis (URA) was utilized to identify potential miRNA regulators upstream of significantly changed gene transcripts and to predict whether these miRNA regulators were activated or inhibited. URA is based on expected causal effects between upstream regulators and targets, which are derived from the literature compiled in the Ingenuity Knowledge Base. We examined the known targets of each upstream regulator in our RNA sequencing data set, compared the targets’ actual direction of change to expectations derived from the literature, and then issued a prediction for each upstream regulator. Two scores were used to address two independent aspects of the inference: an “enrichment” score [Fisher’s exact test (FET) P value] that measures overlap of observed and predicted regulated gene sets, and a Z-score that assesses the match of observed and predicted up-/downregulation patterns. An absolute z-score of ≥ 2 was considered significant. An upstream regulator is predicted to be activated if the z-score is ≥2 and inhibited if the z-score ≤−2. When both the z-score (absolute value >2) and the P value (<10E-3) were significant, the predictions were considered the most reliable. Ingenuity miRNA Target Filter was used to integrate the data sets of differentially expressed miRNAs and gene transcripts of NHE1 transgenic mouse hearts. miRNA-mRNA target relationships were obtained with reciprocal expression patterns and for relevance to cardiovascular signaling pathways.
RESULTS
Cardiac hypertrophy and fibrosis caused by increased NHE1 activation.
NHE1 transgenic hearts have previously been shown to possess approximately threefold increase in NHE1 activity and to have an enlargement in overall size at the age of 2–3 mo old (10, 28, 63). In this study we further characterized the cardiac hypertrophy and fibrosis by examining the cross sections of ventricular cardiomyocytes at the age of 7 mo old. Figure 1, top, represents myocardial cross sections stained with hematoxylin and eosin (H&E) (Fig. 1A) for CSA and with picro-sirius red (Fig. 1B) for IF. Figure 1, bottom, is a quantitative summary of the results, demonstrating that 1) NHE1 activation significantly increased CSA (205.4 ± 11.7%, P < 0.01) as compared with wild-type controls (100.0 ± 10.6%)(Fig. 1A) and 2) no evident fibrosis was detected in the controls, while a significant increase in fibrosis was observed by NHE1 activation (7.6% of area examined, P < 0.01)(Fig. 1B).
Fig. 1.
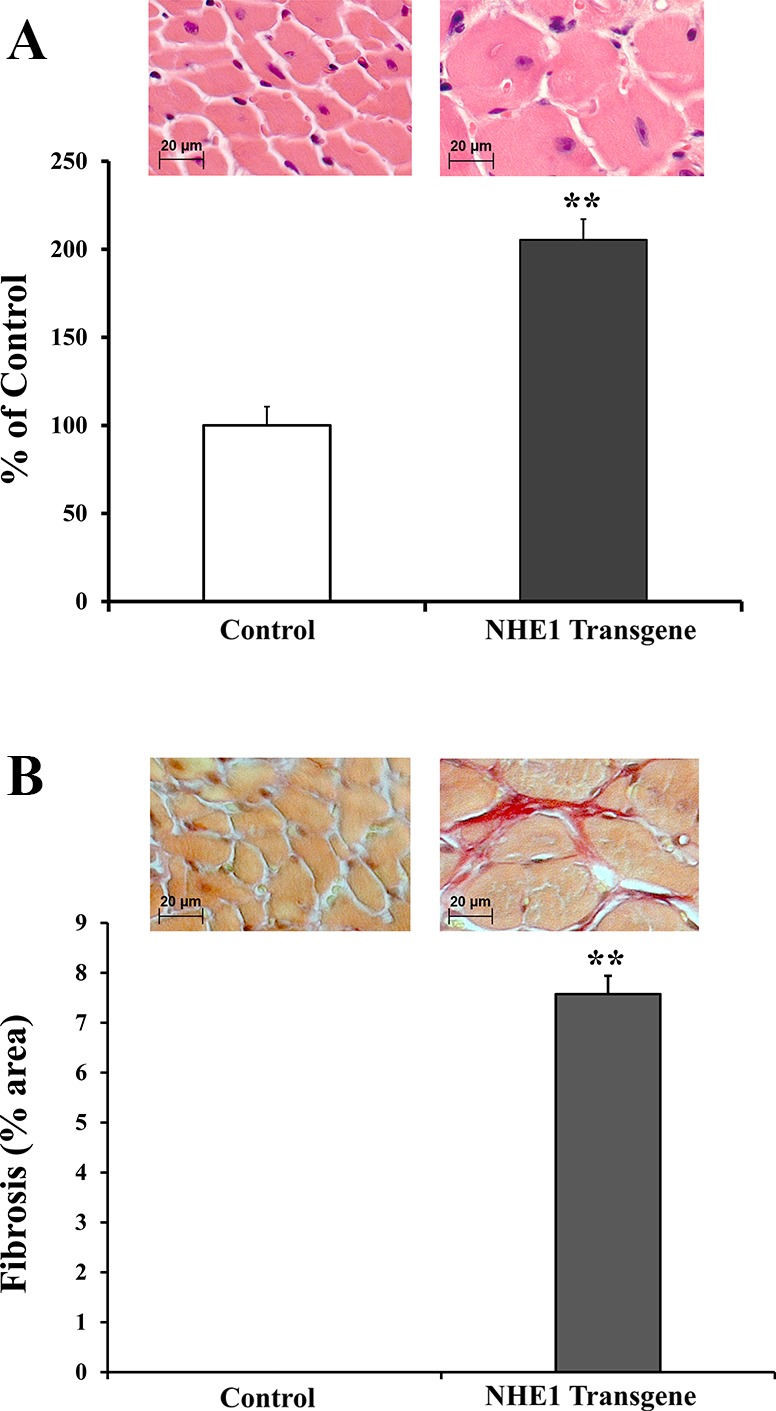
Elevation of activated Na+/H+ exchanger isoform 1 (NHE1) induced larger cardiomyocytes and interstitial fibrosis. Histological analysis of the cross sections (×40) of mouse hearts stained with hematoxylin and eosin for cross-sectional area(CSA) (A) and with picro-sirius red for interstitial fibrosis (IF) (B). Top: examples of cross sections from wild-type control and NHE1 transgenic heart. Bottom: summary of CSAs expressed as % of control and fibrosis as % of area examined. **P < 0.01.
Gene expression profile in NHE1 transgenic mouse hearts.
NHE1 transgene-induced gene expression changes were examined at the age of 7 mo, when cardiac pathologies, including cardiac hypertrophy, interstitial fibrosis, and cardiac dysfunction, have been fully developed (10). The fold change of each gene expression was calculated by the ratio of gene expression in NHE1 transgenic mouse hearts to that of the age-matched controls. NHE1 transgenic mice showed a strong transcriptional response (1,331 upregulations and 553 downregulations, P < 0.05) (Supplemental Table S1). (The online version of this article contains supplemental material.) The magnitude of expression alterations was mostly less than threefold (1,205 up and 476 down) (Fig. 2A). However, some genes showed more than fivefold change (32 up and 16 down) (Fig. 2A). Among them, a marked increase (22-fold up) of Slc9a1 was noted in NHE1 transgenic hearts, indicating that NHE1 gene was indeed overexpressed in the transgenic hearts. In addition, two hypertrophic markers, Nppa and Nppb, were increased 44- and 9-fold separately, correlating with a hypertrophy phenotype in NHE1 transgenic mice.
Fig. 2.

Analysis of gene expression profile in mouse hearts with elevated NHE1. A: number of magnitude of gene changes. B: significant Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways that were enriched with differentially expressed genes.
The differentially expressed genes were further subjected to KEGG pathway analysis to determine which significant pathways were enriched with these gene changes. The results revealed that seven KEGG pathways were significantly enriched in the NHE1 transgenic mouse hearts, including hypertrophic cardiomyopathy (HCM) (Fig. 2B). Table 1 lists the genes altered in NHE1 transgenic hearts that are enriched in KEGG HCM pathway. These genes were further classified into three functional categories (Fig. 3): 1) contraction defects, including Myl2, Myh6, Mybpc3, Actb, Des, and Lmna; 2) impaired intracellular Ca2+ homeostasis, such as SERCA2a, Ryr2, Rcan1, and CaMKII delta; and 3) signaling molecules that lead to HCM and eventually progress to heart failure, such as Itga, Itgb, ACE1, IGF-1, Tgfb2, Tgfb3, Prkaa1, and Prkab2.
Table 1.
Genes altered in NHE1 transgenic hearts was enriched in KEGG HCM pathway
| Symbol | Gene_Name |
|---|---|
| Atp2a2 | ATPase, Ca2+ transporting, cardiac muscle, slow twitch 2 |
| Actb | actin, beta |
| Ace | angiotensin I converting enzyme (peptidyl-dipeptidase A) 1 |
| Des | desmin |
| Igf1 | insulin-like growth factor 1 |
| Itga1 | integrin alpha 1 |
| Itga4 | integrin alpha 4 |
| Itga6 | integrin alpha 6 |
| Itga9 | integrin alpha 9 |
| Itgav | integrin alpha V |
| Itgb1 | integrin beta 1 (fibronectin receptor beta) |
| Itgb5 | integrin beta 5 |
| Lmna | lamin A |
| Mybpc3 | myosin binding protein C, cardiac |
| Myh6 | myosin, heavy polypeptide 6, cardiac muscle, alpha |
| Myl2 | myosin, light polypeptide 2, regulatory, cardiac, slow |
| Prkaa1 | protein kinase, AMP-activated, alpha 1 catalytic subunit |
| Prkab2 | protein kinase, AMP-activated, beta 2 noncatalytic subunit |
| Ryr2 | ryanodine receptor 2, cardiac |
| Tgfb2 | transforming growth factor, beta 2 |
| Tgfb3 | transforming growth factor, beta 3 |
HCM, hypertrophic cardiomyopathy; NHE1, Na+/H+ exchanger isoform 1.
