

Abstract
Recent advances in the field of mineralocorticoid receptor (MR) and its ligand aldosterone expanded the role of this hormone and its receptor far beyond their initial function as a regulator of Na+ and K+ homeostasis in epithelial cells. The symposium “New Roles of Aldosterone and Mineralocorticoid Receptors in Cardiovascular Disease: Translational and Sex-Specific Effects” presented at the 38th World Congress of the International Union of Physiological Sciences (Rio de Janeiro, Brazil) highlighted the contribution of extrarenal MRs to cardiovascular disease. This symposium showcased how MRs expressed in endothelial, vascular smooth muscle, and immune cells plays a critical role in the development of vascular disease associated with aging, obesity, and chronic aldosterone stimulation and demonstrated that MR antagonism prevents the acute renal dysfunction and tubular injury induced by ischemia-reperfusion injury. It was also shown that the adipocyte-derived hormone leptin is a new direct regulator of aldosterone secretion and that leptin-mediated aldosterone production is a major contributor to obesity-associated hypertension in women. Sex differences in the role of aldosterone and of endothelial MR in the cardiovascular outcomes of obesity were highlighted. This review summarizes these important emerging concepts regarding the contribution of aldosterone and cell-specific MR to cardiovascular disease in male and female subjects and further supports sex-specific benefits of MR antagonist drugs to be tested in additional populations.
Keywords: aging, aldosterone, hypertension, kidney, mineralocorticoid receptor
INTRODUCTION
The aldosterone-mineralocorticoid receptor (MR) axis has historically been described as a pathway regulating blood pressure (BP) by controlling Na+ retention in renal epithelial cells (14). However, discoveries from the past two decades reveal that MRs are expressed in nonepithelial cells and exerts numerous additional functions beyond electrolyte handling in the distal tubule of the kidney. MRs are expressed in adipocytes, muscle, the liver, and pancreatic β-cells and in all cells of the cardiovascular and immune systems extending the initial function of the MR to the control of the metabolic, vascular, cardiac, renal, and immune systems (10, 19, 34, 50, 73).
This symposium was organized to present the latest advances regarding the role of the MR and aldosterone in aging, metabolic, and renal disorders at the session titled “New Roles of Aldosterone and Mineralocorticoid Receptors in Cardiovascular Disease: Translational and Sex-Specific Effects” at the 38th World Congress of the International Union of Physiological Sciences at Rio de Janeiro, Brazil, in August 2017. Sponsored by the American Physiological Society, this symposium focused on the newly discovered roles of MR in endothelial, smooth muscle cells (SMCs), and immune cells in the functional and structural vascular remodeling associated with aging, cardiometabolic disease, and kidney injury. This symposium also emphasized the clinical potential of MR antagonism (MRA) for the prevention and treatment of aging and the consequences of metabolic disorders, acute kidney injury (AKI), and chronic kidney disease. The session also highlighted the new mechanisms regulating aldosterone production and demonstrated a role for MRs in the sex specificity of the mechanisms leading to endothelial dysfunction and hypertension in obesity.
The content and conclusions from this session have relevance in understanding the contribution of aldosterone and MRs to cardiovascular disease and highlight the importance of considering MR blockade as a therapeutic avenue for numerous cardiovascular diseases.
A ROLE FOR SMC-MRs IN VASCULAR AGING
Vascular Aging
Aging is a universal and powerful risk factor for cardiovascular disease. The incidence of all forms of cardiovascular disease, including myocardial infarction (MI), stroke, hypertension, heart failure, and cardiovascular death, all increase dramatically with age (32, 44). There are well-described changes in vascular structure and function with aging that contribute to cardiovascular disease (for reviews, see Refs. 98, 100, and 104). Functionally, the aging vasculature produces more reactive oxygen species (ROS) and less nitric oxide (NO), resulting in increased oxidative stress and enhanced vasoconstriction, thereby contributing to hypertension and impaired tissue perfusion. Structurally, SMCs of the aging vasculature are more migratory and proliferative with an altered extracellular matrix resulting in increased fibrosis and progressive vascular stiffening with age. This is important clinically because vascular stiffness directly correlates with the risk of MI, stroke, and cardiovascular death, independent of BP or other risk factors (102). Thus, understanding the mechanisms driving the vascular aging process has substantial clinical significance with potential to lead to novel prevention and treatment strategies for common cardiovascular disorders.
MR Expression in Vascular SMCs Increases with Aging and Contributes to Vascular Remodeling
Data from as early as the 1970s to 1980s suggested that receptors in the arterial wall could directly respond to aldosterone and contribute to hypertension and vascular disease (61, 62). With the identification of the MR as the aldosterone-binding receptor (3), it has since been confirmed that the MR is expressed in human vascular SMCs and regulates expression of genes in SMCs that are involved in vascular fibrosis and calcification (48, 49, 78). Moreover, MR expression in SMCs has been found to increase with age in rat aortic SMCs, in the mouse aorta and mesenteric resistance vessels, and in human saphenous veins after grafting (4, 35, 63). The development of transgenic mice with MRs specifically deleted from SMCs has advanced our understanding of the specific role of SMC-MRs in vascular function and disease (for a review, see Ref. 59). With the use of these mice, SMC-MRs were found to directly contribute to vascular fibrosis in response to wire injury (86) and to vascular stiffening and integrin expression induced by hypertension (41).
Mice Lacking SMC-MRs Are Protected From Vascular Aging
The knowledge that SMC-MRs increase with age and contribute to vascular remodeling in response to injury and hypertension prompted mouse studies to explore the direct role of SMC-MRs in vascular aging. The life expectancy of laboratory mice is ~24 mo, and the aging phenotype in male mice with MRs specifically deleted from SMCs [SMC-MR knockout (KO)] was compared with MR-intact littermates as they aged to determine whether there is a role for SMC-MRs in functional and structural changes with vascular aging (35, 58, 74). In MR-intact mice, BP increased modestly with aging from 3 to 12 mo (35) along with an increase in the resistance vessel contractile response to vasoconstrictors including potassium chloride, phenylephrine, thromboxane, and angiotensin II (ANG II) (74). SMC-MR KO mice were protected from the rise in BP with aging and from the enhanced vasoconstriction to all agonists except the adrenergic agonist phenylephrine. The difference in BP in aged SMC-MR KO mice was independent of changes in renal MR function including the fractional excretion of Na+ under normal or low-Na+ conditions (74). Rather, 12-mo-old male mice lacking SMC-MRs had decreased resistance vessel myogenic tone. This was associated with decreased vascular responsiveness to activation of the L-type Ca2+ channel (LTCC), a channel that is critical to the mechanism of vascular tone and vasoconstriction (35, 74). ANG II-induced vascular oxidative stress and hypertension are also exacerbated with aging (66, 74), and these responses to ANG II were eliminated in aged mice lacking SMC-MRs (74). In addition to ANG II, many other factors, including elevated Na+, can modulate vascular oxidative stress (16, 28, 39). Overall, the new data support that in male mice, the MR, specifically in SMCs, plays a critical role in driving resistance vessel tone, oxidative stress, and vasoconstriction in the aging vasculature thereby contributing to rising BP with aging (66, 74).
