

Abstract
Thioredoxin reductase-1 (TXNRD1) inhibition effectively activates nuclear factor (erythroid-derived 2)-like 2 (Nrf2) responses and attenuates lung injury in acute respiratory distress syndrome (ARDS) and bronchopulmonary dysplasia (BPD) models. Upon TXNRD1 inhibition, heme oxygenase-1 (HO-1) is disproportionally increased compared with Nrf2 target NADPH quinone oxidoreductase-1 (Nqo1). HO-1 has been investigated as a potential therapeutic target in both ARDS and BPD. TXNRD1 is predominantly expressed in airway epithelial cells; however, the mechanism of HO-1 induction by TXNRD1 inhibitors is unknown. We tested the hypothesis that TXNRD1 inhibition induces HO-1 via Nrf2-dependent mechanisms. Wild-type (WT), Nrf2KO1.3, and Nrf2KO2.2 cells were morphologically indistinguishable, indicating that Nrf2 can be deleted from murine-transformed club cells (mtCCs) using CRISPR/Cas9 gene editing. Hemin, a Nrf2-independent HO-1-inducing agent, significantly increased HO-1 expression in WT, Nrf2KO1.3, and Nrf2KO2.2. Auranofin (AFN) (0.5 µM) inhibited TXNRD1 activity by 50% and increased Nqo1 and Hmox1 mRNA levels by 6- and 24-fold, respectively, in WT cells. Despite similar levels of TXNRD1 inhibition, Nqo1 mRNA levels were not different between control and AFN-treated Nrf2KO1.3 and Nrf2KO2.2. AFN slightly increased Hmox1 mRNA levels in Nrf2KO1.3 and Nrf2KO2.2 cells compared with controls. AFN failed to increase HO-1 protein in Nrf2KO1.3 and Nrf2KO2.2 compared with a 36-fold increase in WT mtCCs. Our data indicate that Nrf2 is the primary mechanism by which TXNRD1 inhibitors increase HO-1 in lung epithelia. Future studies will use ARDS and BPD models to define the role of HO-1 in attenuation of lung injury by TXNRD1 inhibitors.
Keywords: acute respiratory distress syndrome, auranofin, bronchopulmonary dysplasia, club cells, heme oxygenase-1, nuclear factor E2-related factor 2, thioredoxin reductase
INTRODUCTION
Thioredoxin reductase-1 (TXNRD1) is an NADPH-dependent selenocysteine-containing oxidoreductase that catalyzes the reduction of oxidized thioredoxin-1 (45). Our group has previously shown that TXNRD1 inhibition attenuates lung injury in newborn and adult murine models of acute respiratory distress syndrome (ARDS) and bronchopulmonary dysplasia (BPD) (5, 25, 41). Our collective data suggest that the protective effects of TXNRD1 inhibition are mediated, at least in part, through the activation of nuclear factor (erythroid-derived 2)-like 2 (Nrf2)-dependent responses (28). These findings are consistent with other studies in which inhibition or genetic deletion of TXNRD1 enhances Nrf2-dependent responses, including increased heme oxygenase-1 (HO-1) expression (3, 6, 8, 15, 18, 29, 38).
HO-1 is the rate-limiting inducible enzyme in the reduction of free heme into iron, carbon monoxide, and biliverdin (12). Hyperoxic exposure of neonatal HO-1 knockout mice elicits a BPD phenotype that is attenuated upon activation or overexpression of HO-1 (43). In models of ARDS, HO-1 overexpression confers resistance to the development of lung injury (10, 22, 26, 35, 42). In contrast to other Nrf2-regulated genes, we have consistently observed a disproportionate increase in HO-1 levels following TXNRD1 inhibition in vitro and in a murine BPD model in vivo (25, 28).
Though Nrf2 is a key regulator of HO-1 expression, HO-1 is not exclusively regulated by Nrf2 (12). HO-1 expression is also regulated by nuclear factor κ-light chain enhancer of activated B cells, activator protein-1, cAMP response binding protein, hypoxia-inducible factor-1, and BTB and CNC homology 1, basic leucine zipper transcription factor 1 (32, 37).
HO-1 is a key regulator of susceptibility to lung injury and has been identified as a therapeutic target to attenuate ARDS and BPD. In addition, there is currently much interest in investigating the use of Nrf2 activating compounds to prevent lung injury. In the adult and newborn lung, TXNRD1 is predominately expressed in airway epithelial cells (40). Our group is actively pursuing TXNRD1 inhibition as a strategy to prevent ARDS and BPD; however, the contribution of Nrf2 toward disproportionate increases in HO-1 following TXNRD1 in lung epithelial cells has not been defined. Thus, the present studies were designed to test the hypothesis that HO-1 induction following TXNRD1 inhibition is Nrf2 dependent.
MATERIALS AND METHODS
Auriothioglucose administration in vivo.
Animal protocols were approved by the Institutional Animal Care and Use Committee at the Research Institute at Nationwide Children’s Hospital. Mice were handled in accordance with the National Institutes of Health guidelines and housed in a pathogen-free facility. Adult C3H/HeN mice (6–8 wk) received a single intraperitoneal injection of 0 or 25 mg/kg auriothioglucose (ATG) (Research Diagnostics, Flanders, NJ) in saline and were euthanized using intraperitoneal ketamine-xylazine (150/15) at 24 h postinjection. Lungs were removed and placed in TRIzol on dry ice. A portion of the left lung was homogenized using TissueLyser II (Qiagen, Valencia, CA).
Cell culture and generation of Nrf2 knockout murine-transformed club cells.
Murine-transformed club cells (mtCCs) (Dr. Franco DeMayo, Baylor University, Houston, TX) were cultured in complete media, Dulbecco's modified Eagle's medium with 4.5 g/l glucose (Thermo Fisher Scientific, Pittsburgh, PA), 10% fetal bovine serum (FBS) (Amizona Scientific, Vestavia Hills, AL), 100 U/ml penicillin, and 100 µg/ml streptomycin (Thermo Fisher Scientific) and were placed in a culture incubator HERAcell Vios 160i (Thermo Fisher Scientific) maintaining 5% CO2 at 37°C.