Fig. 3.

Genes altered in NHE1 transgenic hearts were enriched in KEGG hypertrophic cardiomyopathy (HCM) pathway. Stars, differentially expressed genes in NHE1 transgenic mouse hearts. Red, upregulation; blue, downregulation.
miRNA expression profile in NHE1 transgenic mouse hearts.
NHE1 transgene-induced miRNA expression changes at the age of 7 mo were studied in parallel. The fold change of each miRNA expression was calculated by comparing NHE1 transgenic mouse hearts to their age-matched wild-type controls. miRNA sequencing detected that ~60% of known miRNAs were expressed in the left ventricle of mouse hearts (685 miRNAs in NHE1 transgenic mice and 654 miRNAs in controls). Among them, only 15 miRNAs were differentially expressed (2-fold, P < 0.05) (Table 2) when NHE1 transgenic and control mice were compared by miRanalyzer and miRNAkey. Ten miRNAs were upregulated: miR-208b-3p, miR-206-3p, miR-21a-5p, miR-214-5p, miR-125b-1-3p, miR-21a-3p, miR-146b-5p, miR-379-5p, miR-199a-5p, and miR-34c-5p. Five miRNAs were downregulated: miR-30c-5p, let-7d-3p, miR-9-3p, miR-208a-3p, and miR-499-3p.
Table 2.
Differentially expressed miRNAs in the left ventricle of NHE1 transgenic mouse hearts
| Seq |
qPCR |
|||||||
|---|---|---|---|---|---|---|---|---|
| miRNA Name | RPKM (Control) | RPKM (NHE1ox) | Fold Change | P Value | Corrected P Value (Bonferroni) | Fold Change | P Value | |
| mmu-miR-208b-3p | 2241.66 | 23190.52 | 10.35 | 0.00E +00 | 0.00E +00 | 9.24 | 0.0004 | * |
| mmu-miR-206-3p | 9.49 | 61.22 | 6.45 | 0.00E +00 | 0.00E +00 | 23.75 | 0.0007 | * |
| mmu-miR-21a-5p | 18566.08 | 71506.61 | 3.85 | 0.00E +00 | 0.00E +00 | 2.64 | 0.0393 | * |
| mmu-miR-214-5p | 7.00 | 22.52 | 3.21 | 1.10E-12 | 1.29E-09 | |||
| mmu-miR-125b-1-3p | 7.68 | 24.06 | 3.13 | 4.52E-13 | 5.28E-10 | |||
| mmu-miR-21-a-3p | 22.59 | 68.68 | 3.04 | 0.00E +00 | 0.00E +00 | |||
| mmu-miR-146b-5p | 464.06 | 1267.98 | 2.73 | 0.00E +00 | 0.00E +00 | |||
| mmu-miR-379-5p | 140.53 | 371.39 | 2.64 | 0.00E +00 | 0.00E +00 | |||
| mmu-miR-199a-5p | 235.42 | 599.51 | 2.55 | 0.00E +00 | 0.00E +00 | 2.25 | 0.0569 | |
| mmu-miR-34c-5p | 98.28 | 247.26 | 2.52 | 0.00E +00 | 0.00E +00 | |||
| mmu-miR-30c-5p | 1578.11 | 539.98 | −2.92 | 0.00E +00 | 0.00E +00 | −1.43 | 0.0001 | * |
| mmu-let-7d-3p | 64.16 | 21.95 | −2.92 | 0.00E +00 | 0.00E +00 | −1.67 | 0.3922 | |
| mmu-miR-9-3p | 89.47 | 28.85 | −3.1 | 0.00E +00 | 0.00E +00 | |||
| mmu-miR-208a-3p | 13986.97 | 3826.18 | −3.66 | 0.00E +00 | 0.00E +00 | −4.00 | 0.1649 | |
| mmu-miR-499-3p | 36.60 | 9.01 | −4.06 | 0.00E +00 | 0.00E +00 | |||
miRNA (also miR), microRNA; RPKM, reads per kilobase million; qPCR, quantitative real-time PCR.
P < 0.05.
The data were further analyzed with Ingenuity Pathways Analysis (IPA) to better understand the biological significance of these miRNA changes induced by elevated NHE1. Since NHE1 transgenic mice exhibited cardiac pathology, our focus centered on the miRNAs involved in cardiac pathological processes. Accordingly, six miRNAs that have been previously demonstrated by the experiments in relation to cardiac pathological function were altered by the NHE1 transgene too (Table 3). The majority of these miRNAs showed changes in the same direction as expected from the literature. These miRNAs contribute to several cardiac pathologies, such as cardiac fibrosis, cardiac infarction, cardiac hypertrophy, etc. (Fig. 4), which correlated well with the phenotypes we observed in NHE1 transgenic mice.
Table 3.
Differentially expressed miRNAs in cardiac pathologies in the left ventricle of NHE1 transgenic mouse hearts
| Category | P Value | Molecules |
|---|---|---|
| Cardiac fibrosis | 2.9E-06–1.42E-02 | miR-199a-5p (and other miRNAs w/seed CCAGUGU), miR-30c-5p (and other miRNAs w/seed GUAAACA), miR-208a-3p (and other miRNAs w/seed UAAGACG), miR-21-5p (and other miRNAs w/ seed AGCUUAU) |
| Cardiac infarction | 1.99E-05–1.99E-05 | miR-146a-5p (and other miRNAs w/ seed GAGAACU), miR-1-3p (and other miRNAs w/ seed GGAAUGU), miR-208a-3p (and other miRNAs w/ seed UAAGACG) |
| Cardiac hypertrophy | 4.71E-05–2.64E-03 | miR-1-3p (and other miRNAs w/ seed GGAAUGU), miR-199a-5p (and other miRNAs w/ seed CCAGUGU), miR-208a-3p (and other miRNAs w/ seed UAAGACG), miR-21-5p (and other miRNAs w/ seed AGCUUAU) |
| Cardiac inflammation | 2.11E-03–2.11E-03 | miR-208a-3p (and other miRNAs w/ seed UAAGACG) |
| Cardiac enlargement | 4.74E-03–4.74E-03 | miR-199a-5p (and other miRNAs w/ seed CCAGUGU) |
| Cardiac damage | 6.32E-03–6.32E-03 | miR-21-5p (and other miRNAs w/ seed AGCUUAU) |
| Heart failure | 1.06E-02–1.06E-02 | miR-199a-5p (and other miRNAs w/ seed CCAGUGU), miR-21-5p (and other miRNAs w/seed AGCUUAU) |
| Cardiac arteriopathy | 1.32E-02–1.32E-02 | miR-1-3p (and other miRNAs w/ seed GGAAUGU), miR-208a-3p (and other miRNAs w/seed UAAGACG) |
| Cardiac necrosis/cell death | 1.62E-02–1.62E-02 | miR-21-5p (and other miRNAs w/ seed AGCUUAU) |
Functional categorization of miRNA changes in NHE1 transgenic hearts was created with the Ingenuity Pathway Analysis program. The threshold for significance was P < 0.05.
Fig. 4.

Functional categorization by cardiac pathology. MicroRNAs (miRNAs) significantly altered by NHE1 transgene were classified into associated cardiac pathological functions (as depicted in x-axis) with Ingenuity Pathway Analysis (IPA) software. Functions are listed from most significant to least. y-Axis: –log10 [Benjamini-Hochberg (B-H) P value]. The significance threshold was set to 1.3 (P ≤ 0.05), as delineated by the horizontal line within the bar graph.
Validation of RNA-Seq and miRNA-Seq results with qPCR.
The seven differentially expressed miRNAs and seven differentially expressed mRNA targets of miR-30c were validated by qPCR. As presented in Fig. 5A, miRNAs, four out of seven tested, i.e., miR-208b-3p, miR-206-3p, miR-21a-5p, and miR-30c-5p, were confirmed by qPCR, and the other three miRNAs, i.e., miR-199a-5p, let-7d-3p, and miR-208a-3p, demonstrated the same trend of changes as in the sequencing study with no statistical significance. For mRNA targets of miR-30c, three out of seven tested were confirmed by qPCR with statistical significance: Igf1, Prkar1a, and Plcb4 (Fig. 5B).
Fig. 5.

Verification of sequencing results with quantitative real-time RT-PCR (qPCR). Seven randomly selected, significantly altered miRNAs (A) and seven differentially expressed mRNA targets of miR-30c (B) from the sequencing studies were tested by real-time RT-PCR. The gene expression level was normalized to snoRNA202 for miRNA and to GAPDH for mRNA. The final result represents the mean fold change of three individual transgenic samples over controls. *P < 0.05, Student's t-test.
Integrated analysis of miRNA and mRNA expression in NHE1 transgenic mouse hearts.