SMC-MRs have also recently been found to contribute to cardiovascular structural changes with aging in mice (58). Aortic stiffness, as measured by pulse wave velocity, was found to increase with aging from 3 to 12 or 18 mo of age in male MR-intact mice. This aging-associated increase in vascular stiffness, as well as vascular fibrosis, was prevented in SMC-MR KO mice (58). There is also an aging-associated increase in cardiac stiffness. Interestingly, cardiac stiffness and coronary perivascular fibrosis were both attenuated in aging mice lacking SMC-MRs. The changes in cardiac stiffness correlated with changes in aortic stiffness, suggesting that the cardiac aging effects may in part be secondary to vascular mechanisms. As in humans, cardiovascular aging in mice was associated with a modest decline in cardiac function and exercise capacity, and this decline was attenuated in SMC-MR KO mice (58). Thus, MRs in SMCs contribute to vascular and cardiac structural changes with aging that contribute to tissue stiffness, an important determinant of adverse outcomes in aging humans.
Mechanisms by Which SMC-MRs Contributes to Vascular Aging
The detailed molecular mechanism by which SMC-MRs contribute to vascular aging has begun to be elucidated (Fig. 1). Vascular RNA expression profiling revealed profound global changes in mRNA and microRNA (miR) expression in the aging vasculature that is dramatically altered in the absence of SMC-MRs (35, 58). The most downregulated miR in the aging mouse aorta was miR-155, which did not decline with aging SMC-MR KO vessels. miR-155 has been previously shown to decrease with aging in human peripheral leukocytes (79) and to correlate inversely with BP in humans (24). Mechanistic in vitro studies revealed that the MR negatively regulates transcription of the miR-155 host gene promoter, independent of aldosterone. In aging mice, rising MR mRNA in mesenteric resistance vessels correlated with a decrease in miR-155. The decline in miR-155 with age is further associated with increased vascular expression of predicted miR-155 targets, including the LTCC subunit Cav1.2 and the ANG II type 1 receptor (AT1R). These gene expression changes were further associated with increased LTCC-mediated vasoconstriction and ANG II-induced oxidative stress (35). This aging mechanism is lost in SMC-MR KO mice, implicating a direct role for SMC-MRs in driving the mechanism (Fig. 1).
Fig. 1.

Mechanisms by which the smooth muscle cell mineralocorticoid receptor (SMC-MR) contributes to the adverse outcomes of vascular aging. MR expression rises with aging in vascular SMCs, resulting in suppressed transcription of microRNA (miR)-155 and enhanced expression of profibrotic genes including connective tissue growth factors (CTGF), matrix metaloprotease (MMP)-2, and bone morphogenetic proteins (BMP). Inhibition of miR-155 by MRs leads to an increase in miR-155 targets, including the angiotensin II (ANG II) type 1 receptor (AT1R) and the L-type calcium channel (LTCC) subunit Cav1.2, which together promote vasoconstriction by increasing reactive oxygen species (ROS) and Ca2+ influx in SMCs. Together, these effects of SMC-MRs contribute to changes in vascular function and structure with aging including increased vascular; oxidative stress, vasoconstriction, fibrosis, and stiffness. By blocking these pathways, MR antagonists may improve outcomes be preventing the adverse effect of SMC-MRs on vascular aging pathology including high blood pressure, vascular stiffness, and cardiac fibrosis leading to heart attack, stroke, and heart and renal failure.
Regarding the mechanism for the structural changes with aging, lack of SMC-MRs produced a totally distinct mRNA expression signature with aging that was predicted to oppositely regulate pathways involved in cardiovascular development and function. Specifically, in the absence of SMC-MRs, there was profound suppression of a profibrotic gene expression program, including a downregulation of connective tissue growth factor (CTGF), matrix metalloproteinase (MMP)-2, and bone morphogenetic protein 4 (BMP-4). The mechanism for these global gene expression changes remains to be determined but as the MR is a transcription factor that is known to regulate CTGF (78) and other BMPs (49) in SMCs, transcriptional mechanisms are a focus of investigation.
Translational Implications and Future Directions for the Role of SMC-MRs in Vascular Aging
Alterations in Na+ and K+ metabolism related to renal mineralocorticoid effects may lead to possible confounding effects on the direct vascular actions of mineralocorticoids via the generation of ROS. Additional studies in low-Na+, high-K+ conditions will help address this concern. All of the published studies exploring the role of MRs in aging have been performed in male subjects; thus, future studies are warranted to determine whether there are sex differences in the role of MRs in vascular aging. Substantial investigation is also needed to determine whether this enhanced understanding of the role of SMC-MRs in the mechanisms driving vascular aging can be used therapeutically. Preclinical studies revealed that in aged mice, restoration of vascular miR-155 by a SMC-targeted lentivirus decreased vascular miR-155 target gene expression (Cav1.2 and AT1Rs) and attenuated resistance vessel vasoconstriction (35). Treatment of 12-mo-old mice with the MR antagonist spironolactone for 4 mo decreased vascular fibrosis, stiffness, and expression of CTGF and BMP-4 (58), suggesting that MR inhibition might be used to prevent or slow vascular aging. Whether these pathways contribute to hypertension in aging humans remains to be determined, but there are some suggestive data. A single-nucleotide polymorphism in the AT1R 3′-untranslated region that prevents miR-155 binding is associated with hypertension in humans (24). In a small study in 16 older humans, changes in serum miR-155 levels in response to MRA correlated with an improved BP response to therapy (35). In a study of 11 men (average age: 64 yr), 1 mo of eplerenone treatment also decreased serum MMP-2 with a trend toward decreased CTGF and BMP-4, suggesting an inhibitory effect on fibrosis (58). Larger studies with a longer duration of treatment that also include female subjects are needed to accurately test the potential of MR inhibition as an antiaging therapy.
MR AND ALDOSTERONE IN THE CARDIOVASCULAR CONSEQUENCES OF OBESITY: SEX DIFFERENCES
The recent epidemic of obesity, which affects more women than men worldwide (40), is one of the major risk factors for cardiovascular disease. While women are generally protected from cardiovascular disease until menopause, recent evidence suggests that obesity, particularly with diabetes, eliminates the protective effects of female sex (93, 103) and is the cause of the threefold increase in the risk for cardiovascular disease in premenopausal women over the last three decades (97) as well as of the rising number of schoolgirls diagnosed with hypertension (72, 92). These alarming data raise the urgency of understanding the mechanisms whereby obesity impairs vascular function, raises BP, and induces cardiovascular disease, particularly in female subjects.