Nrf2 knockout mtCCs were generated with the CRISPR-CAS9 system as described (34). Guide sequence 1, GACTTGGAGTTGCCACCGCCAGG, and guide sequence 2, TGGGACTGTAGTCCTGGCGGTGG (Integrated DNA Technologies, Coralville, IA), at exon 1 in the genomic Nrf2 locus (NC_000068.7) were selected using the CRISPR design website (http://crispr.mit.edu/). Guide sequences were annealed into construct plasmids PX 458 SpCas9–2A-EGFP (No. 48138; Addgene, Cambridge, MA) and PX459 SpCas9-2A-Puro (No. 62988; Addgene). Plasmids were transformed into competent Escherichia coli, and the sequences were verified by UAB-Heflin Genomic Science Core (University of Alabama at Birmingham, Birmingham, AL). mtCCs were transfected using Lipofectamine 3000 (Thermo Fisher Scientific). Following 24 h of transfection, transfection media was replaced with selection medium containing 5 μg/ml puromycin (Thermo Fisher Scientific). Clones from guide sequence 1 (n = 3; 1.1, 1.2, and 1.3) and 2 clones from guide sequence 2 (2.1, and 2.2) were selected and expanded in selection media for these studies. Nrf2 deletion was confirmed using quantitative real time polymerase chain reaction (PCR).
Treatment and sample preparation.
Both wild-type and Nrf2 knockout mtCCs were plated on 6-well plates (Denville Scientific, Holliston, MA) at a density of 400,000–450,000 cells per well, adhered for 48 h, and treated with dimethyl sulfoxide (Thermo Fisher Scientific) control, 0.1 or 0.5 µM auranofin (AFN) (Sigma-Aldrich, St. Louis, MO), or 5 µM hemin (Sigma-Aldrich) in 2 ml complete media. Cells were incubated at 37°C under 5% CO2 for 1, 6, or 8 h. Treated cells were washed twice with PBS (Thermo Fisher Scientific) and then collected in 100 µl of lysis buffer (10 mM Tris, 0.1% Triton 100x, and 100 µM diethylenetriamine pentaacetic acid, pH 7.4). Cell lysates were centrifuged at 14,000 g at 4°C for 10 min in Centrifuge 5804 R (Eppendoff, Hamburg, Germany), and supernatant was collected for TXNRD1 activity assays and Western blot. Protein concentrations of the supernatant were determined by bicinchoninic acid assay (Thermo Fisher Scientific).
TXNRD1 activities.
TXNRD1 activities were determined as previously described (7, 36).
Western blot analysis.
Protein (12–20 µg) was separated on 4%–20% Midi-Criterion TGX Stain-Free gels (Bio-Rad Laboratories, Hercules, CA) and transferred to polyvinylidene difluoride membranes (Trans-Blot; Bio-Rad). Membranes were blocked by 5% milk in Tris-buffered saline containing 0.05% Tween 20 and probed with polyclonal rabbit anti-HO-1 antibody (SPA-895, 1:1,000; Santa Cruz Biotechnology, Dallas, TX) followed by goat anti-rabbit-IgG-horseradish peroxidase (sc2004; 1:5,000; Santa Cruz Biotechnology). Membranes were developed using Clarity Western ECL Substrate (Bio-Rad) and imaged using a ChemiDoc MP Imaging System (Bio-Rad). As a loading control, membranes were reprobed with monoclonal mouse anti-β-actin antibody (sc-4777; 5,000; Santa Cruz Biotechnology) or polyclonal rabbit anti-β-tubulin antibody (SC-9104; 5,000; Santa Cruz Biotechnology).
Quantitative real-time PCR.
Treated cells were washed once with PBS, detached by trypsin (Thermo Fisher Scientific), collected in PBS, and centrifuged using Sorvall ST 8 Centrifuge (Thermo Fisher Scientific) for 5 min at 1,000 g. RNA was isolated from the pellets using an RNeasy mini kit (Qiagen). cDNA was synthesized using High Capacity cDNA Reverse Transcription (Thermo Fisher Scientific). Quantitative real time PCR was performed using Premix Taqman master mix (Thermo Fisher Scientific), and cycle threshold (Ct) values were determined using iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad Laboratories). Primer probes for murine 18S (4310893E), HO-1 (Mm00516005_m1) (21), and NQO1 (01253561_m1) (Thermo Fisher Scientific). Ct values were normalized to 18S (ΔCT). Fold-change values were calculated using 2(−∆∆CT).
Statistics.
Data were analyzed using GraphPad Prism 6.0 (La Jolla, CA). All data expressed as means ± SE were tested for homogeneity of variances and were log transformed when indicated. Parametric data were analyzed by one-way analysis of variance followed by Tukey’s multiple-comparison tests. Nonparametric data were analyzed by Kruskal-Wallis followed by Dunn’s post hoc test. Significance was accepted at P < 0.05.
RESULTS
Hmox1 mRNA expression is increased in adult murine lungs following TXNRD1 inhibition.
Our group has established that treatment with the TXNRD1 inhibitor aurothioglucose (ATG) (25 mg/kg) is protective in adult murine lung injury models (5, 40, 41). Though systemic treatment with 25 mg/kg ATG inhibited pulmonary TXNRD1 activity by 80% at 24 h in our prior studies, pulmonary expression of the Nrf2 target NADPH quinone oxidoreductase-1 (NQO1) was not different from control-treated mice (28). We found that treatment with 200 mg/kg ATG was required to elicit a significant increase in Nqo1 mRNA levels (3-fold) in adult murine lungs. At that time, we had also assessed Hmox1 expression in the same lung samples used for our previous publication, although the data were not included because HO-1 was not a focus of that manuscript. Thus, given the current focus of the present manuscript, we now include these assessments of lung Hmox1 mRNA expression in adult murine lung specimens 24 h after treatment with 25 mg/kg ATG or saline. In contrast to our Nqo1 findings, these data reveal that 25 mg/kg ATG significantly increased lung Hmox1 levels by threefold when compared with controls (Fig. 1). These additional data are consistent with our previous reports of disproportionate increases in lung Hmox1 levels (vs. Nqo1) following TXNRD1 inhibition in vitro and in our murine BPD model studies in vivo (25, 28).
Fig. 1.
