

Abstract
Long-acting glucagon-like peptide-1 (GLP-1) receptor (GLP-1R) agonists (GLP-1RA), such as exendin-4 (Ex4), promote weight loss. On the basis of a newly discovered interaction between GLP-1 and oleoylethanolamide (OEA), we tested whether OEA enhances GLP-1RA-mediated anorectic signaling and weight loss. We analyzed the effect of GLP-1+OEA and Ex4+OEA on canonical GLP-1R signaling and other proteins/pathways that contribute to the hypophagic action of GLP-1RA (AMPK, Akt, mTOR, and glycolysis). We demonstrate that OEA enhances canonical GLP-1R signaling when combined with GLP-1 but not with Ex4. GLP-1 and Ex4 promote phosphorylation of mTOR pathway components, but OEA does not enhance this effect. OEA synergistically enhanced GLP-1- and Ex4-stimulated glycolysis but did not augment the hypophagic action of GLP-1 or Ex4 in lean or diet-induced obese (DIO) mice. However, the combination of Ex4+OEA promoted greater weight loss in DIO mice than Ex4 or OEA alone during a 7-day treatment. This was due in part to transient hypophagia and increased energy expenditure, phenotypes also observed in Ex4-treated DIO mice. Thus, OEA augments specific GLP-1RA-stimulated signaling but appears to work in parallel with Ex4 to promote weight loss in DIO mice. Elucidating cooperative mechanisms underlying Ex4+OEA-mediated weight loss could, therefore, be leveraged toward more effective obesity therapies.
Keywords: energy balance, GLP-1, obesity, oleoylethanolamide, weight loss
INTRODUCTION
As a gateway disease, pandemic obesity is paving the way for billions of children and adults to develop type 2 diabetes (T2D), cardiovascular disease, and neurodegenerative diseases in the coming decades (14). Long-acting glucagon-like peptide-1 (GLP-1) receptor (GLP-1R) agonists (GLP-1RA) such as exenatide and liraglutide (Lira) constitute a relatively new class of T2D drugs that stand out among traditional T2D therapies because of their effects on body weight. Whereas most T2D drugs cause weight gain (42), GLP-1RA improve glucose homeostasis while promoting moderate weight loss (36, 56). We and others provide evidence that GLP-1RA promote weight loss by reducing food intake via activation of the GLP-1R in the brain (54, 58, 60). Activation of GLP-1R in the brain subsequently reduces caloric intake via modulation of various molecular pathways including glycolytic, cAMP/cAMP-dependent protein kinase A (PKA), AMP-activated protein kinase (AMPK), Akt (PKB), and mammalian target of rapamycin (mTOR) signaling (5, 7, 50, 72). Therefore, discovering methods for boosting these GLP-1RA-stimulated anorectic pathways is an attractive strategy for developing more effective obesity therapeutics.
To date, strategies to enhance GLP-1R signaling have primarily focused on identifying small molecules that bind to allosteric sites on the GLP-1R and stimulate canonical GLP-1R signaling (e.g., cAMP production) in the presence of GLP-1RA (55, 63, 67). The present studies instead focus on modulating GLP-1R signaling by targeting the GLP-1RA itself. Recently, Cheng et al. reported that the endocannabinoid-like lipid oleoylethanolamide (OEA) binds to GLP-1 and enhances GLP-1-mediated cAMP production without altering the affinity of GLP-1 for the GLP-1R (8). This is intriguing since cAMP signaling and subsequent activation of PKA in the brain have been implicated in the food intake-suppressive effects of GLP-1RA (26, 49). OEA is a lipid-derived messenger that is released postprandially from the small intestine and also promotes satiety through activation of peroxisome proliferator-activated receptor-α (PPARα; 16, 45). Together, these observations proffered the possibility that OEA could enhance other pathways downstream of the GLP-1R that contribute to the anorectic and weight loss effects of GLP-1RA. Furthermore, we posited that if OEA could augment GLP-1RA-mediated anorectic signaling events, then the combination of GLP-1RA+OEA could result in an augmented hypophagia and/or weight loss.
In this study, we first sought to interrogate the effect of OEA on canonical and anorectic GLP-1RA signaling events induced by GLP-1 and another GLP-1RA, exendin-4 (Ex4). The GLP-1R is a Gαs seven-transmembrane receptor that, when activated, stimulates the production of cAMP and recruitment of β-arrestin to the GLP-1R. Since we and others demonstrate that the anorectic potential of GLP-1RA is mediated not only by these canonical pathways but also via brain glycolysis, AMPK, Akt, and mTOR signaling (5, 7, 26, 50, 72), we used cell-based assays to determine the effect of OEA on these signaling pathways in the presence of GLP-1 and Ex4. We then used the results of these in vitro experiments to investigate the physiological relevance of GLP-1RA+OEA with respect to energy balance in vivo in both lean mice and mice in the setting of diet-induced obesity (DIO). Overall, we provide evidence that OEA modulates GLP-1RA signaling in a GLP-1RA-specific manner and that Ex4+OEA promotes greater weight loss than Ex4 or OEA alone, though this latter effect appears to be additive rather than synergistic.
MATERIALS AND METHODS
Cell Culture
cAMP Hunter CHO-K1 GLP-1R Gs cells (hereinafter designated as CHOK1-GLP-1R cells; DiscoverX, Fremont, CA) and PathHunter CHO-K1 GLP-1R β-arrestin cells (DiscoverX) were proliferated and maintained at 37°C (5% CO2, humidified) in Ham’s F-12 complete media (HyClone SH30026.01: 10 mM glucose, 1 mM pyruvate, and 1 mM glutamine) supplemented with 10% fetal bovine serum (FBS), 10 ml/l penicillin-streptomycin (HyClone, Rockford, IL), and Geneticin G418 Sulfate (0.8 mg/ml) antibiotic to preserve the selection of GLP-1R-expressing cells. Cells were split every 48–72 h using TrypLE Select (Life Technologies, Grand Island, NY) for detachment and counted with a Countess automated cell counter (Invitrogen, Carlsbad, CA). GLP-1-(7–36) amide (no. 2082; Tocris), Ex4 (ab120214; Abcam), Lira (H-6724; Bachem), and OEA (O0383; Sigma) were used for cell experiments. For experiments analyzing PKA substrate phosphorylation, nutrient-sensing pathway phosphorylation, and glycolysis, the 20 pM concentration of GLP-1 and Ex4 used was chosen on the basis of the EC50 of cAMP production of GLP-1RA in our studies (GLP-1: 35 pM, Ex4: 0.39 pM) and those of Roed et al. (47; GLP-1: 9.8 pM, Ex4: 23 pM). Furthermore, the concentration of 9.2–10 µM OEA used in all cell studies was based on the findings of Cheng et al. (8).
cAMP Production
cAMP production was measured in CHOK1-GLP-1R cells via time-resolved fluorescence resonance energy transfer immunoassay with the LANCE Ultra cAMP Detection Kit (PerkinElmer, Waltham, MA) per manufacturer’s instructions after stimulation with a range of concentrations of GLP-1 or Ex4 in the absence or presence of OEA (9.2 µM). Stimulation buffer [1X HBSS, HEPES (5 mM), Ro 20-1724 (0.5 mM), and 0.1% BSA, pH 7.4] was made fresh and used for the dilution of reagents and GLP-1RA ± OEA. A volume of 2 µl/well of GLP-1RA ± OEA and controls were added to a 1,536-well plate at two times the final desired concentration, after which cells, resuspended in stimulation buffer, were dispensed (500 cells·2 µl−1·well−1) using a Multidrop Combi reagent dispenser (Thermo Scientific). After centrifugation at 700 rpm for 1 min, the plate was incubated at room temperature for 30 min. Antibody mix (2 µl/well) and 2 µl/well tracer mix were then added to all wells, and the plate was centrifuged at 1,000 rpm for 1 min and then incubated at room temperature for 20 min. Reading of time-resolved fluorescence resonance energy transfer signals at 665 and 615 nm was performed with an EnVision Multilabel plate reader (PerkinElmer).
β-Arrestin Recruitment
CHO-K1 GLP-1R β-arrestin cells were resuspended in Cell Plating 2 reagent (DiscoverX) and seeded (1,000 cells·4 µl−1·well−1) in a 1,536-well tissue culture-treated microplate (Corning, Corning, NY). After an overnight incubation, 1 µl of treatments prepared in ligand buffer (1× HBSS, 10 mM HEPES, and 0.1% BSA) was added to stimulate cells with a range of concentrations of GLP-1 or Ex4 in the absence and presence of OEA (9.2 µM) for 1.5 h at 37°C. With the use of the Multidrop Combi reagent dispenser, 3 µl of PathHunter detection reagent (DiscoverX) were added to all wells and incubated at room temperature for 60 min in the dark. Chemiluminescent signal was detected on an EnVision Multilabel plate reader (PerkinElmer) using a counting time of 1 s/well.
Western Blot for Phosphorylation of PKA Substrates
CHOK1-GLP-1R cells were seeded at 500,000 cells per well in 6-well plates and grown for 48 h. After an 80-min serum starve, cells were treated with vehicle (1× PBS), GLP-1 (20 pM), OEA (10 µM), or GLP-1 (20 pM) + OEA (10 µM) for 15 min in duplicate. After treatment, cells were quickly washed with 1× PBS, snap-frozen on a thin layer of liquid nitrogen, and stored at −80°C until analysis. Cell homogenates were processed by scraping in lysis buffer (25 mM Tris·HCl, 150 mM NaCl, 1 µM EDTA, 1 µM EGTA, 2.5 mM sodium pyrophosphate, 100 mM sodium fluoride, and 1 mM sodium orthovanadate) containing 1× protease and phosphatase inhibitors. Supernatant of combined technical duplicates was collected after centrifugation of cell homogenates at 17,000 g for 10 min at 4°C. Protein concentration was determined with the Bradford assay. After SDS-PAGE (20 µg/well, 200 V for 45 min at 4°C) and transfer of proteins to polyvinylidene difluoride (100 V for 60 min at 4°C), PKA substrate phosphorylation was determined by probing with the Phospho-PKA Substrate monoclonal antibody (1:4,000, 4°C overnight; no. 9624; Cell Signaling Technology) and secondary antibodies (1 h at room temperature, no. 926-32211 and 926-68070; LI-COR). Blots were imaged with an Odyssey Imaging System, and full lanes were analyzed using GelQuant.NET software provided by BiochemLabSolutions.com.