Sex Differences in the Role of Aldosterone in Obesity and Associated Cardiovascular Disease
Several lines of evidence indicate that aldosterone, a major contributor to both cardiovascular and metabolic dysfunctions, is produced in excess in obesity (22) and correlates directly not only with visceral adipose mass and body mass index but also with BP in women only (43). In addition, blockade of aldosterone action via MRA appears more efficacious as a cardiovascular therapy regimen in women compared with men (56), which suggests an important relationship between aldosterone and cardiovascular disease in women. However, the origin of the elevated plasma aldosterone levels in obesity remains incompletely understood. Based on the observations that increases in aldosterone levels in obesity correlate directly with adipose mass and are independent of increases in renin and ANG II levels (15, 45, 57), Ehrhart-Bornstein et al. (36) investigated the contribution of the adipose tissue and identified the adipocyte as a source of aldosterone-releasing factors.
In this context, recent studies have been conducted to determine whether the adipocyte-derived hormone leptin could be the missing link between high adipose mass and elevated circulating aldosterone levels. Studies have shown that, despite obesity, mice, rats (13, 46), and humans (82, 83) deficient in leptin or harboring impaired leptin signaling do not exhibit high circulating aldosterone levels, whereas endogenous increases in leptin levels with obesity, and exogenous leptin supplementation raise adrenal aldosterone synthase [cytochrome P-450 11B2 (CYP11B2)] expression and aldosterone production (46), which suggests a central role for leptin in obesity-associated increases in aldosterone levels. Inhibition of ANG II, the primary regulator of aldosterone production, or of α- and β-adrenergic signaling in vivo does not abolish the stimulating effects of leptin on adrenal CYP11B2 expression and aldosterone production, which suggests that leptin exerts direct action on the adrenal glands. This was confirmed by demonstrating that leptin dose dependently increases CYP11B2 expression and aldosterone production via Ca2+-dependent mechanisms in human adrenal cortical cells in culture (Fig. 2) (46). Therefore, it is suggested that leptin, but not increases in adipose mass per se, is the source of the elevated aldosterone levels in obesity.
Fig. 2.
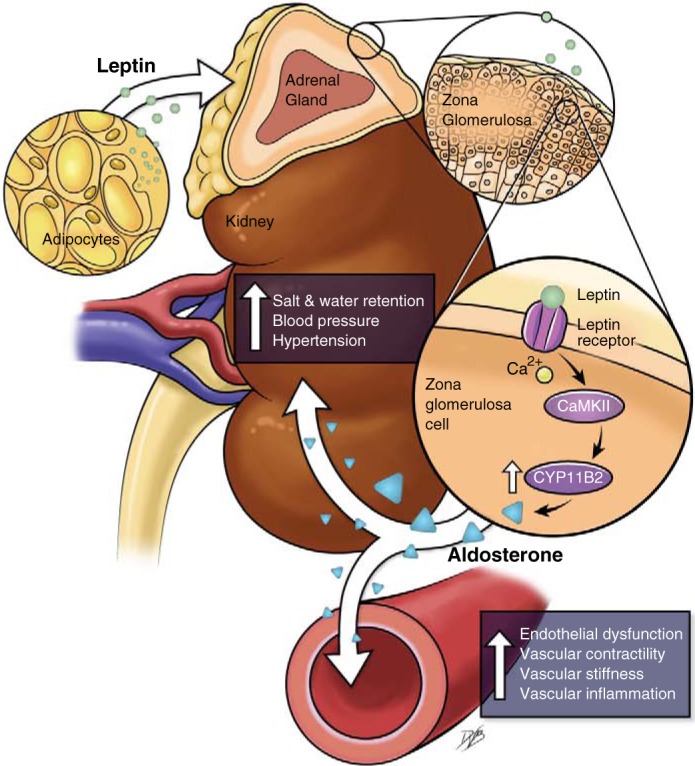
Mechanisms via which leptin increases cytochrome P-450 11B2 (CYP11B2) and aldosterone production in adrenal zona glomerulosa cells. Shown is an illustration of a new mechanism whereby activation of the leptin receptor in adrenal zona glomerulosa cells increases intracellular Ca2+ levels and Ca2+/calmodulin-dependent protein kinase II (CaMKII) expression, leading to an increase in aldosterone synthase expression (CYP11B2) and an increase in aldosterone production.
This newly discovered leptin-aldosterone axis emerges as a key contributor to cardiovascular disease in obese female mice predominantly. Hyperleptinemia in obesity and leptin sensitization in mice deficient in protein tyrosine phosphatase 1B deletion, a molecular “break” on leptin signaling (106), induce hypertension in male and female mice (12, 47). However, female mice only present with marked increases in adrenal aldosterone synthase (CYP11B2) expression and plasma aldosterone levels and blockade of aldosterone action with spironolactone restores BP in female but not male animals (47). Male animals, on the other hand, present with sympathoactivation with obesity and leptin sensitization (11, 47), whereas female animals do not (47). This supports the new concept that leptin induces hypertension via sex-specific mechanisms in obesity: leptin activation of the aldosterone-mineralocorticoid axis in female mice (47) and leptin-induced sympathoactivation in male mice (12) (Fig. 3).
Fig. 3.
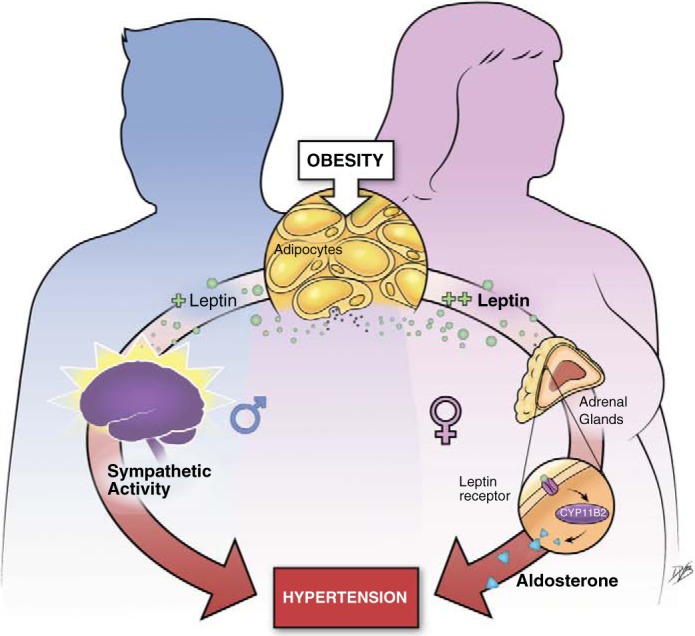
Potential mechanisms via which obesity leads to hypertension in male and female subjects. The illustration shows that leptin elevates blood pressure via sex-specific mechanisms in the context of obesity: elevation in sympathetic activity in male subjects and increased in aldosterone production in female subjects.