To investigate the contribution of miRNA regulation to differential gene expression in NHE1 transgenic mouse hearts, the gene transcripts that were altered by NHE1 transgene were subjected to Ingenuity URA. We identified 30 mature miRNAs as potential upstream regulators that are responsible for the gene expression changes (Table 4). Seven of these miRNAs were actually changed in NHE1 transgenic hearts (P < 0.05, 2-fold, miRNAkey): miR-30c-5p, miR-21-5p, miR-34a-5p, miR-1-3p, miR-199a-5p, miR-153-3p, and miR-133a-3p. Interestingly, miR-30c-5p showed the same directional change as predicted by URA z-score, strongly indicating that it has a key regulatory role in the observed gene expression alterations.
Table 4.
Mature miRNA regulators upstream of changed genes in NHE1 transgenic hearts
| Upstream Regulator | Expr Fold Change | Expr P Value | Predicted Activation State | Bias-corrected z-score | Activation z-score | Flags | P Value of Overlap | Target Molecules in Data Set |
|---|---|---|---|---|---|---|---|---|
| miR-16-5p | −1.825 | 0.00E +00 | inhibited | −5.842 | bias | 1E-08 | ACP2, ACTR1A, BCL2, CAPRIN1, CCND1, CFL2, DMTF1, DNAJB4, GOLPH3L, GRN, GTF2H1, H3F3A/H3F3B, HARS, IGF1, IGF2R, KPNA3, LAMC1, LAMTOR3, MAP2K1, MCL1, MRPL20, NPR3, PAFAH1B2, PHLDB2, PISD, PURA, RARS, RTN4, SERPINE2, SKAP2, SLC12A2, SPTLC1, SQSTM1, TMEM109, TPI1, TPPP3, UGDH, UGP2, VTI1B | |
| miR-124-3p | * | inhibited | −5.168 | bias | 2E-06 | ACAA2, ARFIP1, ARPC1B, ATL3, ATP6V0E1, BLOC1S6, CTDSP2, CTGF, CTNND1, F11R, FCHO2, GSN, HADH, HES1, HTATIP2, ITGB1, KLF15, LAMC1, MDFIC, MMP2, MTMR6, MTPN, NECAP2, PGRMC2, POLR3G, PTTG1IP, R BMS1, ROCK1, RYK, SERPINB6, SLC50A1, SSFA2, STAT3, SWAP70, TLN1, TMBIM1, TMEM109, ZNF367 | ||
| miR-29b-3p | −1.641 | 0.00E +00 | inhibited | −3.429 | bias | 5E-06 | ARPC3, COL15A1, COL3A1, COL4A2, FBN1, GMFB, KLF4, LAMC1, MCL1, PMP22, PPIC, PPM1D, PURA, SPARC, TGFB3, TUBB2A | |
| miR-30c-5p | −2.923 | 0.00E +00 | inhibited | −3.845 | bias | 1E-05 | ATP2A2, BECN1, CTGF, ELMOD2, GALNT1, GNAI2, MAT2A, NPR3, NT5E, PAFAH1B2, PRPF40A, PTGFRN, RBMS1, SEC23A, SEC62, SLC38A2, STRN, STX7, TMED2, TMED7, TMEM59, WDR92 | |
| miR-17-5p | −1.300 | 8.44E-07 | inhibited | −1.456 | −3.275 | bias | 5E-05 | APP, BCL2, BIRC5, BMPR2, CCND1, CRIM1, PURA, RGS5, RHOA, RHOC, S1PR1, STAT3, TGFBR2, VIM |
| miR-192-5p | −1.426 | 0.00E +00 | 1.453 | 0.603 | bias | 9E-05 | BIRC5, DHFR, IGF1, TYMS, ZEB1 | |
| miR-199a-5p | 2.547 | 0.00E +00 | inhibited | −2.376 | bias | 0.0001 | ACTA1, ALOX5AP, BGN, BIRC5, C3, DCN, FN1, HIF1A, ITGA1, KLF4, PIGR, SAMD9L, TGFBR1, TGFBR2, ZBTB16 | |
| miR-155-5p | −1.686 | 8.36E-14 | inhibited | −3.418 | bias | 0.0007 | ARFIP1, ATP6V1C1, CCND1, CYR61, DSG2, FAR1, IL13RA1, KRAS, MATR3, MOSPD2, NARS, NT5E, PDLIM5, PICALM, POLE3, RHOA, SDCBP, SERPINE1, TBCA, TCF7L2, TNFRSF10A, TXNRD1, WDFY1 | |
| miR-542-3p | 1.723 | 3.73E-08 | −1.955 | bias | 0.0008 | ILK, RPL11, RPL22, RPS23 | ||
| miR-199a-3p | inhibited | −2.433 | bias | 0.0009 | CALU, CD44, FN1, ITGA6, PON2, VCAN | |||
| miR-21-5p | 3.851 | 0.00E +00 | inhibited | −2.694 | bias | 0.001 | ACTA2, BMPR2, BTG2, C8orf44-SGK3/SGK3, CFL2, MMP2, PECAM1, SERPINB5, STAT3, TCF21, TGFB2, TGFBR2 | |
| miR-19b-3p | −1.185 | 4.89E-03 | −0.544 | −1.506 | bias | 0.0016 | BIRC5, BMPR2, CCND1, CTGF, THBS1 | |
| miR-291a-3p | inhibited | −3.365 | bias | 0.0016 | ADAM9, APP, C3, CCND1, CD44, CFL2, FAM13B, FYCO1, INSIG2, KLHL12, NUP58, PCGF5, STX11, UBXN1, ZHX1 | |||
| miR-1-3p | 6.451 | 0.00E +00 | inhibited | −4.083 | bias | 0.0019 | ANXA2, AP3B1, AXL, BCL2, CAP1, CNN3, CORO1C, FBLN2, FSTL1, GNPDA2, H3F3A/H3F3B, IGF1, IP6K2, KCNJ2, LRP1, PICALM, PPIB, SDC4, SERPINB5, SLC44A1, SNX6, TAGLN2, THBS1, Tmsb4x (includes others), WDFY1, YWHAQ | |
| miR-293-5p | −1.664 | bias | 0.0031 | AKAP6, GOLPH3, KLF15, MGST1, NR4A1, SSR3 | ||||
| miR-34a-5p | 2.578 | 0.00E +00 | −1.895 | bias | 0.0038 | BCL2, BIRC5, CCND1, CDK1, DHFR, ICAM1, IKBIP, KLF4, MAP2K1, STIM1 | ||
| miR-451a | −1.593 | 0.00E +00 | −1.998 | bias | 0.0041 | BCL2, CCND1, MIF, MMP2 | ||
| miR-1195 | * | 0.0053 | IGF1, STAT3 | |||||
| miR-129-5p | −1.969 | bias | 0.0061 | BMPR2, CAMTA1, GALNT1, PDS5A | ||||
| let-7a-5p | 1.324 | 0.00E +00 | inhibited | −3.219 | bias | 0.0062 | ACTA2, BIRC5, CCND1, CHMP2A, COL3A1, CSDE1, ITGA1, ITGA4, KRAS, MTPN, PGRMC1, SLC25A32, SMOX, SPCS3, STAT3, TGFBR1, THBS1, TLR4, TYMS, VIM | |
| miR-320b | 0.0108 | IGF1, MCL1, VIM | ||||||
| miR-138-5p | * | 0.0108 | RHOC, ROCK2, VCAN | |||||
| miR-153-3p | −2.773 | 4.73E-07 | 0.0151 | BCL2, MCL1 | ||||
| miR-193a-3p | * | 0.0163 | CCND1, MCL1, PTK2 | |||||
| miR-30a-3p | −1.132 | 0.00E +00 | inhibited | −2.236 | bias | 0.0226 | CYR61, RAB8B, RSU1, THBS1, TUBA1A | |
| miR-149-5p | −1.622 | 0.00E +00 | 0.0288 | RAP1A, RAP1B | ||||
| miR-125b-5p | 1.230 | 0.00E +00 | inhibited | −2.224 | bias | 0.0297 | ACSS1, ADAMTS1, GSS, H3F3A/H3F3B, ID2, IGFBP3, LIPA, MAN1A1, MAP2K1, PIGR, SGPL1, VPS4B | |
| miR-133a-3p | −2.692 | 0.00E +00 | −1.875 | bias | 0.0299 | BTBD3, Cdc42, CORO1C, CTGF, KLF15, MCL1, MSN, RHOA | ||
| miR-200b-3p | * | −0.39 | −1.444 | bias | 0.0317 | ERBIN, ERRFI1, VIM, WDR37, ZEB1 | ||
| miR-135a-5p | −1.168 | 3.25E-01 | 0.0408 | ALOX5AP, APC, JAK2 |
Low abundance.
To find out the most biologically relevant mRNA targets of differentially expressed miRNAs, Ingenuity miRNA Target Filter analysis was conducted to explore all the possible miRNA-mRNA interactions that were either experimentally validated from TarBase and miRecords or predicted from TargetScan and Ingenuity Knowledgebase. Among 15 differentially expressed miRNAs, only 10 miRNAs have targeting information available. These 10 miRNAs have 5,385 potential mRNA targets. The expression pairing of our miRNA-mRNA data revealed that eight miRNAs targeting 530 mRNAs did change in NHE1 transgenic mouse hearts. Filtered by inverse expression pattern, it showed eight miRNAs targeting 220 mRNAs. These miRNA-mRNA target relationships were further prioritized either by 1) cardiovascular signaling pathways, six miRNAs targeting 18 mRNAs met the criteria (Table 5) or by 2) cardiovascular disease, the results were narrowed to eight miRNAs targeting 70 mRNAs (Table 6). miR-199a and its mRNA target ITGA4 as well as miR-30c and its nine mRNA targets, CAMK2D, GNAI2, GNAO1, IGF1, KRAS, PPP1R14C, PPKAR1A, RASA1, and TNIP1, have been shown to be involved in cardiovascular signaling and also to be relevant to cardiovascular diseases.