Analysis of ERK1/2 and Nutrient-Sensing Pathway Phosphorylation Events
CHOK1-GLP-1R cells were seeded at 500,000 cells per well in 6-well plates and grown for 48 h. After an 80-min serum starve, cells were treated with vehicle (1× PBS), GLP-1 (20 pM), Ex4 (20 pM), OEA (10 µM), GLP-1 (20 pM) + OEA (10 µM), or Ex4 (20 pM) + OEA (10 µM) for 5, 15, or 60 min in duplicate. For analysis of phosphorylation of PKA substrates, the same procedure was followed, and CHOK1-GLP-1R cells were treated with vehicle (1× PBS), GLP-1 (20 pM), OEA (10 µM), or GLP-1 (20 pM) + OEA (10 µM) for 15 min. After treatment, cells were quickly washed with 1× PBS, snap-frozen on a thin layer of liquid nitrogen, and stored at −80°C until analysis. Cell homogenates were processed by scraping in cell lysis buffer (no. 7018; Cell Signaling Technology) containing protease and phosphatase inhibitors. Supernatant of combined technical duplicates was collected after centrifugation of cell homogenates at 17,000 g for 10 min at 4°C. Protein concentration was determined with the Bradford assay to assure equal loading of protein. The phosphorylated status of AMPK, Akt, ERK1/2, and mTOR pathway targets was determined in four experimental replicates with the PathScan Akt Signaling Antibody Array Kit (no. 9700; Cell Signaling Technology) per manufacturer’s instructions. PKA substrate phosphorylation was determined in three experimental replicates by Western blot using the Phospho-PKA Substrate monoclonal antibody (1:4,000; no. 9624; Cell Signaling Technology). Arrays were imaged with an Odyssey Imaging System and analyzed using Image Studio software (LI-COR).
Seahorse XF Analysis of Glycolysis and Mitochondrial Respiration
Seahorse XF technology (Agilent Technologies, Santa Clara, CA) was used to monitor extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) to measure levels of glycolysis and mitochondrial respiration, respectively. CHOK1-GLP-1R cells were seeded in Seahorse XF96 cell culture microplates at a concentration of 20,000 cells per well and incubated at 37°C overnight. On the experimental day, cells were washed two times with 200 µl of assay media [complete media without sodium bicarbonate, FBS, or Geneticin G418 Sulfate; pH = 7.38–7.42; 37°C] and then incubated in assay media with or without the GLP-1R antagonist exendin-9 (Ex9, 500 nM) for 80 min in a carbon dioxide-free incubator. ECAR and OCR measurements were made at 7-min intervals before (Baseline) and after injection of GLP-1RA (20 pM) ± OEA (10 µM) from the Seahorse XF96 sensor cartridge. To verify that the observed changes in ECAR were due to changes in glycolysis, the same experiment was performed but using the glycolytic inhibitor 2-deoxyglucose (2-DG; 20 mM) instead of Ex9 during the 80-min preincubation. All reagents were brought to a pH of 7.38–7.42 at 37°C before use.
Animal Husbandry
All experiments were performed in male, C57BL/6J mice (Jackson Laboratories) housed in a temperature- and humidity-controlled vivarium with a 12:12-h light (0600–1800)-and-dark schedule. Mice were provided rodent chow (4% fat, 48.5% carbohydrate, and 16.4% protein; no. 2016; Harlan Teklad) and water ad libitum. For DIO experiments, mice were fed a high-fat diet (60% fat, 20% carbohydrate, and 20% protein; D12492; Research Diets) for 9 wk at Jackson Laboratories starting at 6 wk of age and then for an additional 6 wk at Sanford Burnham Prebys Medical Discovery Institute (SBP) before experiments were performed. All procedures were approved by the SBP Institutional Animal Care and Use Committee.
Analysis of Food Intake, Energy Expenditure, and Activity In Vivo
Energy balance experiments were performed using the Promethion metabolic cage system (Sable Systems, Las Vegas, NV) to monitor food intake, body weight, activity, and respiratory gases (O2 and CO2) concurrently throughout the experiment. Mice were individually housed in Promethion cages for 72 h before beginning any experiments. For acute experiments, food was restricted for 5 h before administering treatments within 30 min of the onset of the dark cycle. Vehicle [saline-Tween 80-polyethylene glycol 400 (STP), 90:5:5] and artificial cerebrospinal fluid (ACSF, no. 59-7316; Harvard Apparatus) were used as vehicles for intraperitoneal and intracerebroventricular injections, respectively. Intraperitoneal injection volumes were calculated at 6 µl/g body wt. OEA was prepared by dissolving in STP vehicle at 56°C and inverting to mix. GLP-1 or Ex4 were premixed with OEA in cotreatment groups.
Experiment 1.
Experiment 1 examined whether OEA would augment the hypophagic action of peripherally administered (ip) GLP-1 or Ex4. Food intake was measured for 18 h after intraperitoneal injection of GLP-1 (3 µg/kg), Ex4 (3 µg/kg), OEA (8 mg/kg), GLP-1 (3 µg/kg) + OEA (8 mg/kg), or Ex4 (3 µg/kg) + OEA (8 mg/kg). The dose of GLP-1RA (3 µg/kg) was chosen on the basis of previous studies showing this to be an effective hypophagic dose of Ex4 that does not induce malaise (57). The same dose was used for GLP-1 for consistency. To minimize the effect on food intake by OEA, a lower dose of OEA (8 mg/kg) than what is needed for OEA to elicit a hypophagic response in rodents was used (10–25 mg/kg; 44, 61). Baseline food intake was measured after administration of vehicle the day before treatments.
Experiment 2.
The design for experiment 2 was the same as that for experiment 1 with the addition of an intracerebroventricular injection of the glycolytic inhibitor 2-DG to reduce brain glycolysis. Brain cannulas were implanted in 14–16-wk-old mice under isoflurane (3–5%) anesthesia. The skull was exposed and leveled between bregma and lambda. Guide cannulas (PlasticsOne) were targeted to the lateral ventricle (from bregma: 0.34 mm caudal, 1.0 mm from midline, and 2.4 mm ventral) through a burr hole using a stereotactic apparatus (David Kopf Instruments) and affixed using two jeweler’s screws and dental acrylic. After surgery, mice were individually housed and allowed to recover at least 5 days before intracerebroventricular injections. Two microliters of ACSF or 2-DG (5 mM, D6134; Sigma) were injected intracerebroventricularly 15 min before intraperitoneal injection of treatments: vehicle, OEA (8 mg/kg), Ex4 (3 µg/kg), or premixed Ex4+OEA. An ACSF-vehicle (icv and ip, respectively) dose was given to all mice before experimentation to acclimatize them to both injections. Baseline measurements were obtained after injection of ACSF-vehicle on the day preceding treatments. As intracerebroventricular administration of 2-DG can induce hyperphagia (13, 40, 41, 59), we used a dose of 2-DG that we have previously shown to not induce hyperphagia by itself but that still attenuates the hypophagic action of intracerebroventricularly administered Ex4 (5). Proper placement of cannulae targeting the lateral ventricle was verified postexperimentation if a dipsogenic response was observed in mice after injection of angiotensin II (1 µg/2 µl ACSF, H1705.0025; Bachem).
Experiment 3.
We next investigated the weight loss potential of Ex4+OEA in the context of obesity. After 15 wk on a high-fat diet, mice were housed individually in Promethion metabolic cages for 72 h and then acclimated to intraperitoneal injections and handling for 2 days. Mice were then assigned to treatment groups and injected twice daily (0900–1100 and 1700–1800) with vehicle, OEA (5 mg/kg), Ex4 (100 µg/kg), or Ex4+OEA for 7 days. The dose of OEA (5 mg/kg) was derived from other studies that measured the effect of nonacute (>24 h) OEA administration on body weight in rodents (16, 20, 71). The dose of Ex4 (100 µg/kg) was chosen on the basis of a literature review (1, 34, 43, 69) and pilot studies in our laboratory indicating that a dose higher than that commonly used for nonobese mice (3 µg/kg) is necessary to cause significant weight loss in obese mice. Body weights were measured each morning to calculate treatment doses. Body composition (i.e., lean and fat mass) was measured before and after treatments with a minispec LF90II-TD NMR Analyzer (Bruker, Billerica, MA).
Data Analysis and Statistics
Data were analyzed using GraphPad Prism 7 Software (GraphPad Software, La Jolla, CA) and Excel (Microsoft, Seattle, WA). Two-tailed t-tests, one-way ANOVA, and two-way ANOVA for interaction analysis with Tukey’s multiple-comparisons test were used where appropriate. The energy expenditure analysis of covariance (ANCOVA) done for this work was provided by the National Institute of Diabetes and Digestive and Kidney Diseases Mouse Metabolic Phenotyping Centers (http://www.mmpc.org) using their Energy Expenditure Analysis page (http://www.mmpc.org/shared/regression.aspx). Promethion data were extracted by performing Macro 1, Macro 11, and Macro 13 within ExpeData software (v.1.9.14; Sable Systems) using the UMC10.1.11 macro package for mice. Resting metabolic rate (RMR) is defined as the mean energy expenditure (kcal/h) during the 30 min of lowest activity over a 24-h period (ExpeData readout: QR_EE_30).
RESULTS
OEA Selectively Enhances GLP-1RA-Mediated Intracellular Signaling Events in CHO-K1 Cells Overexpressing the GLP-1R
We first tested the effect of GLP-1RA+OEA combinations on cAMP production and β-arrestin recruitment to the GLP-1R, both of which are canonical Gαs signaling events. As previously shown (8), OEA augmented cAMP production mediated by GLP-1 (GLP-1 EC50 = 34.3 pM vs. GLP-1+OEA EC50 = 6.0 pM, Fig. 1A) but not Ex4 (Ex4 EC50 = 0.39 pM vs. Ex4+OEA EC50 = 0.35 pM, Fig. 1B). Ex4 more potently stimulated cAMP production compared with GLP-1 (Fig. 1C). Furthermore, OEA enhanced β-arrestin recruitment to the GLP-1R specifically in the presence of GLP-1 (GLP-1 EC50 = 53.8 nM vs. GLP-1+OEA EC50 = 10.1 nM, Fig. 1D) but not in the presence of Ex4 (Ex4 EC50 = 20.7 nM vs. Ex4+OEA EC50 = 9.8 nM, Fig. 1E). In the absence of OEA, GLP-1 and Ex4 stimulated similar levels of β-arrestin recruitment (Fig. 1F). To determine whether the enhanced cAMP formation by GLP-1+OEA would lead to increased activity of the cAMP/PKA pathway, we measured the relative phosphorylation of PKA substrates via Western blot. Whereas GLP-1 led to a ~2-fold increase in phosphorylated PKA substrates, the addition of OEA did not alter this response (Fig. 1H). We also measured ERK1/2 phosphorylation since this event can be induced via cAMP and β-arrestin signaling pathways. However, OEA failed to induce ERK1/2 phosphorylation in the presence of 20 pM GLP-1 (Fig. 1I), a concentration that did not elicit any increase in ERK1/2 phosphorylation by itself, or at higher concentrations of GLP-1 (1–100 nM) that all induced an approximately twofold increase in ERK1/2 phosphorylation (data not shown). The concentration of OEA (9.2 µM) used in these studies had no effect on cAMP production, the phosphorylation of PKA substrates, or ERK1/2 phosphorylation by itself (Fig. 1, G–I). These data demonstrate that OEA enhances canonical GLP-1R signaling in a GLP-1RA-dependent manner.