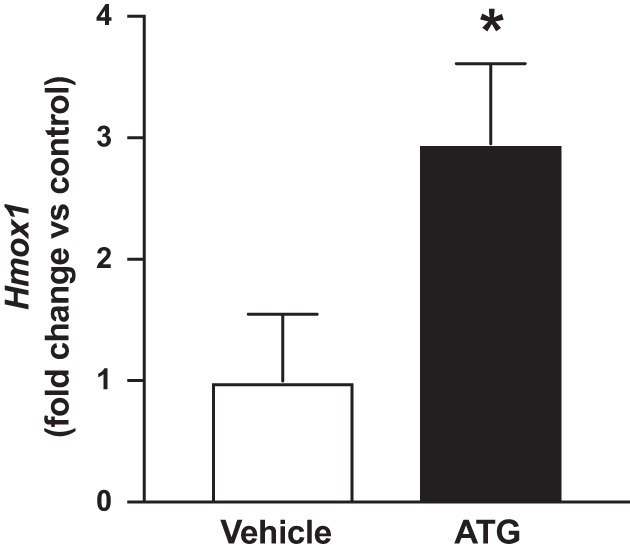
Hmox1 expression in adult murine lungs 24 h after aurothioglucose (ATG) administration. Adult C3H/HeN mice were treated with a single intraperitoneal injection of 0 or 25 mg/kg ATG in saline as described in materials and methods. Lung Hmox1 mRNA levels were increased by threefold in ATG-treated mice. Data (means ± SE, n = 3) were analyzed by t-test. *P = 0.043 vs. control
Optimization of AFN-mediated TXNRD1 inhibition in mtCCs.
To characterize the effects of AFN on TXNRD1 activity, since media contains selenium in serum, mtCCs were cultured in the absence or presence of 10% FBS and were treated with 0, 0.1 or 0.5 µM of AFN for 1 h. Cell lysates were prepared, and TXNRD1 activity was determined as described in materials and methods (Fig. 2). Under serum-free conditions, 0.1 µM AFN decreased TXNRD1 activity by 67% when compared with vehicle-treated cells. TXNRD1 activity was not detectable in mtCCs treated with 0.5 µM AFN under serum-free conditions, and we observed alterations in cell morphology. In mtCCs cultured in the presence of 10% FBS, baseline TXNRD1 activity was not different from serum-free conditions. When compared with control-treated cells, 0.1 µM AFN decreased TXNRD1 activity by 43%, and 0.5 µM AFN decreased TXNRD1 activity by 72%. Based upon these findings, mtCCs were cultured in 10% FBS-containing media, and 0.5 µM AFN was used to inhibit TXNRD1 activity in all subsequent experiments.
Fig. 2.
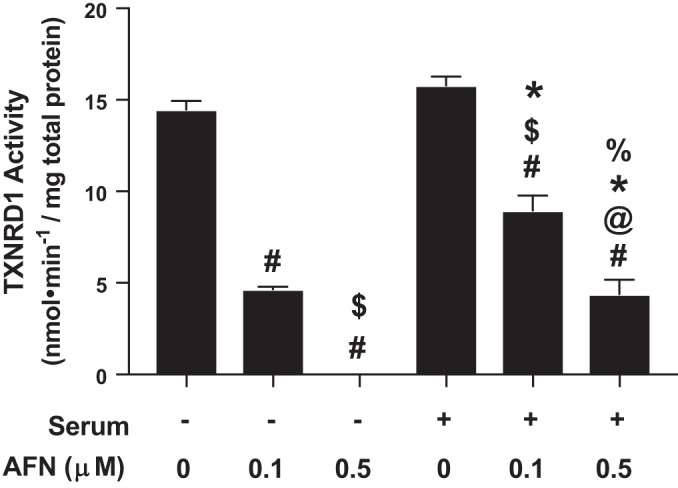
Thioredoxin reductase-1 (TXNRD1) activity in auranofin (AFN)-treated murine-transformed club cells (mtCCs) cultured in the presence and absence of 10% fetal bovine serum (FBS). mtCCs were cultured in medium in the presences/absences of serum containing 0% or 10% FBS and treated with the indicated concentrations of AFN for 1 h. TXNRD1 activity was determined as described in materials and methods. Data (means ± SE, n = 3–9) were analyzed by one-way ANOVA followed by Tukey’s post hoc analysis. #P < 0.006 vs. no serum/0 µM AFN; $P < 0.03 vs. no serum/0.1 µM AFN; @P = 0.0071 vs. no serum/0.5 µM AFN; *P < 0.0001 vs. serum/0 µM AFN; %P = 0.0048 vs. serum/0.1 µM AFN.
We next determined the kinetics of TXNRD1 inhibition and HO-1 expression in AFN-treated mtCCs. When compared with their respective controls, AFN decreased TXNRD1 activity by 72, 55, and 38% at 1, 3, and 6 h, respectively (Fig. 3A). We also detected significant increases in Hmox1 levels by AFN at all three time points (Fig. 3B). Specifically, Hmox1 levels were 7-, 31-, and 10-fold greater in AFN-treated cells at 1, 3, and 6 h, respectively. Our data demonstrated that AFN also significantly enhanced HO-1 protein levels in mtCCs (Fig. 3C). When compared with control-treated mtCCs, HO-1 expression was increased 71-fold after 3 h and 100-fold after 6 h. In light of these findings, we used 3-h AFN treatment to determine the role of Nrf2 on HO-1 induction following TXNRD1 inhibition.
Fig. 3.

Time-dependent effects of auranofin (AFN) on thioredoxin reductase-1 (TXNRD1) activity and heme oxygenase 1 (HO-1) expression in murine-transformed club cells. A: TXNRD1 activity was determined as described in materials and methods. Data (means ± SE, n = 6–15) were analyzed by one-way ANOVA followed by Tukey’s post hoc analysis. *P < 0.0001 vs. 1 h/vehicle; #P < 0.015 vs. 1 h/0.5 µM AFN; %P < 0.0001 vs. 3 h/vehicle; $P = 0.003 vs. 3 h/0.5 µM AFN; and &P = 0.0003 vs. 6 h/vehicle. B: dCt values (means ± SE, n = 5–15) were analyzed by one-way ANOVA followed by Tukey’s post hoc analysis. *P < 0.0001 vs. vehicle; #P < 0.0001 vs. 1 h/0.5 µM AFN; and $P = 0.0005 vs. 3 h/0.5 µM AFN. C: representative Western blot. Relative densitometries were calculated, and data (means ± SE, n = 6) were analyzed by Kruskal-Wallis followed by Dunn’s post hoc analysis. *P < 0.016 vs. vehicle and #P = 0.0256 vs. 1 h /0.5 µM AFN.
CRISPR/Cas9-mediated generation of gene-edited mtCCs.
To test our primary hypothesis, we elected to generate gene-edited mtCCs to ablate Nrf2 expression. We generated seven clones using strategies designed to delete regions of exon 1 in the NFE2L2 gene, as described in materials and methods. We identified three clones that did not express NFE2L2 mRNA (Fig. 4). Cell lines 1.3 (Nrf2KO1.3) and 2.2 (Nrf2KO2.2) were selected for use in subsequent studies.