Further evidence to support the predominance of the leptin-aldosterone axis in the development of cardiovascular disease in obese female mice is presented by demonstrating that leptin deficiency in obese ob/ob female mice protects from obesity-induced endothelial dysfunction and cardiac fibrosis, while restoration of leptin levels in these mice impairs endothelial function and stimulates cardiac collagen deposition (46). Consistent with the work by the Sowers and colleagues (2, 18, 52, 53, 67) in obese diabetic mice, MR blockade with spironolactone abolished leptin-induced endothelial dysfunction and collagen deposition, providing further arguments to support the aldosterone-MR dependence of the mechanisms whereby leptin induces cardiovascular disease in the context of obesity (46).
All of these observations have been made in rodent models of obesity; therefore, additional experiments in humans would be required to confirm the predominance of the aldosterone-MR axis in the development of cardiovascular disease in obese women and determine whether leptin is also a major activator of the aldosterone-MR axis in humans.
Role of Endothelial Cell MRs
In addition to the role of MRs in SMCs in vascular remodeling and aging discussed in the first section of this review, MRs are also expressed in the vascular endothelium (23). Several mechanisms have been associated with endothelial dysfunction induced by obesity (for a review, see Ref. 37), and, recently, attention has been given to MR activation in endothelial cells (ECs) as a potential mediator. Short-term incubation of ECs with aldosterone increases endothelial NO synthase (eNOS) phosphorylation and NO release, but this effect is not observed with long-term application (71, 77). Instead, chronic aldosterone reduces eNOS-derived NO synthesis and increases ROS generation in a dose-dependent manner in ECs, which contributes to a decrease in NO bioavailability (69) and endothelial dysfunction in the presence of several cardiovascular risk factors including obesity (for a review, see Ref. 30). This is consistent with the beneficial effect of MR blockade in coronary microvascular function of individuals with type 2 diabetes (42).
One mechanism involved in the EC-MR-induced endothelial dysfunction involves oxidative stress and reduced NO synthesis and bioavailability (27). MR blockade attenuates eNOS uncoupling and decreased the expression of the NADPH oxidase subunits p22phox and p40phox, which renders oxidative stress and increases NO production in ECs (26, 90). In addition, aldosterone-induced epithelial Na+ channel expression increases EC stiffness, resulting in impaired NO release by inhibition of phosphatidylinositol 3-kinase/Akt/eNOS pathway (38, 65). Enhanced endothelial epithelial Na+ channel activation drives coronary endothelium remodeling and permeability in obese female mice, which was associated with cardiac diastolic dysfunction (51). These experimental data are consistent with the benefit of MR antagonist in patients with reduced ejection fraction (81).
There are sex differences in the mechanisms driving microvascular endothelial dysfunction in response to cardiometabolic risk factors (Fig. 4). This is clinically significant because dysfunction of resistance microvessels, rather than conduit vessels, predicts 5-yr cardiovascular disease risk in humans (70). Resistance vessels dilate in response to environmental changes and physiological needs to modulate blood flow to specific organs. Endothelium-dependent vasodilation is measured experimentally by quantifying the vascular relaxation response to acetylcholine. Two major components contribute to this endothelium-derived vasodilatory response of resistance vessels: eNOS-derived NO and endothelium-derived hyperpolarization (EDH). EDH is mediated by endothelial K+ channels, specifically IK1 and SK3 channels, which account for EDH in resistance microvessels. Microvessels from male subjects exhibit a substantial loss of NO-dependent relaxation in response to obesity, which can be compensated for initially by increased EDH (25, 31). When obesity is associated with hyperlipidemia in male subjects, the compensatory increase in EDH component, with enhanced expression of endothelial SK3 K+ channels, is lost resulting in resistance vessels endothelial dysfunction (31). In female subjects, obesity with or without hyperlipidemia impairs the EDH component of endothelium-dependent vasodilatation in small arteries by reducing expression of endothelial IK1 K+ channels.
Fig. 4.
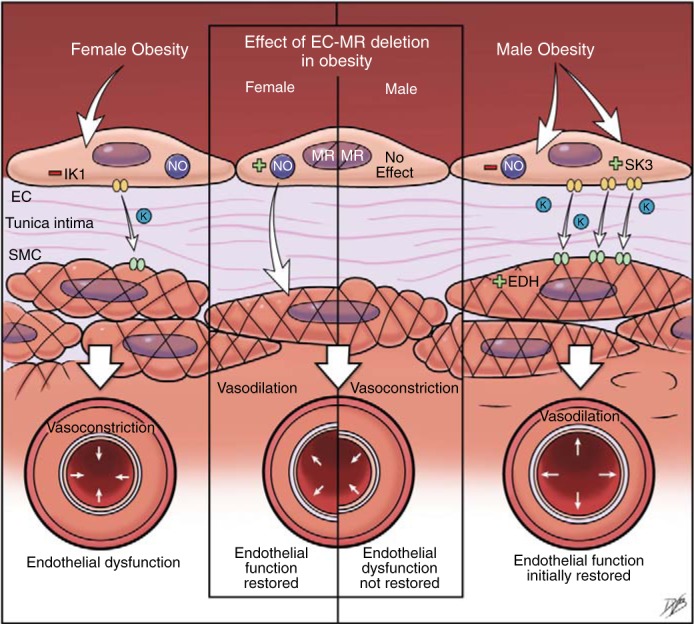
Sex differences in mechanisms of resistance vessel endothelial dysfunction in obesity. Changes in endothelial function and the effect of endothelial mineralocorticoid receptor (EC-MR) deletion in small mesenteric arteries of high fat diet-induced male and female obese mice are shown. Female mice develop endothelial dysfunction associated with reduced expression of endothelial KCa3.1 and intermediate-conductance Ca2+-activated K+ (IK1) channels and impaired endothelium-dependent hyperpolarization (EDH). In male mice, high fat diet-induced obesity leads to a reduction in nitric oxide (NO) levels, which is compensated for by increased expression of endothelial KCa2.3 and small-conductance Ca2+-activated K+ (SK3) channels and EDH. EC-MR deletion restores endothelial function only in female mice by enhancing NO release.