Fig. 1.

Oleoylethanolamide (OEA) enhances glucagon-like peptide-1 (GLP-1)-mediated cAMP production and β-arrestin recruitment to the GLP-1 receptor (GLP-1R) without affecting PKA activity. A–G: cAMP production (A–C and G) was measured in cAMP Hunter CHO-K1 GLP-1R Gs (CHOK1-GLP-1R) cells, and β-arrestin recruitment to the GLP-1R (D–F) was measured in CHOK1-GLP-1R-β-arrestin cells after stimulation with various concentrations of GLP-1 or exendin-4 (Ex4) in the absence and presence of OEA (9.2 µM). Dashed line in G is the concentration at which OEA was used for all cell experiments (9.2 µM). Data points reflect the mean ± SE of 3 experimental replicates containing 4 technical replicates each. *GLP-1R agonist (GLP-1RA) vs. GLP-1RA+OEA, #Ex4 vs. GLP-1 (P < 0.05). The x-axis values represent log [Peptide]. H: phosphorylation of PKA substrates was assessed in CHOK1-GLP-1R cells after stimulation with GLP-1 (20 pM), OEA (10 µM), or GLP-1+OEA. Bars reflect means ± SE of 3 experimental replicates consisting of 2 technical replicates each. †vs. vehicle (P < 0.05). Representative immunoblot is shown. Images obtained from different locations on the original immunoblot are separated by white spaces. I: PathScan (Akt Signaling Antibody Array Kit, no. 9700; Cell Signaling Technology) analysis was used to assess phosphorylation of phospho-ERK1/2 (Thr421/Tyr204) in CHOK1-GLP-1R cells after stimulation with vehicle, GLP-1 (20 pM), OEA (10 µM), or GLP-1+OEA for 5, 15, or 60 min. Bars reflect means ± SE of 4 experimental replicates consisting of 2 technical replicates (P < 0.05).
We next assessed the effect of GLP-1 and Ex4 (20 pM) ± OEA (10 µM) on specific phosphorylation events within the nutrient-sensing pathways of AMPK, Akt, and mTOR in CHOK1-GLP-1R cells (Fig. 2). Both GLP-1 and Ex4 induced mTOR signaling as shown by increased phosphorylation of 70-kDa ribosomal protein S6 kinase (p70S6K; Thr389) and S6 (Ser235/236) at each time point analyzed, but only cells treated with Ex4 expressed increased mTOR (Ser2481) phosphorylation itself at 60 min. OEA alone stimulated phosphorylation of p70S6K (Thr389) by 15 min, but the presence of OEA did not alter GLP-1- or Ex4-stimulated phosphorylation of mTOR pathway targets. Furthermore, neither GLP-1, Ex4, OEA, nor either GLP-1RA+OEA combination altered the phosphorylation of any other targets [AMPK (Thr172), Akt (Thr308 or Ser473), ERK1/2 (Thr202/Tyr204), eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1; Thr37/46), or 40-kDa proline-rich Akt substrate (PRAS40; Thr246)] tested at any time point.
Fig. 2.

Oleoylethanolamide (OEA) does not enhance nutrient-sensing pathways that mediate the hypophagic action of glucagon-like peptide-1 (GLP-1) receptor (GLP-1R) agonists. cAMP Hunter CHO-K1 GLP-1R Gs cells were incubated for 5, 15, or 60 min with either GLP-1 (20 pM; A–C) or exendin-4 (Ex4, 20 pM; D–F) in the absence or presence of OEA (10 µM). PathScan (Akt Signaling Antibody Array Kit, no. 9700; Cell Signaling Technology) analysis was then performed to measure the phosphorylated state of targets within AMPK, Akt, and mTOR signaling pathways. Bars reflect means ± SE of 3 experimental replicates consisting of 2 technical replicates each. 4E-BP1, eukaryotic translation initiation factor 4E-binding protein 1; p70S6K, 70-kDa ribosomal protein S6 kinase; PRAS40, 40-kDa proline-rich Akt substrate. *vs. vehicle (P < 0.05).
We have previously shown that Ex4 moderately stimulates glycolysis in hypothalamic GT1-7 cells (5), and we (5) and others (50) show that reducing brain glycolysis with 2-DG attenuates the hypophagic action of intracerebroventricularly administered Ex4 and GLP-1. We, therefore, assessed the effect of OEA on GLP-1RA-mediated stimulation of intracellular glycolysis in CHOK1-GLP-1R cells. In these experiments, OEA significantly increased ECAR, a readout of glycolysis, when combined with GLP-1 [2-way ANOVA, interaction: F(12,30) = 70.33, P < 0.0001; time: F(3,30) = 213.4, P < 0.0001; treatment: F(4,10) = 238, P < 0.0001; Fig. 3A] or Ex4 [2-way ANOVA, interaction: F(12,30) = 59.71, P < 0.0001; time: F(3,30) = 180.1, P < 0.0001; treatment: F(4,10) = 233.2, P < 0.0001; Fig. 3B]. Pretreatment of CHOK1-GLP-1R cells with the GLP-1R antagonist Ex9 significantly attenuated the enhancement of ECAR by GLP-1+OEA and Ex4+OEA (Fig. 3, A and B) suggesting that this event is mediated via the GLP-1R. Importantly, pretreatment of cells with the glycolytic inhibitor 2-DG blocked the effect of GLP-1RA+OEA on ECAR (data not shown) demonstrating that the observed changes in ECAR reflect changes in glycolysis and not another source of acidification. OCR, an index of mitochondrial respiration, was not altered by GLP-1RA or OEA in GLP-1 [2-way ANOVA, interaction: F(9,21) = 0.6658, P = 0.7300; time: F(3,21) = 1.763, P = 0.1851; treatment: F(3,7) = 0.6962, P = 0.5832; Fig. 3C] or Ex4 [2-way ANOVA, interaction: F(9,21) = 0.7142, P = 0.6904; time: F(3,21) = 1.261, P = 0.3133; treatment: F(3,7) = 0.6737, P = 0.5950; Fig. 3D] studies. Therefore, OEA enhances GLP-1- and Ex4-mediated glycolysis in CHOK1-GLP-1R cells.
Fig. 3.

Oleoylethanolamide (OEA) enhances glycolysis in the presence of glucagon-like peptide-1 (GLP-1) receptor (GLP-1R) agonists (GLP-1RA) in a GLP-1R-dependent manner. Extracellular acidification rate (ECAR; A and B) and oxygen consumption rate (OCR; C and D) were measured in cAMP Hunter CHO-K1 GLP-1R Gs cells with a Seahorse XF96 analyzer to evaluate glycolysis and mitochondrial respiration, respectively, in the presence of GLP-1 (20 pM), exendin-4 (Ex4, 20 pM), OEA (9.2 µM), or GLP-1RA+OEA. For analysis of GLP-1R-mediated effects, cells were exposed to exendin-9 (Ex9, 500 nM) for 80 min before addition of treatments to inhibit GLP-1R signaling. Data points represent the means ± SE of 3 experimental replicates containing 3–5 technical replicates each. Data were normalized to baseline ECAR or OCR and are represented as fold change from vehicle. *GLP-1RA vs. GLP-1RA+OEA. †GLP-1RA+OEA vs. Ex9-GLP-1RA+OEA (P < 0.05).
OEA Does Not Significantly Alter the Acute Hypophagic Action of GLP-1 or Ex4
On the basis of our findings that OEA enhances GLP-1- and Ex4-stimulated glycolysis in cell culture and that brain glycolysis mediates the food intake-suppressive effects of GLP-1RA (5, 50), we tested the hypothesis that OEA augments the hypophagic action of GLP-1 and Ex4 in a brain glycolysis-dependent manner. We first measured food intake in lean, C57BL/6J mice after intraperitoneal injection of GLP-1 or Ex4. In these experiments, food intake was not suppressed by OEA [2-way ANOVA, interaction: F(9,198) = 0.4334, P = 0.9159; time: F(9,198) = 239.8, P < 0.0001; treatment: F(1,22) = 0.4508, P = 0.5089; Fig. 4E], GLP-1 [2-way ANOVA, interaction: F(9,90) = 0.2052, P = 0.9930; time: F(9,90) = 124.9, P < 0.0001; treatment: F(1,10) = 0.1123, P = 0.7444, Fig. 4A], or GLP-1+OEA [2-way ANOVA, interaction: F(9,90) = 0.2644, P = 0.9825; time: F(9,90) = 187.6, P < 0.0001; treatment: F(1,10) = 0.006102, P = 0.9393, Fig. 4B]. The selected dose of Ex4 (3 µg/kg) promoted a moderate, acute hypophagic response [2-way ANOVA, interaction: F(9,252) = 3.095, P = 0.0015; time: F(9,252) = 248.3, P < 0.0001; treatment: F(1,28) = 8.101, P = 0.0082, Fig. 4C]. Ex4+OEA promoted a similar hypophagic response compared with Ex4 [2-way ANOVA, interaction: F(9,279) = 5.404, P < 0.0001; time: F(9, 279) = 447.3, P < 0.0001; treatment: F(1, 31) = 9.253, P = 0.0048, Fig. 4D]. However, statistical comparison of these data demonstrates that Ex4 and Ex4+OEA stimulate similar hypophagic outcomes [2-way ANOVA, interaction: F(27,590) = 0.9349, P = 0.5611; time: F(9,590) = 220.7, P < 0.0001; treatment: F(3,590) = 40.48, P < 0.0001; Fig. 4, C and D].
Fig. 4.