Fig. 4.
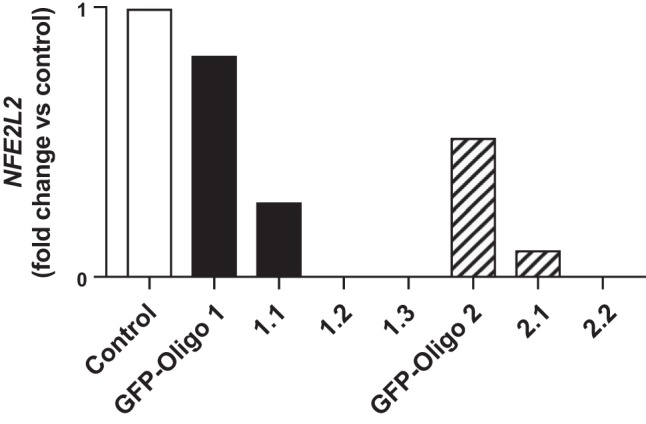
Nuclear factor E2-related factor 2 (NFE2l2) expression in gene-edited murine-transformed club cells. Nrf2 knockout cells were generated via CRISPR/Cas9 technology as described in materials and methods. Based upon these results, lines 1.3 and 2.2 were used for subsequent experiments. GFP, green fluorescent protein.
Hemin, a Nrf2-independent HO-1 inducer, increases HO-1 expression in wild-type and Nrf2 knockout mtCCs.
To confirm that Nrf2 knockout mtCCs maintained the ability to upregulate the expression of Hmox1 and HO-1 protein, we used hemin, an Nrf2-independent HO-1-inducing agent (9, 39, 44). Wild-type, Nrf2KO1.3, and Nrf2KO2.2 mtCCs were cultured for 8 h in the presence and absence of 5 µM hemin and Hmox1 expression, and HO-1 levels were determined (4, 16, 31, 46). Hemin enhanced Hmox1 expression by fivefold in wild-type mtCCs. In Nrf2 knockout cell lines, Hmox1 levels were 1.4-fold greater in Nrf2KO1.3 and 3-fold greater in Nrf2KO2.2, respectively, when compared with respective control-treated cells (Fig. 5A). Similar to mRNA effects, hemin also significantly increased HO-1 protein expression in all three cell lines (Fig. 5B). Specifically, HO-1 expression was 16-fold greater in hemin-treated wild-type mtCCs. In Nrf2KO1.3 and Nrf2KO2.2, hemin treatment increased HO-1 protein levels by six- and threefold, respectively, when compared with respective controls.
Fig. 5.
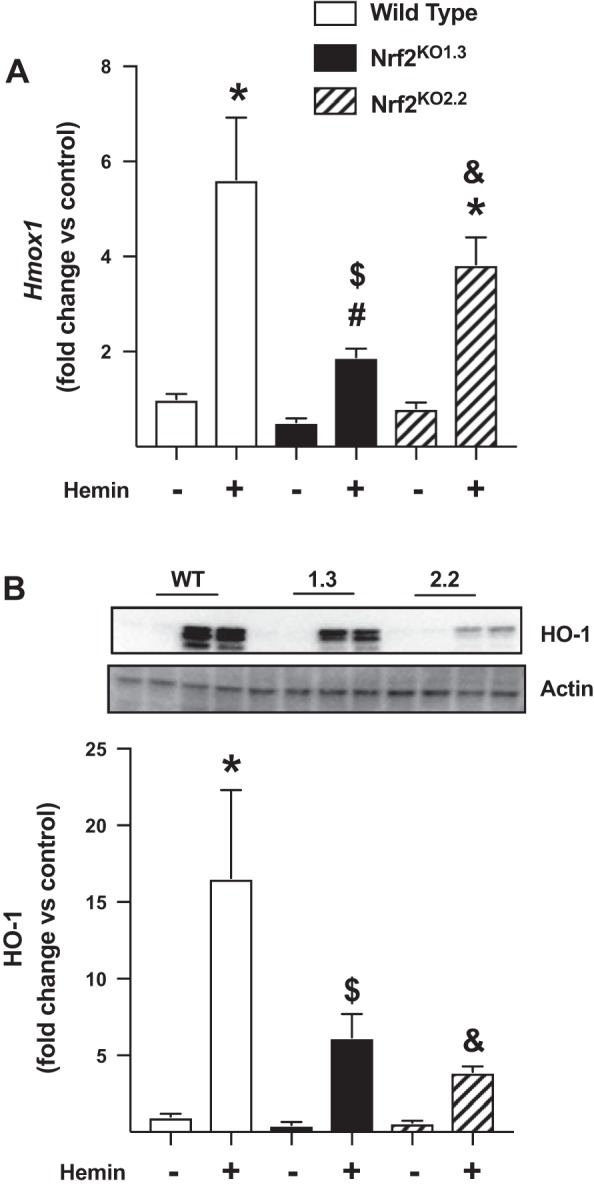
Effects of hemin on heme oxygenase 1 (HO-1) expression in wild-type (WT) and nuclear factor E2-related factor 2 (Nrf2) knockout murine-transformed club cells (mtCCs). mtCCs were cultured in the presence and absence of 5 µM hemin for 8 h, and Hmox1 mRNA and HO-1 protein were determined as described in materials and methods. A: dCt values (means ± SE, n = 5–6) were analyzed by one-way ANOVA followed by Tukey’s post hoc analysis. *P < 0.0012 vs. WT/vehicle; #P = 0.0011 different from WT/hemin; $P = 0.0002 vs. Nrf2 knockout 1.3/vehicle; and &P = 0.0270 vs. Nrf2 knockout 2.2/vehicle. B: representative Western blot. C: relative densitometry. Data (means ± SE, n = 6) were log-transformed and analyzed by one-way ANOVA followed by Tukey’s post hoc analysis. *P = 0.0002 vs. WT/vehicle; $P = 0.0003 vs. Nrf2 knockout 1.3/vehicle; and &P < 0.0001 vs. Nrf2 knockout 2.2/vehicle.
AFN inhibits TXNRD1 activity but does not enhance Nqo1 levels in gene-edited mtCCs.