Table 5.
miRNA-mRNA target relationships with opposite expression and related to cardiovascular signaling
| ID | Fold Change | Confidence | ID | Symbol | Fold Change |
|---|---|---|---|---|---|
| mmu-miR-146b-5p | 2.732 | experimentally observed | 215257 | IL36G | −2.024 |
| mmu-miR-199a-5p | 2.547 | high (predicted) | 16401 | ITGA4 | −1.756 |
| mmu-miR-199a-5p | 2.547 | high (predicted) | 242083 | PPM1L | −1.551 |
| mmu-miR-208b-3p | 10.345 | moderate (predicted) | 236915 | ARHGEF9 | −1.518 |
| mmu-miR-21a-5p | 3.851 | moderate (predicted) | 140491 | PPP1R3A | −1.885 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 108058 | CAMK2D | 1.711 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 71745 | CUL2 | 1.756 |
| mmu-miR-30c-5p | −2.923 | experimentally observed, high (predicted) | 14678 | GNAI2 | 2.210 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 14681 | GNAO1 | 2.769 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 16000 | IGF1 | 2.952 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 16653 | KRAS | 1.558 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 108083 | PIP4K2B | 2.123 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 18798 | PLCB4 | 2.080 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 76142 | PPP1R14C | 1.879 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 19084 | PRKAR1A | 2.492 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 218397 | RASA1 | 1.582 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 57783 | TNIP1 | 2.258 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 56550 | UBE2D2 | 1.698 |
| mmu-miR-34c-5p | 2.516 | moderate (predicted) | 236915 | ARHGEF9 | −1.518 |
Table 6.
miRNA-mRNA target relationships with opposite expression and related to cardiovascular diseases
| ID | Fold Change | Confidence | ID | Symbol | Fold Change |
|---|---|---|---|---|---|
| mmu-miR-206-3p | 6.451 | experimentally observed, high (predicted) | 16518 | KCNJ2 | −1.738 |
| mmu-miR-206-3p | 6.451 | high (predicted) | 243780 | KIAA1147 | −1.615 |
| mmu-miR-206-3p | 6.451 | experimentally observed | 20724 | SERPINB5 | −2.811 |
| mmu-miR-146b-5p | 2.732 | high (predicted) | 223455 | MARCH6 | −1.517 |
| mmu-miR-146b-5p | 2.732 | experimentally observed | 19204 | PTAFR | −1.995 |
| mmu-miR-199a-5p | 2.547 | high (predicted) | 16401 | ITGA4 | −1.756 |
| mmu-miR-199a-5p | 2.547 | moderate (predicted) | 18072 | NHLH2 | −2.017 |
| mmu-miR-199a-5p | 2.547 | high (predicted) | 19017 | PPARGC1A | −2.497 |
| mmu-miR-208b-3p | 10.345 | high (predicted) | 13511 | DSG2 | −1.540 |
| mmu-miR-21a-5p | 3.851 | moderate (predicted) | 26888 | CLEC4A | −1.842 |
| mmu-miR-21a-5p | 3.851 | moderate (predicted) | 15488 | HSD17B4 | −1.693 |
| mmu-miR-21a-5p | 3.851 | moderate (predicted) | 223455 | MARCH6 | −1.517 |
| mmu-miR-21a-5p | 3.851 | experimentally observed, moderate (predicted) | 20724 | SERPINB5 | −2.811 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 54375 | AZIN1 | 3.088 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 12043 | BCL2 | 3.264 |
| mmu-miR-30c-5p | −2.923 | experimentally observed, high (predicted) | 56208 | BECN1 | 1.717 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 12177 | BNIP3L | 2.628 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 12321 | CALU | 1.950 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 108058 | CAMK2D | 1.711 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 100072 | CAMTA1 | 2.314 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 12554 | CDH13 | 2.600 |
| mmu-miR-30c-5p | −2.923 | experimentally observed | 14219 | CTGF | 9.390 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 13371 | DIO2 | 9.348 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 67731 | FBXO32 | 1.552 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 108148 | GALNT2 | 1.621 |
| mmu-miR-30c-5p | −2.923 | experimentally observed, high (predicted) | 14678 | GNAI2 | 2.210 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 14681 | GNAO1 | 2.769 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 15976 | IFNAR2 | 1.921 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 16000 | IGF1 | 2.952 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 16004 | IGF2R | 1.507 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 72999 | INSIG2 | 1.964 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 16653 | KRAS | 1.558 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 84035 | KREMEN1 | 1.611 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 18826 | LCP1 | 2.665 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 74511 | LRRC17 | 4.488 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 17156 | MAN1A2 | 1.772 |
| mmu-miR-30c-5p | −2.923 | experimentally observed, high (predicted) | 232087 | MAT2A | 1.520 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 225164 | MIB1 | 2.080 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 218613 | MIER3 | 1.614 |
| mmu-miR-30c-5p | −2.923 | experimentally observed | 18162 | NPR3 | 2.899 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 14815 | NR3C1 | 1.857 |
| mmu-miR-30c-5p | −2.923 | experimentally observed, high (predicted) | 23959 | NT5E | 2.982 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 56426 | PDCD10 | 2.002 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 18596 | PDGFRB | 1.724 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 233489 | PICALM | 1.849 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 330260 | PON2 | 2.059 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 67738 | PPID | 1.524 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 76142 | PPP1R14C | 1.879 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 19084 | PRKAR1A | 2.492 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 19255 | PTPN2 | 1.875 |
| mmu-miR-30c-5p | −2.923 | moderate (predicted) | 59021 | RAB2A | 1.540 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 215449 | RAP1B | 2.562 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 218397 | RASA1 | 1.582 |
| mmu-miR-30c-5p | −2.923 | experimentally observed | 56878 | RBMS1 | 1.683 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 56437 | RRAD | 1.570 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 18787 | SERPINE1 | 2.606 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 21366 | SLC6A6 | 1.570 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 20843 | STAG2 | 1.552 |
| mmu-miR-30c-5p | −2.923 | experimentally observed | 53331 | STX7 | 1.800 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 320165 | TACC1 | 1.630 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 21826 | THBS2 | 1.561 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 21858 | TIMP2 | 2.602 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 57783 | TNIP1 | 2.258 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 21951 | TNKS | 1.680 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 70892 | TTLL7 | 3.382 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 14479 | USP15 | 1.623 |
| mmu-miR-30c-5p | −2.923 | moderate (predicted) | 109815 | VIMP | 1.613 |
| mmu-miR-30c-5p | −2.923 | high (predicted) | 97064 | WWTR1 | 2.326 |
| mmu-miR-34c-5p | 2.516 | high (predicted) | 14081 | ACSL1 | −2.295 |
| mmu-miR-34c-5p | 2.516 | high (predicted) | 17161 | MAOA | −1.886 |
| mmu-miR-34c-5p | 2.516 | moderate (predicted) | 17318 | MID1 | −2.236 |
| mmu-miR-34c-5p | 2.516 | high (predicted) | 66052 | SDHC | −1.718 |
| mmu-miR-379-5p | 2.643 | moderate (predicted) | 17318 | MID1 | −2.236 |
To study the cardiovascular signaling mediated by these miRNA-mRNA targets that lead to cardiac pathology observed in NHE1 transgenic mouse hearts, IPA core analysis was performed. The role of nuclear factor of activated T cells (NFAT) in cardiac hypertrophy [ratio, 10/179 (0.056); z-score, 2.530; B-H P value, 2.48E-05] and cardiac hypertrophy signaling [ratio, 10/223 (0.045); z-score, 2.333; B-H P value, 1.04E-04] appeared to be top ranked canonical pathways; both predicted activation (Fig. 6). Among 179 genes in NFAT hypertrophy pathway, 31 genes were actually changed in NHE1 transgenic mouse hearts (Table 7, Fig. 7). Ten of 31 genes were targets of four differentially expressed miRNAs, miR-30c-5p, miR-21-5p, miR-199a-5p, and miR-34a-5p (Table 8, Fig. 7). Similarly, among 223 genes in cardiac hypertrophy signaling, 35 genes were altered in NHE1 transgenic mouse hearts (Table 9, Fig. 8). Ten of 35 genes were targets of four differentially expressed miRNAs, the same as those in NFAT hypertrophy (Table 8, Fig. 8). The data suggest that these differentially expressed miRNAs may work via regulation of their mRNA targets expression and stimulate NFAT hypertrophy and cardiac hypertrophy signaling, which in turn result in cardiac hypertrophy.
Fig. 6.