Oleoylethanolamide (OEA) does not alter the hypophagic action of glucagon-like peptide-1 (GLP-1) or exendin-4 (Ex4). A–E: cumulative, 18-h food intake was measured using a PromethION metabolic cage system in lean C57BL/6J mice after intraperitoneal injection of vehicle, OEA (8 mg/kg), GLP-1 receptor agonist (GLP-1RA, 3 µg/kg), or GLP-1RA+OEA at the onset of the dark cycle and after 5 h of food deprivation. Data points reflect the means ± SE (n = 12–17/group) *vs. vehicle at same time point (P < 0.05). F–L: chow-fed C57BL/6J mice received an intracerebroventricular injection of artificial cerebrospinal fluid (ACSF, 2 µl) or 2-deoxyglucose (2-DG, 5 mM, 2 µl) followed 15 min later with intraperitoneal injection of vehicle, OEA (8 mg/kg), Ex4 (3 µg/kg), or Ex4+OEA. Data points reflect the means ± SE (n = 7–8/group). *vs. ACSF-vehicle at same time point (P < 0.05). Black and white bars represent dark and light periods, respectively.
Pretreatment with intracerebroventricular 2-DG demonstrates that the anorectic effect of peripherally administered Ex4 alone is partially, although not significantly, attenuated by inhibition of brain glycolysis [2-way ANOVA, interaction: F(27, 260) = 0.8639, P = 0.6637; time: F(9,260) = 90.98, P < 0.0001; treatment: F(3,260) = 25.25, P < 0.0001; combined comparison of data from Fig. 4, I vs. J]. Similarly, inhibition of brain glycolysis had no effect on the hypophagic action of Ex4+OEA [2-way ANOVA, interaction: F(27,240) = 0.6871, P = 0.8777; time: F(9,240) = 142.5, P < 0.0001; treatment: F(3,240) = 13.5, P < 0.0001; combined comparison of data from Fig. 4, K vs. L]. We demonstrate that the selected dose of OEA (8 mg/kg) did not cause any acute reductions in food intake by itself nor did 2-DG alter this observation (Fig. 4, F and G). Furthermore, since 2-DG delivery to the brain can cause hyperphagia (13, 40, 41, 59), we verified that the dose of 2-DG (2 µl, 5 mM) that we have previously shown to attenuate the hypophagic action of intracerebroventricularly delivered Ex4 (5) did not cause hyperphagia in the present studies (Fig. 4H). From these data, we conclude that brain glycolysis may contribute to reductions in food intake induced by peripherally administered Ex4 but combining OEA with GLP-1 or Ex4 does not enhance the anorectic potential of either of these GLP-1RA.
Chronic Ex4+OEA Causes a Greater Reduction in Body Weight Than Ex4 or OEA Alone in Obese Mice
Although OEA did not enhance the anorectic potential of Ex4 in lean mice, we were interested in testing whether combined administration of Ex4 with OEA would lead to greater weight loss in DIO mice. Herein, we observed that 7-day (twice a day) treatment of DIO mice with either OEA, Ex4, or Ex4+OEA leads to weight loss (Fig. 5, A–C). In support of our hypothesis, Fig. 5, A–C, illustrates that Ex4+OEA (−6.0 ± 0.4 g, −11.6 ± 0.5%) promoted a greater loss in body weight compared with either Ex4 (−4.7 ± 0.4 g, −9.5 ± 0.8%) or OEA (−3.5 ± 0.2 g, −7.1 ± 0.5%) alone [2-way ANOVA, interaction: F(21,266) = 15.39, P < 0.0001; time: F(7,266) = 360.8, P < 0.0001; treatment: F(3,38) = 39.78, P < 0.0001, Fig. 5A]. Whereas the weight loss induced by OEA was gradual, the weight loss observed in Ex4- and Ex4+OEA-treated mice occurred in two phases. The first phase occurred during the first 24 h of treatment (day 1) and was characterized by a −5.1 ± 0.2 and −5.4 ± 0.2% reduction in body weight for Ex4- and Ex4+OEA-treated mice, respectively. The second phase, which occurred over the subsequent 6 treatment days, was characterized by a slower rate of weight loss that yielded a total weight loss of −9.5 ± 0.8 and −11.6 ± 0.5% in Ex4- and Ex4+OEA-treated mice, respectively. Loss of body fat was the main contributor to overall weight loss in mice treated with Ex4 and Ex4+OEA (Fig. 5D); however, there was a trend for lean mass to be significantly lower in Ex4+OEA compared with Ex4 groups (Fig. 5E).
Fig. 5.
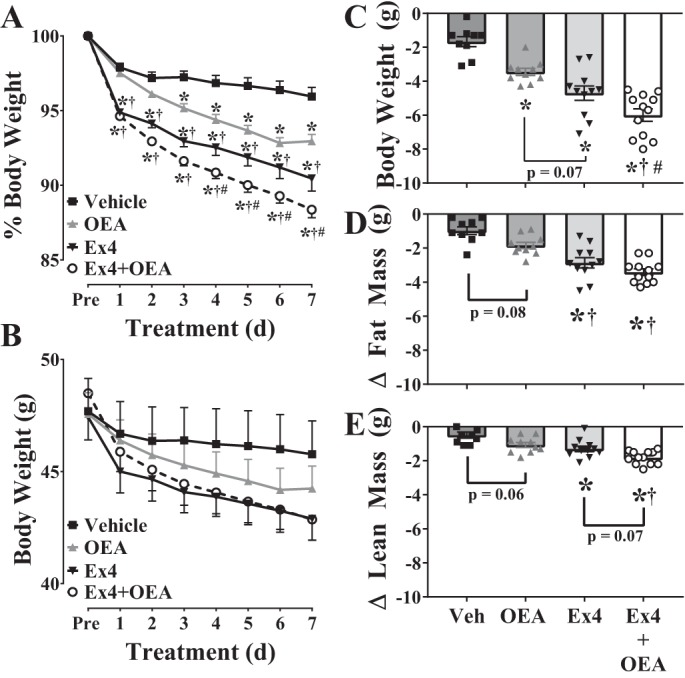
Exendin-4 (Ex4) + oleoylethanolamide (OEA) promotes greater weight loss than Ex4 or OEA alone in diet-induced obese (DIO) mice. A–C: body weights (expressed as %, raw data and delta) of male, C57BL/6J, DIO mice were measured daily during chronic treatment (7 days, intraperitoneal injection, twice a day) with vehicle, OEA (5 mg/kg), Ex4 (100 µg/kg), or Ex4+OEA. D and E: fat (D) and lean mass (E) were measured with NMR before treatments began and after the 7-day treatment course. Values are means ± SE (n = 9–12). d, Days; Pre, preceding treatment; Veh, vehicle. *vs. vehicle; †vs. OEA; #vs. Ex4 (n = 9–12/group, P < 0.05).
Impact of Ex4+OEA Cotherapy on Factors That Regulate Energy Balance
Parameters of energy balance [i.e., food intake, energy expenditure, locomotor activity, and respiratory quotient (RQ)] were interrogated to identify the cause of Ex4+OEA-mediated weight loss in DIO mice. In parallel with the pattern of body weight loss, Ex4 and Ex4+OEA treatment promoted a significant decrease in food intake during the first day of treatment, but interestingly, food intake gradually recovered to match vehicle-treated mice by the fifth day of treatment (Fig. 6A). OEA alone did not significantly alter food intake during this study (Fig. 6A). The fact that Ex4 and Ex4+OEA treatment continued to promote weight loss (Fig. 5A) even as food intake levels were restored to normal (Fig. 6A) suggested to us that energy expenditure may be increasing in response to Ex4 and Ex4+OEA treatment. Analysis of raw energy expenditure failed to discover any difference in daily or cumulative energy expenditure between treatment groups (Fig. 6B). However, comparison of energy expenditure between the first day of treatment (day 1) and the final day of treatment (day 7) within each group revealed that energy expenditure had increased over the treatment period in DIO mice that received either Ex4 or Ex4+OEA but not in vehicle- or OEA-treated mice (Fig. 6C).
Fig. 6.

Exendin-4 (Ex4) and Ex4 + oleoylethanolamide (OEA) transiently reduce 24-h food intake and increase energy expenditure (EE)/resting metabolic rate (RMR). Components of energy balance [food intake, EE, locomotor activity, and respiratory quotient (RQ)] were measured in male, diet-induced obese, C57BL/6J mice with a PromethION metabolic cage system preceding (Pre) and during chronic treatment (7 days, intraperitoneal injection, twice a day) with vehicle, OEA (5 mg/kg), Ex4 (100 µg/kg), or Ex4+OEA. A and B: 24-h averages of food intake (kcal; A) and EE (kcal; B) as well as 7-day averages (Insets) were measured. *vs. vehicle within day (P < 0.05). C: comparison of raw EE in the first 24 h of treatment (day 1) and the final 24 h of treatment (day 7). †Day 1 vs. day 7 within treatment group (P < 0.05). D: analysis of covariance (ANCOVA)-adjusted (covariate: lean mass) EE during the final 24 h of treatment [day 7: light (12 h) and dark (12 h) periods]. #vs. vehicle (P < 0.05). E: ANCOVA-adjusted (covariate: lean mass) RMR during the final 24 h (day 7) of treatment. RMR was calculated as the mean EE (kcal/h) during the 30 min of lowest activity over a 24-h period. #vs. vehicle (P < 0.05). F and G: 24-h averages of RQ (F) and locomotor activity (G) were collected by CO2-to-O2 ratio and beam break, respectively. *vs. vehicle within day (P < 0.05). All values reflect the means ± SE (n = 9–12). d, Days; Veh, vehicle.
Since the differences we observed in lean mass between vehicle-treated mice and Ex4/Ex4+OEA-treated mice (Fig. 5E) could influence energy expenditure, we performed an ANCOVA on energy expenditure data collected during the last day of treatment (day 7) using lean mass as the covariate. Interestingly, Ex4 and Ex4+OEA, but not OEA, treatment increased lean mass-adjusted energy expenditure, particularly during the light cycle (Fig. 6D). There was no significant difference in lean mass-adjusted energy expenditure between Ex4- and Ex4+OEA-treated mice, which means that a yet-unmeasured factor must account for the difference in weight loss by these two therapies. The observation that Ex4 and Ex4+OEA treatments increased energy expenditure during the light cycle, when mice are less active compared with the dark cycle, led us to assess the effects of these treatments on the RMR. This analysis revealed that when adjusted for lean mass, Ex4 and Ex4+OEA treatments promoted an increase, or a tendency to increase, the RMR, respectively (Fig. 6E). OEA alone elicited no effect on energy expenditure or RMR.
Reduced average RQ values were observed in Ex4 and Ex4+OEA groups during the first 3 and 4 days of treatment, respectively (Fig. 6F). This would be expected because of the decreased food intake during this time period (Fig. 6A) and subsequent reliance on fat oxidation. RQ values steadily increased in all groups; however, although not significantly different, RQ values in Ex4- and Ex4+OEA-treated groups remained modestly lower compared with vehicle-treated mice, suggesting an increased reliance on fat oxidation (Fig. 6F). No significant differences in locomotor activity were observed between treatment groups (Fig. 6G), so we do not predict that this was a factor contributing to the increase in lean mass-adjusted energy expenditure in Ex4- or Ex4+OEA-treated mice. Together, these data suggest that the difference in weight loss between Ex4- and Ex4+OEA-treated DIO mice cannot be explained by alterations in food intake, energy expenditure, RQ, or locomotor activity.