To evaluate the effect of loss of Nrf2 on TXNRD1 activity, control and Nrf2 knockout cells were treated with 0.5 µM AFN for 3 h, and TXNRD1 activity was assessed. Baseline TXNRD1 activity was not different between vehicle-treated wild-type and Nrf2KO1.3 cells (Fig. 6A). In contrast, we detected a significant decrease in TXNRD1 activity in Nrf2KO2.2 when compared with both wild-type and Nrf2KO1.3 cells. Following AFN treatment of wild-type mtCCs, TXNRD1 activity was reduced by 46% when compared with vehicle-treated controls. In Nrf2 knockout mtCCs, TXNRD1 activity was 35% lower in Nrf2KO1.3 and 52% lower in Nrf2KO2.2 when compared with respective vehicle treated controls.
Fig. 6.

Thioredoxin reductase-1 (TXNRD1) activity and NAPDH quinone oxidoreductase-1 (Nqo1) expression in auranofin (AFN)-treated wild-type nuclear factor E2-related factor 2 (Nrf2) knockout murine-transformed club cells (mtCCs). mtCCs were cultured in the presence and absence of 0.5 µM AFN for 3 h, and TXNRD1 activity and Nqo1 were determined as described in materials and methods. A: TXNRD1 activity in lysates from wild-type and Nrf2 knockout mtCCs. Data (means ± SE, n = 8–9) were analyzed by one-way ANOVA followed by Tukey’s post hoc analysis. *P < 0.0003 vs. wild-type/vehicle; #P < 0.0001 vs. wild-type/0.5 µM AFN; $P = 0.0002 vs. Nrf2 knockout 1.3/vehicle; and &P < 0.0001 vs. Nrf2 knockout 2.2/vehicle. B: mRNA levels of Nqo1 from wild-type and Nrf2 knockout mtCC lysates following 3 h AFN treatment. Data (means ± SE, n = 5–6) were analyzed by one-way ANOVA followed by Tukey’s post hoc analysis. *P = 0.0455 vs. wild-type/vehicle and #P < 0.0001 vs. wild-type/0.5 µM AFN.
As a surrogate for Nrf2-mediated transcriptional activation, we next evaluated Nqo1 levels in all cell lines cultured in the presence and absence of 0.5 µM AFN for 3 h. When compared with vehicle-treated wild-type cells, Nqo1 expression was significantly lower in Nrf2KO1.3 (0.7-fold) but not in Nrf2KO2.2 (Fig. 6B). In AFN-treated wild-type cells, Nqo1 levels were increased 6-fold when compared with vehicle-treated controls. In contrast, AFN did not significantly enhance Nqo1 levels in Nrf2KO1.3 or Nrf2KO2.2 when compared with respective vehicle-treated controls.
AFN enhances HO-1 expression in wild-type but not Nrf2 knockout mtCCs.
To test the hypothesis that increases in HO-1 levels following TXNRD1 inhibition are Nrf2 dependent, we measured Hmox1 and HO-1 levels in vehicle- and AFN-treated wild-type, Nrf2KO1.3, and Nrf2KO2.2 mtCCs. All cell lines were cultured in the presence or absence of 0.5 µM AFN for 3 h. Baseline Hmox1 levels were significantly lower in Nrf2KO1.3 but not in Nrf2KO2.2 when compared with wild-type mtCCs (Fig. 7A). AFN treatment enhanced Hmox1 levels by 24-fold in wild-type mtCCs when compared with vehicle-treated controls. In Nrf2KO1.3 and Nrf2KO2.2 mtCCs, AFN treatment significantly increased Hmox1 levels, albeit to a lesser degree than in wild-type mtCCs. Compared with wild-type mtCCs, baseline HO-1 protein levels were significantly lower in Nrf2KO1.3 but not in Nrf2KO2.2 (Fig. 7B). Similar to our Hmox1 findings, AFN significantly increased HO-1 protein levels by 36-fold compared with vehicle-treated controls. In contrast, HO-1 protein expression was not different between AFN- and vehicle-treated Nrf2KO1.3 and Nrf2KO2.2.
Fig. 7.

Heme oxygenase 1 (HO-1) expression in auranofin (AFN)-treated wild-type (WT) and nuclear factor E2-related factor 2 (Nrf2) knockout murine-transformed club cells (mtCCs). mtCCs were cultured in the presence and absence of 0.5 µM hemin for 3 h, and Hmox1 mRNA and HO-1 protein were determined as described in materials and methods. A: dCt values (means ± SE, n = 5–15) were analyzed by log-transformed one-way ANOVA followed by Tukey’s post-hoc analysis. *P < 0.0006 vs. WT/vehicle; #P < 0.0001 vs. WT/0.5 µM AFN; $P = 0.0002 vs. Nrf2 knockout 1.3/vehicle; and &P = 0.0046 vs. Nrf2 knockout 2.2/vehicle. B: representative Western blot and relative densitometry. Data (means ± SE, n = 6–9) were analyzed by one-way ANOVA followed by Tukey’s post hoc analysis. *P < 0.002 vs. WT/vehicle; #P < 0.0001 vs. WT/0.5 µM AFN; $P = 0.0005 vs. Nrf2 knockout 1.3/vehicle; and %P < 0.0001 vs. Nrf2 knockout 2.2/0.5 µM AFN.
DISCUSSION
Our past research has consistently demonstrated that TXNRD1 inhibition results in a disproportional increase in Hmox1 expression when compared with other Nrf2-regulated genes, namely Nqo1 (25, 28). The present data extend these findings by revealing that treatment of adult mice with 25 mg/kg ATG increases pulmonary Hmox1 mRNA levels at 24 h, which is in contrast to our previously reported findings with Nqo1 (28). The remainder of our studies focused on defining the role of Nrf2 on HO-1 induction in airway epithelial cells following TXNRD1 inhibition. Our novel data in mtCCs revealed that: 1) the addition of FBS to culture media does not alter baseline TXNRD1 activity but does influence the degree of TXNRD1 inhibition by AFN, 2) Nrf2 can be successfully deleted from mtCCs using CRISPR/Cas9 gene editing, 3) Nrf2 knockout cells are capable of producing HO-1 in response to hemin, 4) AFN inhibits TXNRD1 activity in Nrf2 knockout cells, and 6) AFN does not increase HO-1 in Nrf2 knockout mtCCs.