Use of mice with MRs specifically deleted from ECs (EC-MR KO) revealed the endothelial MR as a key mechanism driving sex differences in the vasodilatory endothelial dysfunction of resistance arteries (31, 52), further supporting a role for EC-MRs in obesity-induced endothelial dysfunction in female subjects. MR deletion in ECs protects female subjects from endothelial dysfunction induced by obesity and dyslipidemia by increasing NO availability, but this mechanism was not observed in male subjects (Fig. 4). We cannot exclude that an interplay between EC-MRs and caveolin-1 prevents an increase in NO in male subjects, as in the absence of caveolin-1 MR activation upregulates eNOS expression and activity (84, 85). However, in obese male subjects, rather than endothelial MR signaling, sympathetic activation appears to be the driver of cardiovascular damage as endothelial dysfunction is attenuated by the treatment with β-blockers (29, 95). These results support the concept that different molecular mechanisms drive endothelial dysfunction in male and female subjects and suggest that sex-specific therapies are likely to be needed to prevent the adverse cardiovascular consequences of obesity.
ROLE OF IMMUNE CELLS IN ALDOSTERONE-INDUCED VASCULAR DAMAGE
Aldosterone has proinflammatory effects in various cell types, including vascular cells and cells of the innate and adaptive immune systems: it increases the DNA-binding activity of transcription factors such as NF-κB and activator protein (AP)-1, increases the expression of adhesion molecules such as ICAM-1 and VCAM-1, and increases the expression of other inflammatory markers (cyclooxygenase-2, macrophage chemoattractant protein-1, osteopontin, TNF-α, IL-1β, and IL-6) (94). Aldosterone (as well as the aldosterone precursor DOCA) also induces accumulation of macrophages and T cells in the kidneys (96), heart (88), and vasculature. These proinflammatory effects of aldosterone usually rely on MR activation and contribute to end-organ damage in many pathological conditions. In addition, many experimental and clinical studies have demonstrated that MRA produces beneficial outcome in patients with cardiovascular and metabolic diseases, mainly due to prevention of inflammatory responses.
Accordingly, cells of the immune system express MRs, and MR activation modulates immune cell functions. For example, MR expression has been reported in primitive blood- and bone marrow-derived progenitor cells (CD34+ hematopoietic stem cells), monocytes/macrophages, neutrophils, dendritic cells, and peripheral T and B lymphocytes from humans and experimental animals. In many of these immune cells, aldosterone-induced MR activation induces cytokine secretion and activates phagocytic/humoral responses. Furthermore, deletion of MRs in macrophages protects from cardiac fibrosis and increased BP induced by DOCA-salt treatment (87), and inhibition of T lymphocytes polarization by MRA reduces mineralocorticoid-induced end-organ damage (1). For more details, please refer to the following excellent and recent review articles in Refs. 6, 21, 76, 91, and 101.
To better understand the mechanisms involved in the proinflammatory phenotype of aldosterone or how the innate immune system contributes to aldosterone-mediated vascular injury, our group investigated whether aldosterone activates the NOD-like receptor (NLR) pyrin-domain-containing protein 3 (NLRP3) inflammasome (20). NLRP3 is a member of the NLR family. It regulates the assembly of a multimolecular complex (the inflammasome) that activates inflammatory caspases and generate proinflammatory cytokines such as IL-1β and IL-18. Cells of the innate immunity express pattern recognition receptors (PRRs), which include the large families of Toll-like receptors, C-type lectin receptors, RIG-I-like receptors, and NLRs. PRRs are activated by pathogen-associated molecular patterns, which are molecules expressed by microbial pathogens and by damage-associated molecular patterns, i.e., cell components that are released during cell damage or death. Activation of PRRs triggers signaling pathways that control the expression and release of cytokines, cell adhesion molecules, and migration. Activation of the NLRP3 occurs in response to a variety of signals that are indicative of damage, including bacterial DNA, ATP, glucose, uric acid and cholesterol crystals, and mitochondria-derived ROS.
In bone marrow-derived macrophages from wild-type mice, aldosterone increases mitochondrial ROS and caspase-1 activation. Caspase-1 activation is blunted in aldosterone-stimulated bone marrow-derived macrophages from NLRP3−/− mice. Aldosterone infusion in wild-type mice activates the NLRP3 inflammasome (it increases NLRP3 expression, caspase-1 activity, and mature IL-1β) in cells from the peritoneal cavity (peritoneal macrophages). Aldosterone also increases plasma IL-1β and induces vascular dysfunction (abnormal vascular reactivity, remodeling, and increased expressed of adhesion molecules). NLRP3 deletion almost completely prevented all effects of aldosterone: changes in vascular reactivity, increased expression of cell adhesion molecules, vascular remodeling (increased cross-sectional area and increased wall-to-lumen ratio), and increased systolic BP (20).
Of clinical importance, patients with aldosterone-producing adenomas and resistant arterial hypertension exhibit high levels of IL-1β and other cytokines (IL-6 and TNF-α), and MR blockade with spironolactone or eplerenone, as well as adrenalectomy, decreases cytokine levels to levels seen in healthy control subjects (64). Our study showed that leukocytes from patients with hyperaldosteronism exhibit NLRP3 inflammasome activation and increased serum IL-1β levels compared with healthy human volunteers (20). Additional data from our laboratory show that aldosterone-induced NLRP3 activation relies on MR activation (N. S. Ferreira, T. Buder-Nascimento, C. A. Pereira, C. Z. Zanotto, D. S. Prado, J. F. Silva, D. M. Rassi, M. C. Foss-Freitas, J. C. F. Alves-Filho, D. C. Sartori, and R. C. Tostes, unpublished observations). Aldosterone seems to be capable of activating both the priming process (it increases the expression of NLRP3 and pro-IL-1β and activates NF-κB) and the assembly of the inflammasome (mainly via generation of mitochondria-derived ROS) (20). However, further studies are needed to clarify how aldosterone/MR activation triggers the activation of the immune system.
Aldosterone-induced NLRP3 activation also contributes to an inflammatory phenotype and end-organ damage in other pathological conditions including kidney injury leading to chronic kidney disease progression/nephrotic syndrome (5, 33, 55) and obesity-associated adipose tissue and liver dysfunction (99), reinforcing that the NLRP3 inflammasome plays a key role on aldosterone/MR-induced inflammation and target-organ abnormalities in cardiovascular, renal, and metabolic diseases.
MRA: A PROMISING THERAPEUTIC APPROACH TO TREAT AKI AND AKI-MEDIATED CHRONIC KIDNEY DISEASE
AKI is a frequent complication in hospitalized patients with higher incidence rates in patients in intensive care units. It is associated with unfavorable outcomes such as increased short- and long-term mortality rates, longer hospital stay, cardiovascular complications and chronic kidney disease development (17).