DISCUSSION
As the twin pandemics of diabetes and obesity continue to expand throughout the world, the GLP-1RA class of T2D drugs is an integral component of preventing the continuation of these diseases because of their ability to improve glucose homeostasis while promoting moderate weight loss. Enhancement of the weight-reducing effects of GLP-1RA is a viable option for the development of more effective antiobesity therapies. With the recent discovery by Cheng et al. (8) that the endocannabinoid-like lipid OEA both binds to and augments cAMP production by GLP-1, we sought to determine whether OEA would also enhance the action of GLP-1RA, namely, GLP-1 and Ex4, on intracellular signaling events that contribute to GLP-1RA-mediated hypophagia. Furthermore, we tested the physiological relevance of a GLP-1RA+OEA combined therapy with respect to energy balance in lean and DIO mice.
Classically, activation of Gαs seven-transmembrane receptors such as the GLP-1R stimulates canonical events of cAMP formation and recruitment of β-arrestin to the receptor. In initial experiments, we utilized two lines of CHO-K1 cells overexpressing the GLP-1R to interrogate the effect of GLP-1+OEA and Ex4+OEA on these pathways. During these analyses we corroborated the finding of Cheng et al. (8) that OEA enhances GLP-1- but not Ex4-mediated cAMP production. This could be due to the fact that cAMP production was significantly enhanced by Ex4 compared with GLP-1, thus potentially not allowing for further stimulation by OEA. It is important to note that this enhanced effect of Ex4 compared with GLP-1 on cAMP production has not been observed by others (8, 47, 68). Nevertheless, similar to our findings, Cheng et al. (8) did not observe any enhancement in Ex4-mediated cAMP production with OEA, suggesting that the OEA effect on cAMP production is specific to GLP-1. cAMP is a second messenger that stimulates both PKA-dependent and PKA-independent signaling (52). To investigate whether the enhancement of cAMP production by GLP-1+OEA would lead to increased PKA activity, we used an antibody that targets the phosphorylated consensus sequence of PKA (RRXS*/T*). Interestingly, GLP-1 and GLP-1+OEA stimulated similar levels of PKA substrate phosphorylation, suggesting that the activity of cAMP receptors other than PKA [e.g., exchange protein directly activated by cAMP (Epac)] could be recruited by GLP-1+OEA. We also provide evidence that OEA augments GLP-1- but not Ex4-mediated β-arrestin recruitment to the GLP-1R. Since both cAMP and β-arrestin pathways can contribute to downstream ERK1/2 phosphorylation (12), we hypothesized that OEA would enhance GLP-1-mediated ERK1/2 phosphorylation. However, we failed to see any increase in GLP-1- or GLP-1+OEA-mediated induction of ERK1/2 phosphorylation. As we also observed that OEA did not enhance Lira-mediated cAMP production or β-arrestin recruitment to the GLP-1R (data not shown), we conclude that OEA alters canonical GLP-1R signaling in a GLP-1RA-specific manner.
It is interesting that OEA can enhance the activation of GLP-1-mediated cAMP and β-arrestin recruitment without affecting the affinity of GLP-1 for the GLP-1R as cited by Cheng et al. (8). There are a few potential explanations for this observation. First, OEA may bias GLP-1RA-mediated signaling toward stronger activation of certain pathways over others. Several studies have now shown that specific amino acid interactions between the GLP-1R and its various ligands are important for GLP-1R signaling events and may be relevant to biased signaling downstream of the GLP-1R (21, 22, 39, 65, 66). Biased signaling refers to the phenomenon where the relative “strength” in the activation of one pathway versus another (e.g., cAMP production vs. β-arrestin recruitment) can differ when different ligands (e.g., GLP-1, Ex4, and Lira) bind to the GLP-1R [see review (31)]. Interestingly, small molecules can modulate the degree to which this signaling bias occurs. For example, flavonoid-like compounds (e.g., quercetin) can shift bias toward cAMP versus Ca2+ signaling via the GLP-1R in a manner that depends on the identity of the GLP-1R ligand (68). Importantly, this signaling modulation by flavonoid-like compounds does not affect ligand affinity for the GLP-1R. We speculate that by affecting the conformation of ligand bound to the GLP-1R, OEA induces a form of signaling bias that enhances certain pathways (e.g., cAMP and glycolysis) downstream of the GLP-1R but not others (e.g., mTOR pathway). Alternatively, GLP-1RA+OEA could be a “superagonist” compared with GLP-1RA if its efficacy to stimulate GLP-1R-mediated signaling cascades is greater than that of GLP-1RA alone. Although we did not see an increase in the efficacy of GLP-1+OEA to promote cAMP formation or β-arrestin recruitment compared with GLP-1, in the present study we did not control for the necessary factors (e.g., test sensitivity and receptor density) to truly determine whether GLP-1+OEA is a superagonist [see review (51)]. Finally, OEA could attenuate the effect of GLP-1RA on the desensitization of the GLP-1R, and therefore the activation of the GLP-1R by GLP-1RA+OEA could persist for a longer duration compared with GLP-1RA alone (28). Further studies are necessary to fully determine the cause of this phenomenon.
Nutrient sensors such as AMPK, Akt, and mTOR within the hypothalamus and hindbrain are key components that facilitate the anorectic action of GLP-1RA (5, 7, 26, 50, 72). In the hypothalamus, we have previously shown that activation of AMPK or inhibition of mTOR in the ventromedial hypothalamus attenuates the anorectic effect of Ex4 (7). In the nucleus tractus solitarius of the hindbrain, Ex4 coordinates its anorectic action by stimulating MAPK signaling (phosphorylation of ERK1/2), inhibiting AMPK (dephosphorylation of AMPKα1/2), and inducing dephosphorylation of membrane-bound Akt signaling in a PKA-dependent manner (26, 49). We, therefore, tested whether OEA would enhance the effect GLP-1 and Ex4 on these signaling events. mTOR signaling was increased in CHOK1-GLP-1R cells treated with GLP-1 or Ex4, but OEA did not alter this response. Phosphorylation of other mTOR pathway components (i.e., 4E-BP1 or PRAS40) was not affected by any treatment. Furthermore, none of the treatments (GLP-1 or Ex4 ± OEA) altered AMPK or Akt phosphorylation at any time point. From these data we posited that neither AMPK, mTOR, nor Akt signaling pathways were viable candidates for further exploration of enhancing GLP-1RA-mediated anorectic signaling in vivo by OEA. However, we did observe that the combination of GLP-1 or Ex4 with OEA caused a synergistic increase in glycolysis (i.e., ECAR) in CHOK1-GLP-1R cells. This was specifically mediated via the known GLP-1R since the ability of GLP-1RA+OEA to enhance glycolysis was blocked by pretreatment with Ex9. We and others have previously shown that intracellular glucose metabolism in the brain is also a key component of the anorectic effect mediated by GLP-1 and Ex4; specifically, reduction of brain glycolysis by 2-DG via intracerebroventricular infusion or direct injection of 2-DG into the ventromedial hypothalamus attenuates Ex4-mediated hypophagia (5, 6, 50).
On the basis of these observations, we tested whether OEA enhances the hypophagic action of GLP-1 and/or Ex4 in a brain glycolysis-dependent manner. This would be particularly useful for native GLP-1 since peripherally administered GLP-1 does not elicit a hypophagic response unless given at doses much higher [e.g., 500 µg/kg (27, 64)] than the dose used in this study (3 µg/kg) because of its rapid degradation by dipeptidyl peptidase-4 (DPP4; 25). As expected, GLP-1 (3 µg/kg) alone did not reduce food intake, but contrary to our hypothesis, the addition of OEA did not enhance the hypophagic response of GLP-1. We cannot exclude the possibility that GLP-1 and OEA dissociate once administered in vivo. However, even if OEA remains associated with GLP-1 in vivo, our results suggest that OEA does not protect GLP-1 from degradation by DPP4. This would be akin to the strategy of covalently linking a fatty acid moiety (i.e., a palmitoyl group) to GLP-1 to produce the DPP4-resistant GLP-1RA Lira (22, 26, 37). In contrast, Ex4 (3 µg/kg) acutely reduced food intake, but OEA exerted no additional effect on this outcome. Additional experiments in control (i.e., icv ACSF pretreatment) mice treated with Ex4+OEA demonstrated that OEA neither consistently nor robustly alters the hypophagic effect of Ex4 (Fig. 4K). Whereas inhibition of brain glycolysis attenuated the anorectic effect of peripherally administered Ex4, it had no effect on Ex4+OEA-induced hypophagia. These findings extend our previous observations implicating brain glucose metabolism in the anorectic effect of centrally administered Ex4 (5) and show that this mechanism may also be involved in mediating the effects of peripherally administered Ex4. However, brain glycolysis does not appear to play a role in Ex4+OEA-induced hypophagia.
On the basis of the weight-reducing potential inherent in both Ex4 and OEA (16, 45, 46, 73), we proceeded to measure the effect of Ex4+OEA on energy balance in the setting of DIO. A 7-day regimen of Ex4+OEA promoted greater weight loss in DIO mice than either Ex4 or OEA treatment alone. Interestingly, following a marked decrease in food intake and body weight after the first day of treatment, mice treated with either Ex4 or Ex4+OEA continued to lose weight even as food intake returned to normal levels. This indicates that focusing on the hypophagic action of Ex4 as the main contributor of the weight loss effects of this compound (54, 58, 60) provides an incomplete mechanism. The present findings provide evidence that chronic Ex4 treatment, in the presence or absence of OEA, incites an elevation in energy expenditure in the setting of obesity. Previous studies have suggested such an effect of chronic GLP-1R activation to elevate energy expenditure in rodents (32, 48, 62) and humans (3); however, in those studies, energy expenditure was only assessed toward the end of GLP-1RA treatment. To our knowledge, our studies are the first to measure these energy balance components continuously throughout the duration of treatment with OEA, Ex4, or the Ex4+OEA combination. Our findings demonstrate that the weight loss effects of Ex4 and Ex4+OEA are composed of two elements: a transient decrease in food intake followed by a sustained elevation of energy expenditure. Moreover, the increased energy expenditure was particularly evident during the light cycle and, as such, was associated with an increase in RMR. Together, these data suggest that reduced food intake contributes to the initial weight loss caused by Ex4 and Ex4+OEA but increased energy expenditure becomes the significant factor promoting long-term weight loss. In humans, there is evidence that GLP-1RA can induce increases in RMR. For example, patients taking a combination of metformin with exenatide (the clinical form of Ex4) or with Lira exhibited an elevated RMR after 1 yr of therapy (3).