In addition to various growth factors, FBS contains the trace mineral selenium (Se). Se is required for the production of selenocysteine, which is present in the active site of selenoproteins, such as TXNRD1 (36). In the absence of Se, cysteine is instead inserted into the active site of selenoproteins, and catalytic activity is significantly compromised. In the present studies, baseline TXNRD1 activity was similar in mtCCs cultured in the presence and absence of FBS, and maximal TXNRD1 inhibition was detected in cells treated with 0 or 0.5 µM AFN (Fig. 2). In contrast to cells cultured in the presence of FBS in which 0.5 µM AFN completely inhibited TXNRD1 activity. Indeed, morphological alterations were also observed in AFN-treated mtCCs cultured in the absence of FBS (data not shown). These findings suggest that Se in FBS dampens AFN toxicity. Our data are consistent with recent findings of AFN-mediated mitochondrial oxidative stress that is rescued upon Se supplementation (13, 30, 33).
Upon determining the kinetics of HO-1 induction following AFN treatment of mtCCs (Fig. 3), we used Nrf2-deficient mtCCs using CRISPR-Cas 9 technology (Fig. 4). To minimize the chance that findings would be due to off-target effects of our targeting constructs, we generated two distinct targeting constructs designed to elicit the deletion of exon 1. To confirm that our Nrf2 knockout cell lines were capable of producing HO-1 in response to treatment with an Nrf2-independent agonist, we used hemin (14, 17). Our data revealed significant increases in Hmox1 and HO-1 levels in wild-type and Nrf2 knockout mtCCs following hemin treatment, though we did observe relative differences between wild-type and knockout lines Nrf2KO1.3 and Nrf2KO2.2 (Fig. 5). The use of a single time point for our analyses limited our ability to evaluate the kinetics of Hmox1 and HO-1 induction by hemin and may be responsible for these observations. Nonetheless, our data indicate that gene-edited mtCCs are capable of HO-1 induction.
To compare the effects of AFN across the three cell lines used to test our primary hypothesis, we measured TXNRD1 activity following AFN treatment (Fig. 6A). Basal TXNRD1 activity was similar between wild-type and Nrf2KO1.3; however, activity was significantly lower in Nrf2KO2.2 mtCCs. Given that TXNRD1 is regulated by Nrf2, this finding is likely to represent altered homeostasis in this cell line. Nqo1 is exclusively regulated by Nrf2. To further evaluate the effects of Nrf2 deletion, we determined Nqo1 levels in response to AFN (Fig. 6B). As expected, Nqo1 levels were increased in AFN-treated wild-type mtCCs. In contrast, AFN did not significantly increase Nqo1 levels in either Nrf2KO1.3 or Nrf2KO2.2.
Lastly, we tested our central hypothesis by measuring Hmox1 and HO-1 expression in wild-type and Nrf2 knockout mtCCs cultured in the presence and absence of AFN. Whereas AFN significantly increased Hmox1 in wild-type cells, this effect was severely attenuated in Nrf2KO1.3 and Nrf2KO2.2 (Fig. 7A). The finding of modestly increased Hmox1 expression in our Nrf2 knockout cells suggests the presence of Nrf2-independent pathways for HO-1 induction in mtCCs. The physiological relevance of this finding is questionable, given the substantial AFN-mediated enhancement of Hmox1 in wild-type cells. As expected, AFN significantly increased HO-1 expression in wild-type mtCCs. In contrast to our mRNA data, AFN did not alter HO-1 protein levels in either Nrf2KO1.3 or Nrf2KO2.2 mtCCs (Fig. 7B). Collectively, our data indicate that increases in HO-1 in the lung following TXNRD1 inhibition are predominantly driven via Nrf2-dependent mechanisms. Our findings in lung epithelia are consistent with Nrf2 dependency of HO-1 induction following induction or stress in other models (1, 2, 9, 11, 23, 27).
HO-1 expression is also regulated by various transcription factors, including nuclear factor κ-light chain enhancer of activated B cells, activator protein-1, cAMP response binding protein, hypoxia-inducible factor-1, and BTB and CNC homology 1, basic leucine zipper transcription factor 1 (32, 37). Hyperoxia, oxidative stress, and pharmacologic agents also induce HO-1 expression (11, 12, 24). The present findings indicate that HO-1 induction following TXNRD1 inhibition is predominately regulated via Nrf2-dependent mechanisms in mtCCs. It should be noted that small but significant increases in Hmox1 mRNA levels were detected in AFN-treated Nrf2KO1.3 and Nrf2KO2.2 mtCCs when compared with respective control-treated cells (Fig. 7A). This finding is in contrast to Nqo1, which was not increased by AFN in either Nrf2KO1.3 and Nrf2KO2.2 cells (Fig. 6B). These data confirm Nrf2-dependent induction of Nqo1 as well as the presence of Nrf2-independent HO-1 induction by AFN. HO-1 inducing agents have been extensively studied as potential therapies to prevent and/or treat ARDS and BPD (10, 22, 26, 35, 42). Our laboratory has established TXNRD1 as a novel therapeutic target to prevent ARDS and BPD (5, 25). Our data do not specifically explain the disproportionate increase in HO-1 upon TXNRD1 inhibition when compared with other Nrf2-dependent genes. Maf proteins modulate Hmox1 transcription by forming heterodimers with Nrf2 and other basic leucine zipper factors and binding to the Maf-ARE sites in distal enhancer regions of the Hmox1 gene (20). AFN and similar gold compounds have been shown to induce Maf heterodimer formation (19). Thus, it is possible that Mafs may contribute to the disproportionate increases in Hmox1 levels by AFN in our studies; however, these investigations are beyond the scope of the present manuscript.
In summary, we used gene editing technology to establish that TXNRD1 inhibitors elicit increases in HO-1 expression in lung epithelial cells, primarily via Nrf2-dependent mechanisms, which is consistent with our hypothesis. Given the key role of HO-1 in modulating susceptibility to ARDS and BPD, we speculate that HO-1 uniquely contributes to the protective effects of TXNRD1 inhibitors in vivo. Future transgenic studies in murine models of ARDS and BPD will define the precise contribution of HO-1 in pulmonary protection by TXNRD1 inhibitors.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01-HL-119280 (to T. E. Tipple) and R01-HL-119280-S1 (to K. Dunigan).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.D., Q.L., M.L.L., and T.E.T. conceived and designed research; K.D., Q.L., R.L., M.L.L., and S.W. performed experiments; K.D., Q.L., R.L., M.L.L., S.W., and T.E.T. analyzed data; K.D., Q.L., R.L., M.L.L., S.W., and T.E.T. interpreted results of experiments; K.D. and T.E.T. prepared figures; K.D., Q.L., and T.E.T. drafted manuscript; K.D., Q.L., and T.E.T. edited and revised manuscript; K.D., Q.L., R.L., M.L.L., S.W., and T.E.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Anupam Agarwal for experimental advice and guidance.