Cardiovascular-related canonical pathways involved by miRNA-mRNA pairs. x-Axis, significant pathways and listed from most significant to least; y-axis, –log10 (B-H P value). The significance threshold was set to 1.3 (P ≤ 0.05), as delineated by the horizontal line within the bar graph. Bar colors: orange, predicted pathway activation; blue, predicted inhibition; white, z-score at or very close to 0; gray, no prediction can currently be made. The orange points connected by a thin line represent the ratio (# of genes in a given pathway that meet cut-off criteria, divided by the total # of genes that make up that pathway and that are in the reference gene set).
Table 7.
Differentially expressed mRNAs in NFAT cardiac hypertrophy pathway from left ventricle of NHE1 transgenic mouse hearts
| Gene Symbol | Entrez Gene ID | Fold Change | Expected Change | Location | Type(s) |
|---|---|---|---|---|---|
| ADCY5 | 224129 | 2.388 | up | plasma membrane | enzyme |
| AKAP5 | 238276 | 2.822 | down | plasma membrane | other |
| AKT3 | 23797 | 1.592 | up | cytoplasm | kinase |
| CAMK1 | 52163 | 1.816 | up | cytoplasm | kinase |
| CAMK2D | 108058 | 1.711 | up | cytoplasm | kinase |
| GNAI2 | 14678 | 2.210 | down | plasma membrane | other |
| GNAS | 14683 | 1.546 | up | plasma membrane | enzyme |
| GNB1 | 14688 | 1.944 | up | plasma membrane | enzyme |
| GNB5 | 14697 | 1.769 | plasma membrane | enzyme | |
| GNB2L1 | 14694 | 1.527 | cytoplasm | enzyme | |
| GNG11 | 66066 | 1.641 | up | plasma membrane | enzyme |
| IGF1 | 16000 | 2.952 | up | extracellular space | growth factor |
| IL11 | 16156 | −1.678 | up | extracellular space | cytokine |
| IL6ST | 16195 | 1.752 | up | plasma membrane | transmembrane receptor |
| KRAS | 16653 | 1.558 | up | cytoplasm | enzyme |
| MAP2K1 | 26395 | 2.255 | up | cytoplasm | kinase |
| MAP2K2 | 26396 | 1.616 | up | cytoplasm | kinase |
| MAP2K6 | 26399 | −1.948 | up | cytoplasm | kinase |
| PDIA3 | 14827 | 1.924 | up | cytoplasm | peptidase |
| PIK3C3 | 225326 | 1.668 | up | cytoplasm | kinase |
| PLCB4 | 18798 | 2.080 | up | cytoplasm | enzyme |
| PRKAR1A | 19084 | 2.492 | up | cytoplasm | kinase |
| PRKAR2A | 19087 | 1.621 | up | cytoplasm | kinase |
| PRKCD | 18753 | 1.999 | up | cytoplasm | kinase |
| RCAN1 | 54720 | 5.226 | down | nucleus | transcription regulator |
| RRAS2 | 66922 | 2.233 | up | plasma membrane | enzyme |
| SHC1 | 20416 | 1.505 | up | cytoplasm | kinase |
| TGFB2 | 21808 | 7.666 | up | extracellular space | growth factor |
| TGFB3 | 21809 | 3.013 | up | extracellular space | growth factor |
| TGFBR1 | 21812 | 1.746 | up | plasma membrane | kinase |
| TGFBR2 | 21813 | 1.787 | up | plasma membrane | kinase |
Ratio, 31/179 (0.173); z-score, 3.528; P value, 3.26E-05. NFAT, nuclear factor of activated T cells.
Fig. 7.

Differentially expressed miRNA-mRNA pairs in nuclear factor of activated T cells (NFAT) cardiac hypertrophy pathway from NHE1 transgenic mouse hearts. Red, upregulation; green, downregulation.
Table 8.
Differentially expressed miRNAs and their mRNA targets in NFAT hypertrophy pathway and cardiac hypertrophy signaling
| Gene Symbol | Fold Change | Expected Change | Target of miRs | Location | Type(s) |
|---|---|---|---|---|---|
| CAMK2D* | 1.711 | up | mmu-miR-30c-5p | cytoplasm | kinase |
| GNAI2 | 2.210 | down | mmu-miR-30c-5p | plasma membrane | other |
| IGF1 | 2.952 | up | mmu-miR-30c-5p | extracellular space | growth factor |
| KRAS | 1.558 | up | mmu-miR-30c-5p | cytoplasm | enzyme |
| MAP2K1 | 2.255 | up | mmu-miR-34a-5p | cytoplasm | kinase |
| PLCB4 | 2.080 | up | mmu-miR-30c-5p | cytoplasm | enzyme |
| PRKAR1A | 2.492 | up | mmu-miR-30c-5p | cytoplasm | kinase |
| TGFB2 | 7.666 | up | mmu-miR-21–5p | extracellular space | growth factor |
| TGFBR1 | 1.746 | up | mmu-miR-199a-5p | plasma membrane | kinase |
| TGFBR2 | 1.787 | up | mmu-miR-21-5p, mmu-miR-199a-5p | plasma membrane | kinase |
| GNAO1† | 2.769 | mmu-miR-30c-5p | plasma membrane | enzyme |
In NFAT hypertrophy pathway only;
in cardiac hypertrophy signaling only.
Table 9.
Differentially expressed mRNAs in cardiac hypertrophy signaling from the left ventricle of NHE1 transgenic mouse hearts
| Gene Symbol | Entrez Gene ID | Fold Change | Expected Change | Location | Type(s) |
|---|---|---|---|---|---|
| ADCY5 | 224129 | 2.388 | up | plasma membrane | enzyme |
| EIF2B5 | 224045 | 1.504 | down | cytoplasm | translation regulator |
| GNAI2 | 14678 | 2.210 | down | plasma membrane | other |
| GNAO1 | 14681 | 2.769 | plasma membrane | enzyme | |
| GNAS | 14683 | 1.546 | up | plasma membrane | enzyme |
| GNB1 | 14688 | 1.944 | up | plasma membrane | enzyme |
| GNB5 | 14697 | 1.769 | plasma membrane | enzyme | |
| GNB2L1 | 14694 | 1.527 | cytoplasm | enzyme | |
| GNG11 | 66066 | 1.641 | up | plasma membrane | enzyme |
| HSPB1 | 15507 | 1.836 | up | cytoplasm | other |
| IGF1 | 16000 | 2.952 | up | extracellular space | growth factor |
| KRAS | 16653 | 1.558 | up | cytoplasm | enzyme |
| MAP2K1 | 26395 | 2.255 | up | cytoplasm | kinase |
| MAP2K2 | 26396 | 1.616 | up | cytoplasm | kinase |
| MAP2K6 | 26399 | −1.948 | up | cytoplasm | kinase |
| MYL1 | 17901 | 4.363 | up | cytoplasm | other |
| MYL2 | 17906 | −1.755 | up | cytoplasm | other |
| MYL6 | 17904 | 2.373 | up | cytoplasm | other |
| MYL7 | 17898 | −2.259 | up | cytoplasm | enzyme |
| MYL12B | 67268 | 1.776 | up | cytoplasm | other |
| PDIA3 | 14827 | 1.924 | up | cytoplasm | peptidase |
| PIK3C3 | 225326 | 1.668 | up | cytoplasm | kinase |
| PLCB4 | 18798 | 2.080 | up | cytoplasm | enzyme |
| PRKAR1A | 19084 | 2.492 | up | cytoplasm | kinase |
| PRKAR2A | 19087 | 1.621 | up | cytoplasm | kinase |
| RHOA | 11848 | 1.888 | up | cytoplasm | enzyme |
| RHOC | 11853 | 2.482 | up | plasma membrane | enzyme |
| RHOQ | 104215 | 1.609 | up | plasma membrane | enzyme |
| ROCK1 | 19877 | 1.851 | up | cytoplasm | kinase |
| ROCK2 | 19878 | 1.902 | up | cytoplasm | kinase |
| RRAS2 | 66922 | 2.233 | up | plasma membrane | enzyme |
| TGFB2 | 21808 | 7.666 | up | extracellular space | growth factor |
| TGFB3 | 21809 | 3.013 | up | extracellular space | growth factor |
| TGFBR1 | 21812 | 1.746 | up | plasma membrane | kinase |
| TGFBR2 | 21813 | 1.787 | up | plasma membrane | kinase |
Ratio, 35/223 (0.157); z-score, 3.889; P value: 8.69E-05.
Fig. 8.

Differentially expressed miRNA-mRNA pairs in cardiac hypertrophy signaling from NHE1 transgenic mouse hearts. Red, upregulation; green, downregulation.
DISCUSSION
NHE1 protein expression and activity are elevated in various cardiac diseases, including ischemia-reperfusion (I/R) injury, cardiac hypertrophy, and heart failure. Two transgenic mouse lines with cardiac-specific overexpression of activated NHE1 develop cardiac hypertrophy, fibrosis and cardiac dysfunction as demonstrated by our group (63) and Nakamura et al. (47). Conversely, NHE1 inhibition has been proven to prevent or induce regression of hypertrophy in different models of cardiac hypertrophy (5, 6, 15, 16, 33, 38, 42). Substantial evidence has accumulated to indicate a key role of NHE1 in the promotion of cardiomyocyte growth. A number of signaling pathways have been proposed to mediate NHE1-induced hypertrophic response (17, 47). However, which mRNAs and miRNAs get involved in this process and how they contribute to cardiac hypertrophy have not been thoroughly investigated. The questions we aimed to answer by the present study were 1) what are the mRNAs and miRNAs that are altered by activated NHE1? 2) what are the functional pairs of miRNAs-mRNAs among these NHE1-induced changes? and 3) what are the signaling pathways that miRNA-mRNA pairs work through to result in cardiac hypertrophy?