Potential mechanisms that could mediate this activity-independent increase in energy expenditure are beginning to be explored with specific emphasis on the effect of GLP-1RA on adipose tissue and skeletal muscle. For instance, activation of the GLP-1R in the brain has been associated with increased sympathetic outflow to brown (BAT) and white (WAT) adipose tissue (30) as well as increased BAT activity and WAT browning (3). Furthermore, knockdown of the GLP-1R in the dorsomedial hypothalamus results in decreased BAT activity, increased weight gain, and increased adiposity (38). GLP-1RA also promotes thermogenic gene expression (9) upregulation of uncoupling genes in mouse skeletal muscle (11). The possibility of direct effects of GLP-1RA on either adipose tissue or skeletal muscle is controversial given that it is not known whether the GLP-1R, or another GLP-1R-like receptor (70), is expressed in these tissues. This makes a brain-peripheral tissue axis the likely route by which GLP-1RA could alter metabolism within adipose and skeletal muscle. Still, studies have shown that Ex4 can exert direct effects on adipocytes (18, 69) and skeletal muscle cells (2, 9). Thus, further investigation into the target tissue(s) and axes mediating the effects of GLP-1RA on energy expenditure is needed.
Interestingly, the enhanced weight loss in Ex4+OEA-treated mice occurred in the absence of significant differences in food intake or energy expenditure between Ex4- and Ex4+OEA-treated mice over the 7-day treatment period. However, Ex4 and OEA were premixed before administration and we can only speculate that they physically interact like GLP-1+OEA. Even if Ex4 and OEA physically interact, we cannot exclude the possibility that these molecules dissociate in vivo and act via parallel pathways. The GLP-1R is essential for the anorectic action of GLP-1RA (54). Whereas OEA is a ligand for both PPARα and G protein-coupled receptor 119 (GPR119), evidence suggests that the anorectic action of OEA is mediated via PPARα (15, 20, 24) and not GPR119 (35). Thus, the weight loss effect of Ex4+OEA treatment could be attributed to additive effects of Ex4 and OEA working separately through these pathways. Though our initial hypothesis was that OEA would enhance the anorectic action of GLP-1 and Ex4 through a synergistic relationship, the mechanism by which chronic Ex4+OEA treatment promotes greater body weight loss compared with Ex4 treatment in the absence of significant differences in food intake or energy expenditure remains unknown. It is possible that the reduction in food intake is not the only means by which OEA reduces body weight. OEA promotes lipolysis, and this has been suggested as a contributor to OEA-mediated weight loss (20); however, reductions in body fat were similar between Ex4- and Ex4+OEA-treated mice. In the absence of differences between Ex4 and Ex4+OEA in food intake, energy expenditure, or locomotor activity, there is a possibility that part of the weight loss effect of Ex4+OEA may be due to malabsorption of ingested food. In normal chow-fed rats, OEA increases lipid transporters and absorption in the gut (71), but these data cannot be extrapolated to DIO rodents since high-fat feeding alters normal gut absorption (10, 53). Therefore, future studies should examine whether long-term OEA-mediated weight loss as well as the additional weight loss observed in Ex4+OEA mice is due to nutrient malabsorption in DIO mice.
Perspectives and Significance
OEA carries great potential for obesity therapy as there is evidence that it promotes weight loss in obese rodents and humans (4, 23, 33). To this end, OEA is presently marketed as a weight loss supplement called RiduZone (23). The possibility that it could be used therapeutically in conjunction with GLP-1RA to promote greater weight loss than either compound alone merits additional study. Because OEA stimulates GLP-1 secretion via GPR119 activation on L cells (35, 37), there is also an intriguing possibility that because of the proximity of these ligands in vivo, endogenous OEA could bind to and alter the signaling of endogenous GLP-1. This could lead to OEA enhancing other outcomes associated with GLP-1RA action not measured in this series of experiments such as insulin secretion, a physiological event that is mediated by an increase in cAMP signaling and glycolysis (17, 19).
In sum, an important aspect of this study is the finding that the interaction between the lipid OEA and GLP-1RA modulates signaling pathways downstream of the GLP-1R (i.e., cAMP production, β-arrestin recruitment to the GLP-1R, and glycolysis). This suggests that the strategy of combining lipid moieties with GLP-1RA has consequences beyond an originally intended purpose of increasing binding to albumin and extending the half-life of GLP-1RA (29, 36). Our data support the strategy of combining bioactive lipids with GLP-1RA as an innovative approach for modulating GLP-1R signaling; however, the physiological relevance of this observation remains to be shown. We conclude that elevations in energy expenditure are a major contributor to the long-term weight loss stimulated by Ex4 and Ex4+OEA. Furthermore, whereas most GLP-1RA research focuses on the hypophagic action of these drugs with respect to weight loss, these data reveal an important point that the hypophagic action of Ex4 is transient and that increased energy expenditure may play a larger role in sustaining Ex4-mediated weight loss than is presently appreciated. Though Ex4+OEA treatment leads to more potent weight loss than Ex4 or OEA alone, additional studies into the effect of this combination on mechanisms that affect energy balance (e.g., nutrient absorption) are needed. Present use of GLP-1RA and OEA as weight loss drugs and supplements, respectively, merit further study into the mechanisms regulated by a combination of these two compounds.
GRANTS
These studies were supported by American Diabetes Association Career Development Award 1-14-CD-01 (principal investigator, Julio E. Ayala) and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) F32 NRSA Postdoctoral Fellowship Award F32-DK-112603 (principal investigator, J. D. Brown). The energy expenditure ANCOVA done for this work was supported by NIDDK Grants DK-076169 and DK-115255.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.D.B., L.S., and Julio E. Ayala conceived and designed research; J.D.B., D.M., Jennifer E. Ayala, M.A.B., and C.M. performed experiments; J.D.B., D.M., Jennifer E. Ayala, C.M., and Julio E. Ayala analyzed data; J.D.B., D.M., Jennifer E. Ayala, L.S., and Julio E. Ayala interpreted results of experiments; J.D.B., D.M., and Jennifer E. Ayala prepared figures; J.D.B. and Julio E. Ayala drafted manuscript; J.D.B., Jennifer E. Ayala, M.A.B., and Julio E. Ayala edited and revised manuscript; J.D.B., D.M., Jennifer E. Ayala, M.A.B., C.M., L.S., and Julio E. Ayala approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Owen McGuinness for expert help with ANCOVA analysis.
Present addresses of J. E. Ayala and J. D. Brown: Dept. of Molecular Physiology and Biophysics, Vanderbilt University, 2200 Pierce Ave., 702 Light Hall, Nashville, TN 37232.
REFERENCES
- 1.Abdesselam I, Pepino P, Troalen T, Macia M, Ancel P, Masi B, Fourny N, Gaborit B, Giannesini B, Kober F, Dutour A, Bernard M. Time course of cardiometabolic alterations in a high fat high sucrose diet mice model and improvement after GLP-1 analog treatment using multimodal cardiovascular magnetic resonance. J Cardiovasc Magn Reson 17: 95, 2015. doi: 10.1186/s12968-015-0198-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andreozzi F, Raciti GA, Nigro C, Mannino GC, Procopio T, Davalli AM, Beguinot F, Sesti G, Miele C, Folli F. The GLP-1 receptor agonists exenatide and liraglutide activate Glucose transport by an AMPK-dependent mechanism. J Transl Med 14: 229, 2016. doi: 10.1186/s12967-016-0985-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beiroa D, Imbernon M, Gallego R, Senra A, Herranz D, Villarroya F, Serrano M, Fernø J, Salvador J, Escalada J, Dieguez C, Lopez M, Frühbeck G, Nogueiras R. GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes 63: 3346–3358, 2014. doi: 10.2337/db14-0302. [DOI] [PubMed] [Google Scholar]
- 4.Brown JD, Karimian Azari E, Ayala JE. Oleoylethanolamide: a fat ally in the fight against obesity. Physiol Behav 176: 50–58, 2017. doi: 10.1016/j.physbeh.2017.02.034. [DOI] [PubMed] [Google Scholar]
- 5.Burmeister MA, Ayala J, Drucker DJ, Ayala JE. Central glucagon-like peptide 1 receptor-induced anorexia requires glucose metabolism-mediated suppression of AMPK and is impaired by central fructose. Am J Physiol Endocrinol Metab 304: E677–E685, 2013. doi: 10.1152/ajpendo.00446.2012. [DOI] [PubMed] [Google Scholar]
- 6.Burmeister MA, Ayala JE, Smouse H, Landivar-Rocha A, Brown JD, Drucker DJ, Stoffers DA, Sandoval DA, Seeley RJ, Ayala JE. The hypothalamic glucagon-like peptide 1 receptor is sufficient but not necessary for the regulation of energy balance and glucose homeostasis in mice. Diabetes 66: 372–384, 2017. doi: 10.2337/db16-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burmeister MA, Brown JD, Ayala JE, Stoffers DA, Sandoval DA, Seeley RJ, Ayala JE. The glucagon-like peptide-1 receptor in the ventromedial hypothalamus reduces short-term food intake in male mice by regulating nutrient sensor activity. Am J Physiol Endocrinol Metab 313: E651–E662, 2017. doi: 10.1152/ajpendo.00113.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng YH, Ho MS, Huang WT, Chou YT, King K. Modulation of glucagon-like peptide-1 (GLP-1) potency by endocannabinoid-like lipids represents a novel mode of regulating GLP-1 receptor signaling. J Biol Chem 290: 14302–14313, 2015. doi: 10.1074/jbc.M115.655662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choung JS, Lee YS, Jun HS. Exendin-4 increases oxygen consumption and thermogenic gene expression in muscle cells. J Mol Endocrinol 58: 79–90, 2017. doi: 10.1530/JME-16-0078. [DOI] [PubMed] [Google Scholar]