REFERENCES
- 1.Aggarwal S, Lam A, Bolisetty S, Carlisle MA, Traylor A, Agarwal A, Matalon S. Heme attenuation ameliorates irritant gas inhalation-induced acute lung injury. Antioxid Redox Signal 24: 99–112, 2016. doi: 10.1089/ars.2015.6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aodengqimuge LS, Liu S, Mai S, Li X, Li Y, Hu M, Yuan S, Song L. AP-1 activation attenuates the arsenite-induced apoptotic response in human bronchial epithelial cells by up-regulating HO-1 expression. Biotechnol Lett 36: 1927–1936, 2014. doi: 10.1007/s10529-014-1560-z. [DOI] [PubMed] [Google Scholar]
- 3.Arnér ES. Focus on mammalian thioredoxin reductases–important selenoproteins with versatile functions. Biochim Biophys Acta 1790: 495–526, 2009. doi: 10.1016/j.bbagen.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 4.Basireddy M, Lindsay JT, Agarwal A, Balkovetz DF. Epithelial cell polarity and hypoxia influence heme oxygenase-1 expression by heme in renal epithelial cells. Am J Physiol Renal Physiol 291: F790–F795, 2006. doi: 10.1152/ajprenal.00402.2005. [DOI] [PubMed] [Google Scholar]
- 5.Britt RD Jr, Velten M, Locy ML, Rogers LK, Tipple TE. The thioredoxin reductase-1 inhibitor aurothioglucose attenuates lung injury and improves survival in a murine model of acute respiratory distress syndrome. Antioxid Redox Signal 20: 2681–2691, 2014. doi: 10.1089/ars.2013.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cebula M, Schmidt EE, Arnér ES. TrxR1 as a potent regulator of the Nrf2-Keap1 response system. Antioxid Redox Signal 23: 823–853, 2015. doi: 10.1089/ars.2015.6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cenas N, Prast S, Nivinskas H, Sarlauskas J, Arnér ES. Interactions of nitroaromatic compounds with the mammalian selenoprotein thioredoxin reductase and the relation to induction of apoptosis in human cancer cells. J Biol Chem 281: 5593–5603, 2006. doi: 10.1074/jbc.M511972200. [DOI] [PubMed] [Google Scholar]
- 8.Chew EH, Nagle AA, Zhang Y, Scarmagnani S, Palaniappan P, Bradshaw TD, Holmgren A, Westwell AD. Cinnamaldehydes inhibit thioredoxin reductase and induce Nrf2: potential candidates for cancer therapy and chemoprevention. Free Radic Biol Med 48: 98–111, 2010. doi: 10.1016/j.freeradbiomed.2009.10.028. [DOI] [PubMed] [Google Scholar]
- 9.Chi X, Guo N, Yao W, Jin Y, Gao W, Cai J, Hei Z. Induction of heme oxygenase-1 by hemin protects lung against orthotopic autologous liver transplantation-induced acute lung injury in rats. J Transl Med 14: 35, 2016. doi: 10.1186/s12967-016-0793-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi AM, Alam J. Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am J Respir Cell Mol Biol 15: 9–19, 1996. doi: 10.1165/ajrcmb.15.1.8679227. [DOI] [PubMed] [Google Scholar]
- 11.Dennery PA. Heme oxygenase in neonatal lung injury and repair. Antioxid Redox Signal 21: 1881–1892, 2014. doi: 10.1089/ars.2013.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dennery PA. Signaling function of heme oxygenase proteins. Antioxid Redox Signal 20: 1743–1753, 2014. doi: 10.1089/ars.2013.5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du Y, Zhang H, Lu J, Holmgren A. Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J Biol Chem 287: 38210–38219, 2012. doi: 10.1074/jbc.M112.392225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrándiz ML, Devesa I. Inducers of heme oxygenase-1. Curr Pharm Des 14: 473–486, 2008. doi: 10.2174/138161208783597399. [DOI] [PubMed] [Google Scholar]
- 15.Fourquet S, Guerois R, Biard D, Toledano MB. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J Biol Chem 285: 8463–8471, 2010. doi: 10.1074/jbc.M109.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hock TD, Liby K, Wright MM, McConnell S, Schorpp-Kistner M, Ryan TM, Agarwal A. JunB and JunD regulate human heme oxygenase-1 gene expression in renal epithelial cells. J Biol Chem 282: 6875–6886, 2007. doi: 10.1074/jbc.M608456200. [DOI] [PubMed] [Google Scholar]
- 17.Hualin C, Wenli X, Dapeng L, Xijing L, Xiuhua P, Qingfeng P. The anti-inflammatory mechanism of heme oxygenase-1 induced by hemin in primary rat alveolar macrophages. Inflammation 35: 1087–1093, 2012. doi: 10.1007/s10753-011-9415-4. [DOI] [PubMed] [Google Scholar]
- 18.Iverson SV, Eriksson S, Xu J, Prigge JR, Talago EA, Meade TA, Meade ES, Capecchi MR, Arnér ES, Schmidt EE. A Txnrd1-dependent metabolic switch alters hepatic lipogenesis, glycogen storage, and detoxification. Free Radic Biol Med 63: 369–380, 2013. doi: 10.1016/j.freeradbiomed.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kassovska-Bratinova S, Yang G, Igarashi K, Dennery PA. Bach1 modulates heme oxygenase-1 expression in the neonatal mouse lung. Pediatr Res 65: 145–149, 2009. doi: 10.1203/PDR.0b013e318191eedc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kataoka K, Handa H, Nishizawa M. Induction of cellular antioxidative stress genes through heterodimeric transcription factor Nrf2/small Maf by antirheumatic gold(I) compounds. J Biol Chem 276: 34074–34081, 2001. doi: 10.1074/jbc.M105383200. [DOI] [PubMed] [Google Scholar]
- 21.Kim C, Jang JS, Cho MR, Agarawal SR, Cha YN. Taurine chloramine induces heme oxygenase-1 expression via Nrf2 activation in murine macrophages. Int Immunopharmacol 10: 440–446, 2010. doi: 10.1016/j.intimp.2009.12.018. [DOI] [PubMed] [Google Scholar]
- 22.Lam A, Vetal N, Matalon S, Aggarwal S. Role of heme in bromine-induced lung injury. Ann N Y Acad Sci 1374: 105–110, 2016. doi: 10.1111/nyas.13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, Choi AM. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem 272: 5375–5381, 1997. doi: 10.1074/jbc.272.9.5375. [DOI] [PubMed] [Google Scholar]