Diverse stimuli, such as hormones (endothelin-1, angiotensin II, and α1-adrenergic agonists), growth factors, stretch, I/R injury, and sustained acidosis, activate NHE1 through ERK1/2 (extracellular signal-regulated kinase 1 and 2), p90RSK (p90 ribosomal S6 kinase), and CaMKII (Ca2+/calmodulin-dependent protein kinase) (8, 19, 24, 35, 41, 43, 45, 55, 59, 60). NHE1 activation results in elevation of intracellular Na+ concentration ([Na+]i) and subsequent increase in [Ca2+]i via reverse mode of Na+/Ca2+ exchanger (NCX). Elevated [Ca2+]i in turn activates two important Ca2+-dependent prohypertrophic signaling molecules, calcineurin and CaMKII, and causes nuclear translocation of NFAT and nuclear exclusion of histone deacetylase (HDAC)4, thus promoting hypertrophy-associated gene expression (59). For the first time, our study comprehensively profiled expression changes of mRNAs and miRNAs induced by activated NHE1. Moreover, we classified hypertrophic genes into distinct functional categories, which help us better understand their impacts in cardiac hypertrophy and heart failure.
First, our current results showed that activated NHE1 downregulated myosin light polypeptide (Myl2) and heavy polypeptide (Myh6) gene expression in 7 mo old NHE1 transgenic hearts, contrasting with an increased expression at postnatal 2 wk (63). Myosin is a main component of sarcomeres, which is a basic contractile unit of striated muscle tissue. At the younger age of 2 wk, activated NHE1 promotes the synthesis of new contractile proteins and assembly of sarcomeres that ultimately increases contractile force and preserves heart function. But with the aging of the NHE1 transgenic mice, reduced expression of myosin suggests that the hearts lose the ability to maintain contractility and might evolve from cardiac hypertrophy to heart failure.
Second, reduced gene expression of SERCA2a and Ryr2 were detected in the 7 mo old NHE1 transgenic hearts. SERCA2a and ryanodine receptor (RyR)2 are two Ca2+ handling molecules located in the sarcoplasmic reticulum (SR). In cardiac myocytes, depolarization elicits a small Ca2+ influx through L-type Ca2+ channels localized in the t-tubules and subsequently triggers an additional release of Ca2+ via RyRs in SR. The amplitude and kinetics of Ca2+ release determine cardiac contractile force. Impairments in this release process cause systolic dysfunction. Following contraction, relaxation occurs when Ca2+ is recycled into the SR via SR Ca2+ ATPase (SERCA) and extruded from the cell via NCX and the plasma membrane Ca2+ ATPase. Disrupted Ca2+ removal leads to diastolic dysfunction (40, 51). Decreased expression of SERCA2a and Ryr2 correlated well with systolic and diastolic dysfunction observed in NHE1 transgenic mice at the age of 7 mo (failing heart). Our previous study on 2 wk old NHE1 transgenic mice showed no change in expression of SERCA2a and Ryr2 (28, 63), which was in agreement with a study by Nakamura et al. (47) in their NHE1 transgenic mice at the age of 40 days. The difference could be related to age. At an earlier age, NHE1 transgenic hearts are still under compensated hypertrophic phase.
In addition to stimulation of myocyte contraction, Ca2+ also plays an important role in regulating the expression of hypertrophy-associated genes. Calcineurin and CaMKII delta appear to be two principal mediators (17, 47). Our data revealed that expression of Rcan1 (regulator of calcineurin 1) and CaMKII delta were upregulated in NHE1 transgenic mice. It is possible therefore that Ca2+-dependent hypertrophic signaling is modulated not only at a posttranscriptional level but also at a transcriptional one. A recent study found that CAMTA2 (CaM-binding transcription activator 2) stimulates cardiac growth by opposing class II histone deacetylases (54). Our data showed that CAMTA1 was increased by activated NHE1. Whether CAMTA1 plays a similar hypertrophic role as CAMTA2 needs further investigation.
Third, a myriad of signaling pathways have been implicated in the development of cardiac hypertrophy, including calcineurin-NFAT, MAPKs (ERK, p38, and JNK), RSK, and PI3K/Akt/GSK-3 (20). Our findings are the first to demonstrate that hypertrophic signaling molecules, such as CaMKII, Ras, MEK1/2, PI3K, Akt, PLC, PKA, and PKC, were transcriptionally induced by activated NHE1. A previous study on the same NHE1 transgenic hearts found no differences in phosphorylated ERK, p38, JNK, and RSK at postnatal 2 and 12 wk old as compared with wild-type controls (46). In contrast, Nakamura et al. (47) showed that p38 and ERK were significantly activated in their NHE1 transgenic hearts. The discrepancy is probably caused by a large deletion of NHE1 COOH terminus (amino acid 637–656) in Nakamura’s model, which leads to a more extreme activation of NHE1 protein and an earlier onset of cardiac pathology. Our present data support this concept because these hypertrophic kinases were increased in our model of transgenic hearts at the age of 7 mo old.
Expression patterns of miRNAs were investigated in parallel in NHE1 transgenic hearts in our current study. Upregulation of miR-21, miR-125b, miR-199a, miR-214, miR-208, and miR-146, as well as downregulation of miR-30c, was found not only by our data but also reported in other cardiac hypertrophy models (7, 56, 58), including thoracic aortic-banded hearts, the calcineurin-overexpressed transgenic mice, and phenylephrine-treated neonatal cardiomyocytes, suggesting that they may share a similarity in hypertrophic mechanisms controlled by miRNAs. In particular, the role of certain miRNAs (such as miR-208 and miR-1) has been demonstrated in cardiac hypertrophy. miR-208a and miR-208b are encoded within an intron of alpha- and beta- cardiac muscle myosin heavy chain genes (Myh6, α-MHC and Myh7, β-MHC) respectively. These miRNAs are cotranscribed with their host MHC genes. During normal development, the switch from fetal isoform β-MHC to the adult isoform α-MHC in the mouse occurs shortly after birth (4). Re-induction of β-MHC gene is a well-documented marker of pathological cardiac hypertrophy and normal aging in many experimental models (48). Reduced expression of miR-208a and myh6 and elevated expression of miR-208b and myh7 were noted in NHE1 transgenic hearts. miR-1 is considered as an antihypertrophic miRNA because it negatively regulates CaM, Mef2 (muscle enhancer factor 2), and Gata 4 (27), all of which are components of Ca2+-dependent hypertrophic signaling. Our NHE1 transgenic mice showed an increase of miR-1 expression. Whether this miR-1 elevation is a fine modulator of hypertrophy or it is a later consequence of heart failure is not known at present. Future studies will need to examine these possibilities.
miRNAs have profound impact on physiological functions and pathogenesis of diseases through regulating functionally related mRNA targets. We paired expression data of mRNAs and miRNAs in this study to identify important miRNA-mRNA targets. Our integrative analysis uncovered the possibility that miR-30c and its mRNA targets may be key players in NHE1-induced cardiac hypertrophy. Reduced miR-30c in NHE1 transgenic hearts led to increased expression of their mRNA targets. These mRNA targets act in NFAT hypertrophic and cardiac hypertrophic pathways and promote cardiac hypertrophy. miR-30c is abundantly expressed in both cardiac myocytes and cardiac fibroblasts. Reduced expression of miRNA-30c was observed in pathological conditions, such as hypertrophic hearts, diabetic cardiomyocytes, and cancers (25, 29, 49, 50). Downregulation of miR-30c contributes to cardiac hypertrophy, apoptosis, and extracellular matrix remodeling via upregulation of its mRNA targets p53 and p21, Cdc42 and Pak1, as well as CTGF, respectively (13, 29, 49, 50). Our study is the first to link miR-30c with NFAT hypertrophic and cardiac hypertrophic pathways. Future studies are needed to evaluate the therapeutic value of miR-30c in CVD by either gain- and loss-of-function strategy or miR mimic and anti-miR approach.
In summary, our study is the first to perform a global comparative study of mRNA and miRNA expression induced by activated NHE1. Moreover, we categorized the differentially expressed genes in terms of contractility, Ca2+ handling/signaling, as well as hypertrophic signaling and found the expression pattern was determined by mouse age examined and severity of cardiac pathology. The current results suggest that NHE1-mediated hypertrophic signaling can be modulated not only at the activity and protein levels but also at the mRNA level. NHE1 activates distinct signaling cascades in response to various stimuli, and hypertrophic gene transcription works as a coincidence detector of many signaling inputs and controls specific sets of genes. The expression of these genes in turn acts along the pathways to fine-tune the hypertrophic response. miR-30c and their mRNA targets were highlighted by integrative analysis of present mRNA and miRNA data. mRNA targets of miR-30c are mainly involved in NFAT and cardiac hypertrophy signaling. We hypothesize that miR-30c may be a promising therapeutic target for cardiac hypertrophy.