- 10.Clara R, Schumacher M, Ramachandran D, Fedele S, Krieger JP, Langhans W, Mansouri A. Metabolic adaptation of the small intestine to short- and medium-term high-fat diet exposure. J Cell Physiol 232: 167–175, 2017. doi: 10.1002/jcp.25402. [DOI] [PubMed] [Google Scholar]
- 11.Decara J, Rivera P, Arrabal S, Vargas A, Serrano A, Pavón FJ, Dieguez C, Nogueiras R, Rodríguez de Fonseca F, Suárez J. Cooperative role of the glucagon-like peptide-1 receptor and β3-adrenergic-mediated signalling on fat mass reduction through the downregulation of PKA/AKT/AMPK signalling in the adipose tissue and muscle of rats. Acta Physiol (Oxf) 222: e13008, 2018. doi: 10.1111/apha.13008. [DOI] [PubMed] [Google Scholar]
- 12.Eishingdrelo H, Kongsamut S. Minireview: Targeting GPCR activated ERK pathways for drug discovery. Curr Chem Genomics Transl Med 7: 9–15, 2013. doi: 10.2174/2213988501307010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engeset RM, Ritter RC. Intracerebroventricular 2-DG causes feeding in the absence of other signs of glucoprivation. Brain Res 202: 229–233, 1980. doi: 10.1016/S0006-8993(80)80051-5. [DOI] [PubMed] [Google Scholar]
- 14.Frühbeck G, Yumuk V. Obesity: a gateway disease with a rising prevalence. Obes Facts 7, Suppl 2: 33–36, 2014. doi: 10.1159/000361004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodríguez De Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α. Nature 425: 90–93, 2003. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- 16.Fu J, Oveisi F, Gaetani S, Lin E, Piomelli D. Oleoylethanolamide, an endogenous PPAR-α agonist, lowers body weight and hyperlipidemia in obese rats. Neuropharmacology 48: 1147–1153, 2005. doi: 10.1016/j.neuropharm.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Göke R, Wagner B, Fehmann HC, Göke B. Glucose-dependency of the insulin stimulatory effect of glucagon-like peptide-1 (7–36) amide on the rat pancreas. Res Exp Med (Berl) 193: 97–103, 1993. doi: 10.1007/BF02576216. [DOI] [PubMed] [Google Scholar]
- 18.Góralska J, Śliwa A, Gruca A, Raźny U, Chojnacka M, Polus A, Solnica B, Malczewska-Malec M. Glucagon-like peptide-1 receptor agonist stimulates mitochondrial bioenergetics in human adipocytes. Acta Biochim Pol 64: 423–429, 2017. doi: 10.18388/abp.2017_1634. [DOI] [PubMed] [Google Scholar]
- 19.Gromada J, Ding WG, Barg S, Renström E, Rorsman P. Multisite regulation of insulin secretion by cAMP-increasing agonists: evidence that glucagon-like peptide 1 and glucagon act via distinct receptors. Pflügers Arch 434: 515–524, 1997. doi: 10.1007/s004240050431. [DOI] [PubMed] [Google Scholar]
- 20.Guzmán M, Lo Verme J, Fu J, Oveisi F, Blázquez C, Piomelli D. Oleoylethanolamide stimulates lipolysis by activating the nuclear receptor peroxisome proliferator-activated receptor α (PPAR-α). J Biol Chem 279: 27849–27854, 2004. doi: 10.1074/jbc.M404087200. [DOI] [PubMed] [Google Scholar]
- 21.Hager MV, Clydesdale L, Gellman SH, Sexton PM, Wootten D. Characterization of signal bias at the GLP-1 receptor induced by backbone modification of GLP-1. Biochem Pharmacol 136: 99–108, 2017. doi: 10.1016/j.bcp.2017.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hager MV, Johnson LM, Wootten D, Sexton PM, Gellman SH. β-arrestin-biased agonists of the GLP-1 receptor from β-amino acid residue incorporation into GLP-1 analogues. J Am Chem Soc 138: 14970–14979, 2016. doi: 10.1021/jacs.6b08323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hallmark BD, Rose K, Yepuri JN, Marulappa S.. Dietary supplementation of RiduZone (oleoylethanolamide/OEA) results in weight loss in humans: the first-in-human case studies (Abstract). Overcoming Obesity 2016 Conference Chicago, IL, September 23 2016, p. CHI19. [Google Scholar]
- 24.Hankir MK, Seyfried F, Hintschich CA, Diep TA, Kleberg K, Kranz M, Deuther-Conrad W, Tellez LA, Rullmann M, Patt M, Teichert J, Hesse S, Sabri O, Brust P, Hansen HS, de Araujo IE, Krügel U, Fenske WK. Gastric bypass surgery recruits a gut ppar-α-striatal D1R pathway to reduce fat appetite in obese rats. Cell Metab 25: 335–344, 2017. doi: 10.1016/j.cmet.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 25.Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon-like peptide-1-(7–36)amide is transformed to glucagon-like peptide-1-(9–36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 140: 5356–5363, 1999. doi: 10.1210/endo.140.11.7143. [DOI] [PubMed] [Google Scholar]
- 26. Hayes MR, Leichner TM, Zhao S, Lee GS, Chowansky A, Zimmer D, De Jonghe BC, Kanoski SE, Grill HJ, Bence KK. Intracellular signals mediating the food intake-suppressive effects of hindbrain glucagon-like peptide-1 receptor activation. Cell Metab 13: 320–330, 2011. [Erratum in Cell Metab 23:745, 2016.] doi: 10.1016/j.cmet.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jessen L, Aulinger BA, Hassel JL, Roy KJ, Smith EP, Greer TM, Woods SC, Seeley RJ, D’Alessio DA. Suppression of food intake by glucagon-like peptide-1 receptor agonists: relative potencies and role of dipeptidyl peptidase-4. Endocrinology 153: 5735–5745, 2012. doi: 10.1210/en.2012-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones B, Buenaventura T, Kanda N, Chabosseau P, Owen BM, Scott R, Goldin R, Angkathunyakul N, Corrêa IR Jr, Bosco D, Johnson PR, Piemonti L, Marchetti P, Shapiro AM, Cochran BJ, Hanyaloglu AC, Inoue A, Tan T, Rutter GA, Tomas A, Bloom SR. Targeting GLP-1 receptor trafficking to improve agonist efficacy. Nat Commun 9: 1602, 2018. doi: 10.1038/s41467-018-03941-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, Thøgersen H, Wilken M, Agersø H. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem 43: 1664–1669, 2000. doi: 10.1021/jm9909645. [DOI] [PubMed] [Google Scholar]
- 30.Kooijman S, Wang Y, Parlevliet ET, Boon MR, Edelschaap D, Snaterse G, Pijl H, Romijn JA, Rensen PC. Central GLP-1 receptor signalling accelerates plasma clearance of triacylglycerol and glucose by activating brown adipose tissue in mice. Diabetologia 58: 2637–2646, 2015. doi: 10.1007/s00125-015-3727-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koole C, Savage EE, Christopoulos A, Miller LJ, Sexton PM, Wootten D. Minireview: Signal bias, allosterism, and polymorphic variation at the GLP-1R: implications for drug discovery. Mol Endocrinol 27: 1234–1244, 2013. doi: 10.1210/me.2013-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krieger JP, Langhans W, Lee SJ. Novel role of GLP-1 receptor signaling in energy expenditure during chronic high fat diet feeding in rats. Physiol Behav 192: 194–199, 2018. doi: 10.1016/j.physbeh.2018.03.037. [DOI] [PubMed] [Google Scholar]
- 33.Laleh P, Yaser K, Abolfazl B, Shahriar A, Mohammad AJ, Nazila F, Alireza O. Oleoylethanolamide increases the expression of PPAR-Α and reduces appetite and body weight in obese people: a clinical trial. Appetite 128: 44–49, 2018. doi: 10.1016/j.appet.2018.05.129. [DOI] [PubMed] [Google Scholar]
- 34.Lamont BJ, Drucker DJ. Differential antidiabetic efficacy of incretin agonists versus DPP-4 inhibition in high fat fed mice. Diabetes 57: 190–198, 2008. doi: 10.2337/db07-1202. [DOI] [PubMed] [Google Scholar]
- 35.Lan H, Vassileva G, Corona A, Liu L, Baker H, Golovko A, Abbondanzo SJ, Hu W, Yang S, Ning Y, Del Vecchio RA, Poulet F, Laverty M, Gustafson EL, Hedrick JA, Kowalski TJ. GPR119 is required for physiological regulation of glucagon-like peptide-1 secretion but not for metabolic homeostasis. J Endocrinol 201: 219–230, 2009. doi: 10.1677/JOE-08-0453. [DOI] [PubMed] [Google Scholar]
- 36.Larsen PJ, Fledelius C, Knudsen LB, Tang-Christensen M. Systemic administration of the long-acting GLP-1 derivative NN2211 induces lasting and reversible weight loss in both normal and obese rats. Diabetes 50: 2530–2539, 2001. doi: 10.2337/diabetes.50.11.2530. [DOI] [PubMed] [Google Scholar]
- 37.Lauffer LM, Iakoubov R, Brubaker PL. GPR119 is essential for oleoylethanolamide-induced glucagon-like peptide-1 secretion from the intestinal enteroendocrine L-cell. Diabetes 58: 1058–1066, 2009. doi: 10.2337/db08-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee SJ, Sanchez-Watts G, Krieger JP, Pignalosa A, Norell PN, Cortella A, Pettersen KG, Vrdoljak D, Hayes MR, Kanoski SE, Langhans W, Watts AG. Loss of dorsomedial hypothalamic GLP-1 signaling reduces BAT thermogenesis and increases adiposity. Mol Metab 11: 33–46, 2018. doi: 10.1016/j.molmet.2018.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lei S, Clydesdale L, Dai A, Cai X, Feng Y, Yang D, Liang YL, Koole C, Zhao P, Coudrat T, Christopoulos A, Wang MW, Wootten D, Sexton PM. Two distinct domains of the glucagon-like peptide-1 receptor control peptide-mediated biased agonism. J Biol Chem 293: 9370–9387, 2018. doi: 10.1074/jbc.RA118.003278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller CC, Martin RJ, Whitney ML, Edwards GL. Intracerebroventricular injection of fructose stimulates feeding in rats. Nutr Neurosci 5: 359–362, 2002. doi: 10.1080/1028415021000033839. [DOI] [PubMed] [Google Scholar]