During the past decade, evidence indicating that MRA may be a useful strategy to protect against AKI has been accumulating (54). The prophylactic administration of spironolactone prevented the acute renal dysfunction and tubular injury induced by ischemia-reperfusion (I/R) (75). The beneficial effects of spironolactone were associated with the preservation of a normal renal blood flow and reduction in oxidative stress. Importantly spironolactone is also able to efficiently treat kidney I/R injury (IRI) when administered up to 3 h after reperfusion (89). This protective effect was also observed with nonsteroidal MRAs (7, 9), which have been suggested to have a better therapeutic index for hyperkalemia in patients with renal dysfunction (60). The mechanisms underlying the deleterious effects of MR activation during IRI highlight the critical role of MR-mediated oxidative stress; MR-mediated oxidative stress has been shown to lead to a specific imbalance of vascular endothelin signaling through an oxidative stress-dependent posttranslational modification of the vasodilatory endothelin B receptor, leading to functional inactivation of the endothelin B receptor and sustained decrease of the renal blood flow (9, 68). MRAs can prevent oxidative stress and its deleterious consequences (7, 68). The genetic deletion of the MR in SMCs was associated with weaker Rac1 signaling and oxidative stress production (7), and the role of Rac1 was confirmed by the in vivo deletion of Rac-1 in SMCs, which also protected against AKI (7). Decreased oxidative stress production in mice with genetic deletion of the MR in SMCs was associated with reduced posttranslational modification of the endothelin B receptor leading to increased NO production and improved renal perfusion (7). Importantly, the data obtained in both mice and rats can be translated to the Large White pig: soludactone, a soluble MRA, prevented acute IRI after bilateral IRI. This was associated to decreased urinary excretion of an oxidative stress marker (7).
Episodes of AKI lead to increased risk of chronic kidney disease progression and renal failure (105). MRA administration during the acute phase of IRI also prevents the decline of long-term renal function and tubulo-interstitial fibrosis in rats (8, 68) and mice 7a. Steroidal and nonsteroidal MRAs are equally efficient in preventing the progression of chronic kidney disease (8, 68). The underlying mechanism of the benefit of MRAs relies on the prevention of low-grade inflammation and polarization of macrophages toward an M2 repair phenotype (7a). Whether prevention of the progression of chronic kidney disease after IRI using MRAs also on living-donor renal transplantation (80) demonstrated a beneficial effect of spironolactone on renal oxidative stress when administered to the recipients 1 day before and 3 days after transplantation. However, this was not associated with improved short-term renal function since renal function was already good in the placebo group, as expected for living-donor transplantation. A multicenter clinical trial is ongoing in France to study the impact of short-term administration of eplerenone (25 mg twice a day, just before and 4 days after the transplant) on 3-mo renal graft function in patients receiving a graft from an expanded-criteria donor, which is more susceptible to ischemia insult [Eplerenone in Patients Undergoing Renal Transplant (EPURE): ClinicalTrials.gov NCT02490904, supported by the French Programme Hospitalier de Recherche Clinique].
In conclusion, MRA is a promising therapeutic approach to treat AKI and AKI-mediated CKD. The benefit relies on decreased oxidative stress and improved renal perfusion associated with reduced low-grade inflammation and increased macrophage-mediated repair, leading to reduced progression to chronic kidney disease.
CONCLUSIONS
In summary, the symposium presentations reflected growing insights into the new roles for extrarenal MR signaling in aging and cardiovascular diseases. The focus was on the sex differences in MR activation in SMCs and ECs. There are two main conclusions from the presentations. First, MR activation in SMCs plays a critical role driving vascular aging phenotype and AKI in male subjects. SMC-MR deletion attenuates aging-induced aortic stiffness, increased resistance vessel tone and vasoconstriction, and hypertension. Moreover, mice lacking SMC-MRs are protected against AKI associated with weaker Rac1 signaling and oxidative stress, improving NO production and renal perfusion. Second, a possible role of SMC-MR in female subjects still need to be addressed: endothelial MR signaling is a key mechanism for sex differences in obesity-induced endothelial dysfunction, mediating endothelial stiffness and impairing endothelium-dependent relaxation of conduit and resistance arteries of obese female but not male subjects. Leptin is a major activator of the aldosterone-MR axis, contributing to the cardiovascular damage in female obesity. MR antagonist drugs are efficient in preventing cardiovascular injury associated with EC- and SMC-MR signaling and may be beneficial in additional populations that remain to be tested in clinical trials.
GRANTS
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo 2017/10771-5 and 2014/26192-6 (to A. P. Davel), National Heart, Lung, and Blood Institute (NHLBI) Grant 1R0-1HL-130301-01 and American Heart Association (AHA) Grant 16IRG27770047 (to E. J. Belin de Chantemèle), and NHLBI Grants R01-HL-095590 and R01-HL-119290 and AHA Grant EIA-18290005 (to I. Z. Jaffe).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.P.D., I.Z.J., R.C.T., F.J., and E.J.B.d.C. conceived and designed research; A.P.D., I.Z.J., R.C.T., F.J., and E.J.B.d.C. performed experiments; A.P.D., I.Z.J., R.C.T., F.J., and E.J.B.d.C. analyzed data; A.P.D., I.Z.J., R.C.T., F.J., and E.J.B.d.C. interpreted results of experiments; A.P.D., I.Z.J., R.C.T., F.J., and E.J.B.d.C. prepared figures; A.P.D., I.Z.J., R.C.T., F.J., and E.J.B.d.C. drafted manuscript; A.P.D., I.Z.J., R.C.T., F.J., and E.J.B.d.C. edited and revised manuscript; A.P.D., I.Z.J., R.C.T., F.J., and E.J.B.d.C. approved final version of manuscript.