GRANTS
This study was supported by National Institutes of Health Grants PO1HD-32573 and RO1NS-037756 to G. G. Haddad.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.X., D.Z., and G.G.H. conceived and designed research; J.X., D.Z., O.P., T.I., and E.X.X. performed experiments; J.X., D.Z., O.P., I.H., T.I., and E.X.X. analyzed data; J.X. and D.Z. interpreted results of experiments; J.X., D.Z., and O.P. prepared figures; J.X. and D.Z. drafted manuscript; J.X., D.Z., O.P., I.H., and G.G.H. edited and revised manuscript; J.X., D.Z., O.P., I.H., T.I., E.X.X., and G.G.H. approved final version of manuscript.
Supplemental Data
Table S1: Differentially-expressed genes in the left ventricle of NHE1 transgenic mouse hearts - .xlsx (147 KB)
ACKNOWLEDGMENTS
We thank Travis Smith for technical assistance.
REFERENCES
- 1.Avkiran M, Cook AR, Cuello F. Targeting Na+/H+ exchanger regulation for cardiac protection: a RSKy approach? Curr Opin Pharmacol 8: 133–140, 2008. doi: 10.1016/j.coph.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 2.Baczkó I, Mraiche F, Light PE, Fliegel L. Diastolic calcium is elevated in metabolic recovery of cardiomyocytes expressing elevated levels of the Na+/H+ exchanger. Can J Physiol Pharmacol 86: 850–859, 2008. doi: 10.1139/Y08-092. [DOI] [PubMed] [Google Scholar]
- 3.Bertrand B, Wakabayashi S, Ikeda T, Pouysségur J, Shigekawa M. The Na+/H+ exchanger isoform 1 (NHE1) is a novel member of the calmodulin-binding proteins. Identification and characterization of calmodulin-binding sites. J Biol Chem 269: 13703–13709, 1994. [PubMed] [Google Scholar]
- 4.Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, Willis MS, Selzman CH, Wang DZ. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest 119: 2772–2786, 2009. doi: 10.1172/JCI36154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L, Chen CX, Gan XT, Beier N, Scholz W, Karmazyn M. Inhibition and reversal of myocardial infarction-induced hypertrophy and heart failure by NHE-1 inhibition. Am J Physiol Heart Circ Physiol 286: H381–H387, 2004. doi: 10.1152/ajpheart.00602.2003. [DOI] [PubMed] [Google Scholar]
- 6.Chen L, Gan XT, Haist JV, Feng Q, Lu X, Chakrabarti S, Karmazyn M. Attenuation of compensatory right ventricular hypertrophy and heart failure following monocrotaline-induced pulmonary vascular injury by the Na+-H+ exchange inhibitor cariporide. J Pharmacol Exp Ther 298: 469–476, 2001. [PubMed] [Google Scholar]
- 7.Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol 170: 1831–1840, 2007. doi: 10.2353/ajpath.2007.061170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cingolani HE, Pérez NG, Pieske B, von Lewinski D, Camilión de Hurtado MC. Stretch-elicited Na+/H+ exchanger activation: the autocrine/paracrine loop and its mechanical counterpart. Cardiovasc Res 57: 953–960, 2003. doi: 10.1016/S0008-6363(02)00768-X. [DOI] [PubMed] [Google Scholar]
- 9.Cingolani HE, Rebolledo OR, Portiansky EL, Pérez NG, Camilión de Hurtado MC. Regression of hypertensive myocardial fibrosis by Na(+)/H(+) exchange inhibition. Hypertension 41: 373–377, 2003. doi: 10.1161/01.HYP.0000051502.93374.1C. [DOI] [PubMed] [Google Scholar]
- 10.Coccaro E, Mraiche F, Malo M, Vandertol-Vanier H, Bullis B, Robertson M, Fliegel L. Expression and characterization of the Na+/H+ exchanger in the mammalian myocardium. Mol Cell Biochem 302: 145–155, 2007. doi: 10.1007/s11010-007-9436-3. [DOI] [PubMed] [Google Scholar]
- 11.Da Costa Martins PA, De Windt LJ. MicroRNAs in control of cardiac hypertrophy. Cardiovasc Res 93: 563–572, 2012. doi: 10.1093/cvr/cvs013. [DOI] [PubMed] [Google Scholar]
- 12.Ding SL, Zhou LY, Li PF. MicroRNAs in cardiac hypertrophy: angels or devils. Wiley Interdiscip Rev RNA 2: 124–134, 2011. doi: 10.1002/wrna.61. [DOI] [PubMed] [Google Scholar]
- 13.Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, Herias V, van Leeuwen RE, Schellings MW, Barenbrug P, Maessen JG, Heymans S, Pinto YM, Creemers EE. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res 104: 170–178, 2009. doi: 10.1161/CIRCRESAHA.108.182535. [DOI] [PubMed] [Google Scholar]
- 14.Dyck JR, Maddaford TG, Pierce GN, Fliegel L. Induction of expression of the sodium-hydrogen exchanger in rat myocardium. Cardiovasc Res 29: 203–208, 1995. doi: 10.1016/S0008-6363(96)88571-3. [DOI] [PubMed] [Google Scholar]
- 15.Engelhardt S, Hein L, Keller U, Klämbt K, Lohse MJ. Inhibition of Na(+)-H(+) exchange prevents hypertrophy, fibrosis, and heart failure in beta(1)-adrenergic receptor transgenic mice. Circ Res 90: 814–819, 2002. doi: 10.1161/01.RES.0000014966.97486.C0. [DOI] [PubMed] [Google Scholar]
- 16.Ennis IL, Escudero EM, Console GM, Camihort G, Dumm CG, Seidler RW, Camilión de Hurtado MC, Cingolani HE. Regression of isoproterenol-induced cardiac hypertrophy by Na+/H+ exchanger inhibition. Hypertension 41: 1324–1329, 2003. doi: 10.1161/01.HYP.0000071180.12012.6E. [DOI] [PubMed] [Google Scholar]
- 17.Ennis IL, Garciarena CD, Escudero EM, Pérez NG, Dulce RA, Camilión de Hurtado MC, Cingolani HE. Normalization of the calcineurin pathway underlies the regression of hypertensive hypertrophy induced by Na+/H+ exchanger-1 (NHE-1) inhibition. Can J Physiol Pharmacol 85: 301–310, 2007. doi: 10.1139/y06-072. [DOI] [PubMed] [Google Scholar]
- 18.Fliegel L. Regulation of the Na(+)/H(+) exchanger in the healthy and diseased myocardium. Expert Opin Ther Targets 13: 55–68, 2009. doi: 10.1517/14728220802600707. [DOI] [PubMed] [Google Scholar]
- 19.Fliegel L, Walsh MP, Singh D, Wong C, Barr A. Phosphorylation of the C-terminal domain of the Na+/H+ exchanger by Ca2+/calmodulin-dependent protein kinase II. Biochem J 282: 139–145, 1992. doi: 10.1042/bj2820139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol 65: 45–79, 2003. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- 21.Gan XT, Chakrabarti S, Karmazyn M. Modulation of Na+/H+ exchange isoform 1 mRNA expression in isolated rat hearts. Am J Physiol Heart Circ Physiol 277: H993–H998, 1999. [DOI] [PubMed] [Google Scholar]
- 22.Gurha P. MicroRNAs in cardiovascular disease. Curr Opin Cardiol 31: 249–254, 2016. doi: 10.1097/HCO.0000000000000280. [DOI] [PubMed] [Google Scholar]
- 23.Hackenberg M, Rodríguez-Ezpeleta N, Aransay AM. miRanalyzer: an update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic Acids Res 39, Suppl: W132–W138, 2011. doi: 10.1093/nar/gkr247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haworth RS, McCann C, Snabaitis AK, Roberts NA, Avkiran M. Stimulation of the plasma membrane Na+/H+ exchanger NHE1 by sustained intracellular acidosis. Evidence for a novel mechanism mediated by the ERK pathway. J Biol Chem 278: 31676–31684, 2003. doi: 10.1074/jbc.M304400200. [DOI] [PubMed] [Google Scholar]
- 25.Hirt MN, Werner T, Indenbirken D, Alawi M, Demin P, Kunze AC, Stenzig J, Starbatty J, Hansen A, Fiedler J, Thum T, Eschenhagen T. Deciphering the microRNA signature of pathological cardiac hypertrophy by engineered heart tissue- and sequencing-technology. J Mol Cell Cardiol 81: 1–9, 2015. doi: 10.1016/j.yjmcc.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 26.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57, 2009. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 27.Ikeda S, He A, Kong SW, Lu J, Bejar R, Bodyak N, Lee KH, Ma Q, Kang PM, Golub TR, Pu WT. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol Cell Biol 29: 2193–2204, 2009. doi: 10.1128/MCB.01222-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imahashi K, Mraiche F, Steenbergen C, Murphy E, Fliegel L. Overexpression of the Na+/H+ exchanger and ischemia-reperfusion injury in the myocardium. Am J Physiol Heart Circ Physiol 292: H2237–H2247, 2007. doi: 10.1152/ajpheart.00855.2006. [DOI] [PubMed] [Google Scholar]
- 29.Irani S, Hussain MM. Role of microRNA-30c in lipid metabolism, adipogenesis, cardiac remodeling and cancer. Curr Opin Lipidol 26: 139–146, 2015. doi: 10.1097/MOL.0000000000000162. [DOI] [PubMed] [Google Scholar]
- 30.Jandeleit-Dahm K, Hannan KM, Farrelly CA, Allen TJ, Rumble JR, Gilbert RE, Cooper ME, Little PJ. Diabetes-induced vascular hypertrophy is accompanied by activation of Na(+)-H(+) exchange and prevented by Na(+)-H(+) exchange inhibition. Circ Res 87: 1133–1140, 2000. doi: 10.1161/01.RES.87.12.1133. [DOI] [PubMed] [Google Scholar]