- 41.Miselis RR, Epstein AN. Feeding induced by intracerebroventricular 2-deoxy-d-glucose in the rat. Am J Physiol 229: 1438–1447, 1975. doi: 10.1152/ajplegacy.1975.229.5.1438. [DOI] [PubMed] [Google Scholar]
- 42.Mitri J, Hamdy O. Diabetes medications and body weight. Expert Opin Drug Saf 8: 573–584, 2009. doi: 10.1517/14740330903081725. [DOI] [PubMed] [Google Scholar]
- 43.Müller TD, Sullivan LM, Habegger K, Yi CX, Kabra D, Grant E, Ottaway N, Krishna R, Holland J, Hembree J, Perez-Tilve D, Pfluger PT, DeGuzman MJ, Siladi ME, Kraynov VS, Axelrod DW, DiMarchi R, Pinkstaff JK, Tschöp MH. Restoration of leptin responsiveness in diet-induced obese mice using an optimized leptin analog in combination with exendin-4 or FGF21. J Pept Sci 18: 383–393, 2012. doi: 10.1002/psc.2408. [DOI] [PubMed] [Google Scholar]
- 44.Proulx K, Cota D, Castañeda TR, Tschöp MH, D’Alessio DA, Tso P, Woods SC, Seeley RJ. Mechanisms of oleoylethanolamide-induced changes in feeding behavior and motor activity. Am J Physiol Regul Integr Comp Physiol 289: R729–R737, 2005. doi: 10.1152/ajpregu.00029.2005. [DOI] [PubMed] [Google Scholar]
- 45.Rodríguez de Fonseca F, Navarro M, Gómez R, Escuredo L, Nava F, Fu J, Murillo-Rodríguez E, Giuffrida A, LoVerme J, Gaetani S, Kathuria S, Gall C, Piomelli D. An anorexic lipid mediator regulated by feeding. Nature 414: 209–212, 2001. doi: 10.1038/35102582. [DOI] [PubMed] [Google Scholar]
- 46.Rodriquez de Fonseca F, Navarro M, Alvarez E, Roncero I, Chowen JA, Maestre O, Gómez R, Muñoz RM, Eng J, Blázquez E. Peripheral versus central effects of glucagon-like peptide-1 receptor agonists on satiety and body weight loss in Zucker obese rats. Metabolism 49: 709–717, 2000. doi: 10.1053/meta.2000.6251. [DOI] [PubMed] [Google Scholar]
- 47.Roed SN, Wismann P, Underwood CR, Kulahin N, Iversen H, Cappelen KA, Schäffer L, Lehtonen J, Hecksher-Soerensen J, Secher A, Mathiesen JM, Bräuner-Osborne H, Whistler JL, Knudsen SM, Waldhoer M. Real-time trafficking and signaling of the glucagon-like peptide-1 receptor. Mol Cell Endocrinol 382: 938–949, 2014. doi: 10.1016/j.mce.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 48.Rozo AV, Babu DA, Suen PA, Groff DN, Seeley RJ, Simmons RA, Seale P, Ahima RS, Stoffers DA. Neonatal GLP1R activation limits adult adiposity by durably altering hypothalamic architecture. Mol Metab 6: 748–759, 2017. doi: 10.1016/j.molmet.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rupprecht LE, Mietlicki-Baase EG, Zimmer DJ, McGrath LE, Olivos DR, Hayes MR. Hindbrain GLP-1 receptor-mediated suppression of food intake requires a PI3K-dependent decrease in phosphorylation of membrane-bound Akt. Am J Physiol Endocrinol Metab 305: E751–E759, 2013. doi: 10.1152/ajpendo.00367.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sandoval D, Barrera JG, Stefater MA, Sisley S, Woods SC, D’Alessio DD, Seeley RJ. The anorectic effect of GLP-1 in rats is nutrient dependent. PLoS One 7: e51870, 2012. doi: 10.1371/journal.pone.0051870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schrage R, De Min A, Hochheiser K, Kostenis E, Mohr K. Superagonism at G protein-coupled receptors and beyond. Br J Pharmacol 173: 3018–3027, 2016. doi: 10.1111/bph.13278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev 85: 1303–1342, 2005. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- 53.Singh A, Balint JA, Edmonds RH, Rodgers JB. Adaptive changes of the rat small intestine in response to a high fat diet. Biochim Biophys Acta 260: 708–715, 1972. doi: 10.1016/0005-2760(72)90019-7. [DOI] [PubMed] [Google Scholar]
- 54.Sisley S, Gutierrez-Aguilar R, Scott M, D’Alessio DA, Sandoval DA, Seeley RJ. Neuronal GLP1R mediates liraglutide’s anorectic but not glucose-lowering effect. J Clin Invest 124: 2456–2463, 2014. doi: 10.1172/JCI72434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sloop KW, Willard FS, Brenner MB, Ficorilli J, Valasek K, Showalter AD, Farb TB, Cao JX, Cox AL, Michael MD, Gutierrez Sanfeliciano SM, Tebbe MJ, Coghlan MJ. Novel small molecule glucagon-like peptide-1 receptor agonist stimulates insulin secretion in rodents and from human islets. Diabetes 59: 3099–3107, 2010. doi: 10.2337/db10-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Szayna M, Doyle ME, Betkey JA, Holloway HW, Spencer RG, Greig NH, Egan JM. Exendin-4 decelerates food intake, weight gain, and fat deposition in Zucker rats. Endocrinology 141: 1936–1941, 2000. doi: 10.1210/endo.141.6.7490. [DOI] [PubMed] [Google Scholar]
- 57.Talsania T, Anini Y, Siu S, Drucker DJ, Brubaker PL. Peripheral exendin-4 and peptide YY3–36 synergistically reduce food intake through different mechanisms in mice. Endocrinology 146: 3748–3756, 2005. doi: 10.1210/en.2005-0473. [DOI] [PubMed] [Google Scholar]
- 58.Tang-Christensen M, Larsen PJ, Göke R, Fink-Jensen A, Jessop DS, Møller M, Sheikh SP. Central administration of GLP-1-(7-36) amide inhibits food and water intake in rats. Am J Physiol Regul Integr Comp Physiol 271: R848–R856, 1996. doi: 10.1152/ajpregu.1996.271.4.R848. [DOI] [PubMed] [Google Scholar]
- 59.Tsujii S, Bray GA. Effects of glucose, 2-deoxyglucose, phlorizin, and insulin on food intake of lean and fatty rats. Am J Physiol Endocrinol Metab 258: E476–E481, 1990. 10.1152/ajpendo.1990.258.3.E476. . [DOI] [PubMed] [Google Scholar]
- 60.Turton MD, O’Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, Choi SJ, Taylor GM, Heath MM, Lambert PD, Wilding JP, Smith DM, Ghatei MA, Herbert J, Bloom SR. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 379: 69–72, 1996. doi: 10.1038/379069a0. [DOI] [PubMed] [Google Scholar]
- 61.Wang X, Miyares RL, Ahern GP. Oleoylethanolamide excites vagal sensory neurones, induces visceral pain and reduces short-term food intake in mice via capsaicin receptor TRPV1. J Physiol 564: 541–547, 2005. doi: 10.1113/jphysiol.2004.081844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wei Q, Li L, Chen JA, Wang SH, Sun ZL. Exendin-4 improves thermogenic capacity by regulating fat metabolism on brown adipose tissue in mice with diet-induced obesity. Ann Clin Lab Sci 45: 158–165, 2015. [PubMed] [Google Scholar]
- 63.Willard FS, Bueno AB, Sloop KW. Small molecule drug discovery at the glucagon-like peptide-1 receptor. Exp Diabetes Res 2012: 709893, 2012. doi: 10.1155/2012/709893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Williams DL, Baskin DG, Schwartz MW. Evidence that intestinal glucagon-like peptide-1 plays a physiological role in satiety. Endocrinology 150: 1680–1687, 2009. doi: 10.1210/en.2008-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wootten D, Reynolds CA, Koole C, Smith KJ, Mobarec JC, Simms J, Quon T, Coudrat T, Furness SG, Miller LJ, Christopoulos A, Sexton PM. A hydrogen-bonded polar network in the core of the glucagon-like peptide-1 receptor is a fulcrum for biased agonism: lessons from class b crystal structures. Mol Pharmacol 89: 335–347, 2016. doi: 10.1124/mol.115.101246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wootten D, Reynolds CA, Smith KJ, Mobarec JC, Koole C, Savage EE, Pabreja K, Simms J, Sridhar R, Furness SG, Liu M, Thompson PE, Miller LJ, Christopoulos A, Sexton PM. The extracellular surface of the GLP-1 receptor is a molecular trigger for biased agonism. Cell 165: 1632–1643, 2016. doi: 10.1016/j.cell.2016.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wootten D, Savage EE, Willard FS, Bueno AB, Sloop KW, Christopoulos A, Sexton PM. Differential activation and modulation of the glucagon-like peptide-1 receptor by small molecule ligands. Mol Pharmacol 83: 822–834, 2013. doi: 10.1124/mol.112.084525. [DOI] [PubMed] [Google Scholar]
- 68.Wootten D, Simms J, Koole C, Woodman OL, Summers RJ, Christopoulos A, Sexton PM. Modulation of the glucagon-like peptide-1 receptor signaling by naturally occurring and synthetic flavonoids. J Pharmacol Exp Ther 336: 540–550, 2011. doi: 10.1124/jpet.110.176362. [DOI] [PubMed] [Google Scholar]
- 69.Xu F, Lin B, Zheng X, Chen Z, Cao H, Xu H, Liang H, Weng J. GLP-1 receptor agonist promotes brown remodelling in mouse white adipose tissue through SIRT1. Diabetologia 59: 1059–1069, 2016. doi: 10.1007/s00125-016-3896-5. [DOI] [PubMed] [Google Scholar]
- 70.Yang H, Egan JM, Wang Y, Moyes CD, Roth J, Montrose MH, Montrose-Rafizadeh C. GLP-1 action in L6 myotubes is via a receptor different from the pancreatic GLP-1 receptor. Am J Physiol Cell Physiol 275: C675–C683, 1998. doi: 10.1152/ajpcell.1998.275.3.C675. [DOI] [PubMed] [Google Scholar]
- 71.Yang Y, Chen M, Georgeson KE, Harmon CM. Mechanism of oleoylethanolamide on fatty acid uptake in small intestine after food intake and body weight reduction. Am J Physiol Regul Integr Comp Physiol 292: R235–R241, 2007. doi: 10.1152/ajpregu.00270.2006. [DOI] [PubMed] [Google Scholar]
- 72.Yang Y, Choi PP, Smith WW, Xu W, Ma D, Cordner ZA, Liang NC, Moran TH. Exendin-4 reduces food intake via the PI3K/AKT signaling pathway in the hypothalamus. Sci Rep 7: 6936, 2017. doi: 10.1038/s41598-017-06951-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Young AA, Gedulin BR, Bhavsar S, Bodkin N, Jodka C, Hansen B, Denaro M. Glucose-lowering and insulin-sensitizing actions of exendin-4: studies in obese diabetic (ob/ob, db/db) mice, diabetic fatty Zucker rats, and diabetic rhesus monkeys (Macaca mulatta). Diabetes 48: 1026–1034, 1999. doi: 10.2337/diabetes.48.5.1026. [DOI] [PubMed] [Google Scholar]