REFERENCES
- 1.Amador CA, Barrientos V, Peña J, Herrada AA, González M, Valdés S, Carrasco L, Alzamora R, Figueroa F, Kalergis AM, Michea L. Spironolactone decreases DOCA-salt-induced organ damage by blocking the activation of T helper 17 and the downregulation of regulatory T lymphocytes. Hypertension 63: 797–803, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02883. [DOI] [PubMed] [Google Scholar]
- 2.Aroor AR, Jia G, Sowers JR. Cellular mechanisms underlying obesity-induced arterial stiffness. Am J Physiol Regul Integr Comp Physiol 314: R387–R398, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 237: 268–275, 1987. doi: 10.1126/science.3037703. [DOI] [PubMed] [Google Scholar]
- 4.Bafford R, Sui XX, Park M, Miyahara T, Newfell BG, Jaffe IZ, Romero JR, Adler GK, Williams GH, Khalil RA, Conte MS. Mineralocorticoid receptor expression in human venous smooth muscle cells: a potential role for aldosterone signaling in vein graft arterialization. Am J Physiol Heart Circ Physiol 301: H41–H47, 2011. doi: 10.1152/ajpheart.00637.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bai M, Chen Y, Zhao M, Zhang Y, He JC, Huang S, Jia Z, Zhang A. NLRP3 inflammasome activation contributes to aldosterone-induced podocyte injury. Am J Physiol Renal Physiol 312: F556–F564, 2017. doi: 10.1152/ajprenal.00332.2016. [DOI] [PubMed] [Google Scholar]
- 6.Barbaro NR, Kirabo A, Harrison DG. A new role of mister (MR) T in hypertension: mineralocorticoid receptor, immune system, and hypertension. Circ Res 120: 1527–1529, 2017. doi: 10.1161/CIRCRESAHA.117.310985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrera-Chimal J, André-Grégoire G, Nguyen Dinh Cat A, Lechner SM, Cau J, Prince S, Kolkhof P, Loirand G, Sauzeau V, Hauet T, Jaisser F. Benefit of mineralocorticoid receptor antagonism in AKI: role of vascular smooth muscle Rac1. J Am Soc Nephrol 28: 1216–1226, 2017. doi: 10.1681/ASN.2016040477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7a. Barrera-Chimal J, Estrela G, Lechner SM, Giraud S, El Moghrabi S, Kaaki S, Kolkhof P, Hauet H, Jaisser F. Myeloid mineralocorticoid receptor controls inflammatory and fibrotic responses after renal injury via macrophage interleukin-4 receptor signaling. Kidney Int 93: 1344–1355, 2018. doi: 10.1016/j.kint.2017.12.016. [DOI] [PubMed] [Google Scholar]
- 8.Barrera-Chimal J, Pérez-Villalva R, Rodríguez-Romo R, Reyna J, Uribe N, Gamba G, Bobadilla NA. Spironolactone prevents chronic kidney disease caused by ischemic acute kidney injury. Kidney Int 83: 93–103, 2013. doi: 10.1038/ki.2012.352. [DOI] [PubMed] [Google Scholar]
- 9.Barrera-Chimal J, Prince S, Fadel F, El Moghrabi S, Warnock DG, Kolkhof P, Jaisser F. Sulfenic acid modification of endothelin B receptor is responsible for the benefit of a nonsteroidal mineralocorticoid receptor antagonist in renal ischemia. J Am Soc Nephrol 27: 398–404, 2016. doi: 10.1681/ASN.2014121216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belden Z, Deiuliis JA, Dobre M, Rajagopalan S. The role of the mineralocorticoid receptor in inflammation: focus on kidney and vasculature. Am J Nephrol 46: 298–314, 2017. doi: 10.1159/000480652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belin de Chantemèle EJ, Mintz JD, Rainey WE, Stepp DW. Impact of leptin-mediated sympatho-activation on cardiovascular function in obese mice. Hypertension 58: 271–279, 2011. doi: 10.1161/HYPERTENSIONAHA.110.168427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belin de Chantemèle EJ, Muta K, Mintz J, Tremblay ML, Marrero MB, Fulton DJ, Stepp DW. Protein tyrosine phosphatase 1B, a major regulator of leptin-mediated control of cardiovascular function. Circulation 120: 753–763, 2009. doi: 10.1161/CIRCULATIONAHA.109.853077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bender SB, DeMarco VG, Padilla J, Jenkins NT, Habibi J, Garro M, Pulakat L, Aroor AR, Jaffe IZ, Sowers JR. Mineralocorticoid receptor antagonism treats obesity-associated cardiac diastolic dysfunction. Hypertension 65: 1082–1088, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhargava A, Wang J, Pearce D. Regulation of epithelial ion transport by aldosterone through changes in gene expression. Mol Cell Endocrinol 217: 189–196, 2004. doi: 10.1016/j.mce.2003.10.020. [DOI] [PubMed] [Google Scholar]
- 15.Bochud M, Nussberger J, Bovet P, Maillard MR, Elston RC, Paccaud F, Shamlaye C, Burnier M. Plasma aldosterone is independently associated with the metabolic syndrome. Hypertension 48: 239–245, 2006. doi: 10.1161/01.HYP.0000231338.41548.fc. [DOI] [PubMed] [Google Scholar]
- 16.Boegehold MA, Drenjancevic I, Lombard JH. Salt, angiotensin ii, superoxide, and endothelial function. Compr Physiol 6: 215–254, 2015. doi: 10.1002/cphy.c150008. [DOI] [PubMed] [Google Scholar]
- 17.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121: 4210–4221, 2011. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bostick B, Habibi J, DeMarco VG, Jia G, Domeier TL, Lambert MD, Aroor AR, Nistala R, Bender SB, Garro M, Hayden MR, Ma L, Manrique C, Sowers JR. Mineralocorticoid receptor blockade prevents Western diet-induced diastolic dysfunction in female mice. Am J Physiol Heart Circ Physiol 308: H1126–H1135, 2015. doi: 10.1152/ajpheart.00898.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat Rev Nephrol 9: 459–469, 2013. doi: 10.1038/nrneph.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bruder-Nascimento T, Ferreira NS, Zanotto CZ, Ramalho F, Pequeno IO, Olivon VC, Neves KB, Alves-Lopes R, Campos E, Silva CA, Fazan R, Carlos D, Mestriner FL, Prado D, Pereira FV, Braga T, Luiz JP, Cau SB, Elias PC, Moreira AC, Câmara NO, Zamboni DS, Alves-Filho JC, Tostes RC. NLRP3 inflammasome mediates aldosterone-induced vascular damage. Circulation 134: 1866–1880, 2016. doi: 10.1161/CIRCULATIONAHA.116.024369. [DOI] [PubMed] [Google Scholar]
- 21.Caillon A, Schiffrin EL. Role of inflammation and immunity in hypertension: recent epidemiological, laboratory, and clinical evidence. Curr Hypertens Rep 18: 21, 2016. doi: 10.1007/s11906-016-0628-7. [DOI] [PubMed] [Google Scholar]