- 31.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28: 27–30, 2000. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karmazyn M, Kilić A, Javadov S. The role of NHE-1 in myocardial hypertrophy and remodelling. J Mol Cell Cardiol 44: 647–653, 2008. doi: 10.1016/j.yjmcc.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Karmazyn M, Liu Q, Gan XT, Brix BJ, Fliegel L. Aldosterone increases NHE-1 expression and induces NHE-1-dependent hypertrophy in neonatal rat ventricular myocytes. Hypertension 42: 1171–1176, 2003. doi: 10.1161/01.HYP.0000102863.23854.0B. [DOI] [PubMed] [Google Scholar]
- 34.Karmazyn M, Sawyer M, Fliegel L. The Na(+)/H(+) exchanger: a target for cardiac therapeutic intervention. Curr Drug Targets Cardiovasc Haematol Disord 5: 323–335, 2005. doi: 10.2174/1568006054553417. [DOI] [PubMed] [Google Scholar]
- 35.Khandoudi N, Ho J, Karmazyn M. Role of Na(+)-H+ exchange in mediating effects of endothelin-1 on normal and ischemic/reperfused hearts. Circ Res 75: 369–378, 1994. doi: 10.1161/01.RES.75.2.369. [DOI] [PubMed] [Google Scholar]
- 36.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36, 2013. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumarswamy R, Thum T. Non-coding RNAs in cardiac remodeling and heart failure. Circ Res 113: 676–689, 2013. doi: 10.1161/CIRCRESAHA.113.300226. [DOI] [PubMed] [Google Scholar]
- 38.Kusumoto K, Haist JV, Karmazyn M. Na(+)/H(+) exchange inhibition reduces hypertrophy and heart failure after myocardial infarction in rats. Am J Physiol Heart Circ Physiol 280: H738–H745, 2001. doi: 10.1152/ajpheart.2001.280.2.H738. [DOI] [PubMed] [Google Scholar]
- 39.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25: 402–408, 2001. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 40.Louch WE, Stokke MK, Sjaastad I, Christensen G, Sejersted OM. No rest for the weary: diastolic calcium homeostasis in the normal and failing myocardium. Physiology (Bethesda) 27: 308–323, 2012. doi: 10.1152/physiol.00021.2012. [DOI] [PubMed] [Google Scholar]
- 41.Malo ME, Li L, Fliegel L. Mitogen-activated protein kinase-dependent activation of the Na+/H+ exchanger is mediated through phosphorylation of amino acids Ser770 and Ser771. J Biol Chem 282: 6292–6299, 2007. doi: 10.1074/jbc.M611073200. [DOI] [PubMed] [Google Scholar]
- 42.Marano G, Vergari A, Catalano L, Gaudi S, Palazzesi S, Musumeci M, Stati T, Ferrari AU. Na+/H+ exchange inhibition attenuates left ventricular remodeling and preserves systolic function in pressure-overloaded hearts. Br J Pharmacol 141: 526–532, 2004. doi: 10.1038/sj.bjp.0705631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsui H, Barry WH, Livsey C, Spitzer KW. Angiotensin II stimulates sodium-hydrogen exchange in adult rabbit ventricular myocytes. Cardiovasc Res 29: 215–221, 1995. doi: 10.1016/S0008-6363(96)88573-7. [DOI] [PubMed] [Google Scholar]
- 44.Meima ME, Mackley JR, Barber DL. Beyond ion translocation: structural functions of the sodium-hydrogen exchanger isoform-1. Curr Opin Nephrol Hypertens 16: 365–372, 2007. doi: 10.1097/MNH.0b013e3281bd888d. [DOI] [PubMed] [Google Scholar]
- 45.Moor AN, Gan XT, Karmazyn M, Fliegel L. Activation of Na+/H+ exchanger-directed protein kinases in the ischemic and ischemic-reperfused rat myocardium. J Biol Chem 276: 16113–16122, 2001. doi: 10.1074/jbc.M100519200. [DOI] [PubMed] [Google Scholar]
- 46.Mraiche F, Oka T, Gan XT, Karmazyn M, Fliegel L. Activated NHE1 is required to induce early cardiac hypertrophy in mice. Basic Res Cardiol 106: 603–616, 2011. doi: 10.1007/s00395-011-0161-4. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura TY, Iwata Y, Arai Y, Komamura K, Wakabayashi S. Activation of Na+/H+ exchanger 1 is sufficient to generate Ca2+ signals that induce cardiac hypertrophy and heart failure. Circ Res 103: 891–899, 2008. doi: 10.1161/CIRCRESAHA.108.175141. [DOI] [PubMed] [Google Scholar]
- 48.Pandya K, Kim HS, Smithies O. Fibrosis, not cell size, delineates beta-myosin heavy chain reexpression during cardiac hypertrophy and normal aging in vivo. Proc Natl Acad Sci USA 103: 16864–16869, 2006. doi: 10.1073/pnas.0607700103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raut SK, Kumar A, Singh GB, Nahar U, Sharma V, Mittal A, Sharma R, Khullar M. miR-30c Mediates Upregulation of Cdc42 and Pak1 in Diabetic Cardiomyopathy. Cardiovasc Ther 33: 89–97, 2015. doi: 10.1111/1755-5922.12113. [DOI] [PubMed] [Google Scholar]
- 50.Raut SK, Singh GB, Rastogi B, Saikia UN, Mittal A, Dogra N, Singh S, Prasad R, Khullar M. miR-30c and miR-181a synergistically modulate p53-p21 pathway in diabetes induced cardiac hypertrophy. Mol Cell Biochem 417: 191–203, 2016. doi: 10.1007/s11010-016-2729-7. [DOI] [PubMed] [Google Scholar]
- 51.Roderick HL, Higazi DR, Smyrnias I, Fearnley C, Harzheim D, Bootman MD. Calcium in the heart: when it’s good, it’s very very good, but when it’s bad, it’s horrid. Biochem Soc Trans 35: 957–961, 2007. doi: 10.1042/BST0350957. [DOI] [PubMed] [Google Scholar]
- 52.Ronen R, Gan I, Modai S, Sukacheov A, Dror G, Halperin E, Shomron N. miRNAkey: a software for microRNA deep sequencing analysis. Bioinformatics 26: 2615–2616, 2010. doi: 10.1093/bioinformatics/btq493. [DOI] [PubMed] [Google Scholar]
- 53.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675, 2012. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song K, Backs J, McAnally J, Qi X, Gerard RD, Richardson JA, Hill JA, Bassel-Duby R, Olson EN. The transcriptional coactivator CAMTA2 stimulates cardiac growth by opposing class II histone deacetylases. Cell 125: 453–466, 2006. doi: 10.1016/j.cell.2006.02.048. [DOI] [PubMed] [Google Scholar]
- 55.Takahashi E, Abe J, Gallis B, Aebersold R, Spring DJ, Krebs EG, Berk BC. p90(RSK) is a serum-stimulated Na+/H+ exchanger isoform-1 kinase. Regulatory phosphorylation of serine 703 of Na+/H+ exchanger isoform-1. J Biol Chem 274: 20206–20214, 1999. doi: 10.1074/jbc.274.29.20206. [DOI] [PubMed] [Google Scholar]
- 56.Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, Rojas M, Hammond SM, Wang DZ. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol 42: 1137–1141, 2007. doi: 10.1016/j.yjmcc.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578, 2012. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA 103: 18255–18260, 2006. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wakabayashi S, Hisamitsu T, Nakamura TY. Regulation of the cardiac Na+/H+ exchanger in health and disease. J Mol Cell Cardiol 61: 68–76, 2013. doi: 10.1016/j.yjmcc.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 60.Wallert MA, Fröhlich O. Alpha 1-adrenergic stimulation of Na-H exchange in cardiac myocytes. Am J Physiol 263: C1096–C1102, 1992. doi: 10.1152/ajpcell.1992.263.5.C1096. [DOI] [PubMed] [Google Scholar]
- 61.Wang J, Liew OW, Richards AM, Chen YT. Overview of MicroRNAs in Cardiac Hypertrophy, Fibrosis, and Apoptosis. Int J Mol Sci 17: E749, 2016. doi: 10.3390/ijms17050749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Writing Group Members; Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jiménez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER III, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB; American Heart Association Statistics Committee; Stroke Statistics Subcommittee . Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 133: e38–e360, 2016. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 63.Xue J, Mraiche F, Zhou D, Karmazyn M, Oka T, Fliegel L, Haddad GG. Elevated myocardial Na+/H+ exchanger isoform 1 activity elicits gene expression that leads to cardiac hypertrophy. Physiol Genomics 42: 374–383, 2010. doi: 10.1152/physiolgenomics.00064.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yokoyama H, Gunasegaram S, Harding SE, Avkiran M. Sarcolemmal Na+/H+ exchanger activity and expression in human ventricular myocardium. J Am Coll Cardiol 36: 534–540, 2000. doi: 10.1016/S0735-1097(00)00730-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Differentially-expressed genes in the left ventricle of NHE1 transgenic mouse hearts - .xlsx (147 KB)