- 24.Lever JM, Boddu R, George JF, Agarwal A. Heme oxygenase-1 in kidney health and disease. Antioxid Redox Signal 25: 165–183, 2016. doi: 10.1089/ars.2016.6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Q, Wall SB, Ren C, Velten M, Hill CL, Locy ML, Rogers LK, Tipple TE. Thioredoxin reductase inhibition attenuates neonatal hyperoxic lung injury and enhances nuclear factor E2-related factor 2 activation. Am J Respir Cell Mol Biol 55: 419–428, 2016. doi: 10.1165/rcmb.2015-0228OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin XH, Pan JB, Zhang XJ. WITHDRAWN: anti-inflammatory and anti-oxidant effects of apigetrin on LPS-induced acute lung injury by regulating Nrf2 and AMPK pathways. Biochem Biophys Res Commun pii: S0006-291X(17)31413-4, 2017. doi: 10.1016/j.bbrc.2017.07.071. [DOI] [PubMed] [Google Scholar]
- 27.Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci 73: 3221–3247, 2016. doi: 10.1007/s00018-016-2223-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Locy ML, Rogers LK, Prigge JR, Schmidt EE, Arnér ES, Tipple TE. Thioredoxin reductase inhibition elicits Nrf2-mediated responses in Clara cells: implications for oxidant-induced lung injury. Antioxid Redox Signal 17: 1407–1416, 2012. doi: 10.1089/ars.2011.4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandal PK, Schneider M, Kölle P, Kuhlencordt P, Förster H, Beck H, Bornkamm GW, Conrad M. Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res 70: 9505–9514, 2010. doi: 10.1158/0008-5472.CAN-10-1509. [DOI] [PubMed] [Google Scholar]
- 30.Ouyang Y, Peng Y, Li J, Holmgren A, Lu J. Modulation of thiol-dependent redox system by metal ions via thioredoxin and glutaredoxin systems. Metallomics 10: 218–228, 2018. doi: 10.1039/C7MT00327G. [DOI] [PubMed] [Google Scholar]
- 31.Park EJ, Kim YM, Chang KC. Hemin reduces HMGB1 release by UVB in an AMPK/HO-1-dependent pathway in human keratinocytes HaCaT cells. Arch Med Res 48: 423–431, 2017. doi: 10.1016/j.arcmed.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 32.Prawan A, Kundu JK, Surh YJ. Molecular basis of heme oxygenase-1 induction: implications for chemoprevention and chemoprotection. Antioxid Redox Signal 7: 1688–1703, 2005. doi: 10.1089/ars.2005.7.1688. [DOI] [PubMed] [Google Scholar]
- 33.Radenkovic F, Holland O, Vanderlelie JJ, Perkins AV. Selective inhibition of endogenous antioxidants with Auranofin causes mitochondrial oxidative stress which can be countered by selenium supplementation. Biochem Pharmacol 146: 42–52, 2017. doi: 10.1016/j.bcp.2017.09.009. [DOI] [PubMed] [Google Scholar]
- 34.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8: 2281–2308, 2013. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raval CM, Lee PJ. Heme oxygenase-1 in lung disease. Curr Drug Targets 11: 1532–1540, 2010. doi: 10.2174/1389450111009011532. [DOI] [PubMed] [Google Scholar]
- 36.Rengby O, Cheng Q, Vahter M, Jörnvall H, Arnér ES. Highly active dimeric and low-activity tetrameric forms of selenium-containing rat thioredoxin reductase 1. Free Radic Biol Med 46: 893–904, 2009. doi: 10.1016/j.freeradbiomed.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 37.Ryter SW, Choi AM. Heme oxygenase-1: redox regulation of a stress protein in lung and cell culture models. Antioxid Redox Signal 7: 80–91, 2005. doi: 10.1089/ars.2005.7.80. [DOI] [PubMed] [Google Scholar]
- 38.Suvorova ES, Lucas O, Weisend CM, Rollins MF, Merrill GF, Capecchi MR, Schmidt EE. Cytoprotective Nrf2 pathway is induced in chronically txnrd 1-deficient hepatocytes. PLoS One 4: e6158, 2009. doi: 10.1371/journal.pone.0006158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tahara T, Sun J, Igarashi K, Taketani S. Heme-dependent up-regulation of the alpha-globin gene expression by transcriptional repressor Bach1 in erythroid cells. Biochem Biophys Res Commun 324: 77–85, 2004. doi: 10.1016/j.bbrc.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 40.Tipple TE. The thioredoxin system in neonatal lung disease. Antioxid Redox Signal 21: 1916–1925, 2014. doi: 10.1089/ars.2013.5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tipple TE, Welty SE, Rogers LK, Hansen TN, Choi YE, Kehrer JP, Smith CV. Thioredoxin-related mechanisms in hyperoxic lung injury in mice. Am J Respir Cell Mol Biol 37: 405–413, 2007. doi: 10.1165/rcmb.2006-0376OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu D, Wang Y, Zhang H, Du M, Li T. Acacetin attenuates mice endotoxin-induced acute lung injury via augmentation of heme oxygenase-1 activity. Inflammopharmacology 26: 635–643, 2017. doi: 10.1007/s10787-017-0398-0. [DOI] [PubMed] [Google Scholar]
- 43.Yang G, Biswasa C, Lin QS, La P, Namba F, Zhuang T, Muthu M, Dennery PA. Heme oxygenase-1 regulates postnatal lung repair after hyperoxia: role of β-catenin/hnRNPK signaling. Redox Biol 1: 234–243, 2013. doi: 10.1016/j.redox.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zenke-Kawasaki Y, Dohi Y, Katoh Y, Ikura T, Ikura M, Asahara T, Tokunaga F, Iwai K, Igarashi K. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol Cell Biol 27: 6962–6971, 2007. doi: 10.1128/MCB.02415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang B, Zhang J, Peng S, Liu R, Li X, Hou Y, Han X, Fang J. Thioredoxin reductase inhibitors: a patent review. Expert Opin Ther Pat 27: 547–556, 2017. doi: 10.1080/13543776.2017.1272576. [DOI] [PubMed] [Google Scholar]
- 46.Zhu Y, Sun Y, Jin K, Greenberg DA. Hemin induces neuroglobin expression in neural cells. Blood 100: 2494–2498, 2002. doi: 10.1182/blood-2002-01-0280. [DOI] [PubMed] [Google Scholar]