- 22.Calhoun DA, Sharma K. The role of aldosteronism in causing obesity-related cardiovascular risk. Cardiol Clin 28: 517–527, 2010. doi: 10.1016/j.ccl.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caprio M, Newfell BG, la Sala A, Baur W, Fabbri A, Rosano G, Mendelsohn ME, Jaffe IZ. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circ Res 102: 1359–1367, 2008. doi: 10.1161/CIRCRESAHA.108.174235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ceolotto G, Papparella I, Bortoluzzi A, Strapazzon G, Ragazzo F, Bratti P, Fabricio AS, Squarcina E, Gion M, Palatini P, Semplicini A. Interplay between miR-155, AT1R A1166C polymorphism, and AT1R expression in young untreated hypertensives. Am J Hypertens 24: 241–246, 2011. doi: 10.1038/ajh.2010.211. [DOI] [PubMed] [Google Scholar]
- 25.Chadderdon SM, Belcik JT, Bader L, Peters DM, Kievit P, Alkayed NJ, Kaul S, Grove KL, Lindner JR. Temporal changes in skeletal muscle capillary responses and endothelial-derived vasodilators in obesity-related insulin resistance. Diabetes 65: 2249–2257, 2016. doi: 10.2337/db15-1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen L, Ding ML, Wu F, He W, Li J, Zhang XY, Xie WL, Duan SZ, Xia WH, Tao J. Impaired endothelial repair capacity of early endothelial progenitor cells in hypertensive patients with primary hyperaldosteronemia: role of 5,6,7,8-tetrahydrobiopterin oxidation and endothelial nitric oxide synthase uncoupling. Hypertension 67: 430–439, 2016. [DOI] [PubMed] [Google Scholar]
- 27.Cooper SA, Whaley-Connell A, Habibi J, Wei Y, Lastra G, Manrique C, Stas S, Sowers JR. Renin-angiotensin-aldosterone system and oxidative stress in cardiovascular insulin resistance. Am J Physiol Heart Circ Physiol 293: H2009–H2023, 2007. doi: 10.1152/ajpheart.00522.2007. [DOI] [PubMed] [Google Scholar]
- 28.Cosic A, Jukic I, Stupin A, Mihalj M, Mihaljevic Z, Novak S, Vukovic R, Drenjancevic I. Attenuated flow-induced dilatation of middle cerebral arteries is related to increased vascular oxidative stress in rats on a short-term high salt diet. J Physiol 594: 4917–4931, 2016. doi: 10.1113/JP272297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.da Silva Franco N, Lubaczeuski C, Guizoni DM, Victorio JA, Santos-Silva JC, Brum PC, Carneiro EM, Davel AP. Propranolol treatment lowers blood pressure, reduces vascular inflammatory markers and improves endothelial function in obese mice. Pharmacol Res 122: 35–45, 2017. doi: 10.1016/j.phrs.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 30.Davel AP, Anwar IJ, Jaffe IZ. The endothelial mineralocorticoid receptor: mediator of the switch from vascular health to disease. Curr Opin Nephrol Hypertens 26: 97–104, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davel AP, Lu Q, Moss ME, Rao S, Anwar IJ, DuPont JJ, Jaffe IZ. Sex-specific mechanisms of resistance vessel endothelial dysfunction induced by cardiometabolic risk factors. J Am Heart Assoc 7: e007675, 2018. doi: 10.1161/JAHA.117.007675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dhingra R, Vasan RS. Age as a risk factor. Med Clin North Am 96: 87–91, 2012. doi: 10.1016/j.mcna.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding W, Liu T, Bi X, Zhang Z. Mitochondria-targeted antioxidant mito-tempo protects against aldosterone-induced renal injury in vivo. Cell Physiol Biochem 44: 741–750, 2017. doi: 10.1159/000485287. [DOI] [PubMed] [Google Scholar]
- 34.DuPont JJ, Jaffe IZ. 30 YEARS OF THE MINERALOCORTICOID RECEPTOR: The role of the mineralocorticoid receptor in the vasculature. J Endocrinol 234: T67–T82, 2017. doi: 10.1530/JOE-17-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DuPont JJ, McCurley A, Davel AP, McCarthy J, Bender SB, Hong K, Yang Y, Yoo JK, Aronovitz M, Baur WE, Christou DD, Hill MA, Jaffe IZ. Vascular mineralocorticoid receptor regulates microRNA-155 to promote vasoconstriction and rising blood pressure with aging. JCI Insight 1: e88942, 2016. doi: 10.1172/jci.insight.88942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ehrhart-Bornstein M, Lamounier-Zepter V, Schraven A, Langenbach J, Willenberg HS, Barthel A, Hauner H, McCann SM, Scherbaum WA, Bornstein SR. Human adipocytes secrete mineralocorticoid-releasing factors. Proc Natl Acad Sci USA 100: 14211–14216, 2003. doi: 10.1073/pnas.2336140100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engin A. Endothelial dysfunction in obesity. Adv Exp Med Biol 960: 345–379, 2017. doi: 10.1007/978-3-319-48382-5_15. [DOI] [PubMed] [Google Scholar]
- 38.Fels J, Oberleithner H, Kusche-Vihrog K. Ménage à trois: aldosterone, sodium and nitric oxide in vascular endothelium. Biochim Biophys Acta 1802: 1193–1202, 2010. doi: 10.1016/j.bbadis.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 39.Feng W, Dell’Italia LJ, Sanders PW. Novel Paradigms of salt and hypertension. J Am Soc Nephrol 28: 1362–1369, 2017. doi: 10.1681/ASN.2016080927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flegal KM, Kruszon-Moran D, Carroll MD, Fryar CD, Ogden CL. Trends in obesity among adults in the United States, 2005 to 2014. JAMA 315: 2284–2291, 2016. doi: 10.1001/jama.2016.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galmiche G, Pizard A, Gueret A, El Moghrabi S, Ouvrard-Pascaud A, Berger S, Challande P, Jaffe IZ, Labat C, Lacolley P, Jaisser F. Smooth muscle cell mineralocorticoid receptors are mandatory for aldosterone-salt to induce vascular stiffness. Hypertension 63: 520–526, 2014. doi: 10.1161/HYPERTENSIONAHA.113.01967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garg R, Rao AD, Baimas-George M, Hurwitz S, Foster C, Shah RV, Jerosch-Herold M, Kwong RY, Di Carli MF, Adler GK. Mineralocorticoid receptor blockade improves coronary microvascular function in individuals with type 2 diabetes. Diabetes 64: 236–242, 2015. doi: 10.2337/db14-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goodfriend TL, Kelley DE, Goodpaster BH, Winters SJ. Visceral obesity and insulin resistance are associated with plasma aldosterone levels in women. Obes Res 7: 355–362, 1999. doi: 10.1002/j.1550-8528.1999.tb00418.x. [DOI] [PubMed] [Google Scholar]
- 44.Heidenreich PA, Trogdon JG, Khavjou OA, Butler J, Dracup K, Ezekowitz MD, Finkelstein EA, Hong Y, Johnston SC, Khera A, Lloyd-Jones DM, Nelson SA, Nichol G, Orenstein D, Wilson PW, Woo YJ; American Heart Association Advocacy Coordinating Committee; Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Clinical Cardiology; Council on Epidemiology and Prevention; Council on Arteriosclerosis; Thrombosis and Vascular Biology; Council on Cardiopulmonary; Critical Care; Perioperative and Resuscitation; Council on Cardiovascular Nursing; Council on the Kidney in Cardiovascular Disease; Council on Cardiovascular Surgery and Anesthesia, and Interdisciplinary Council on Quality of Care and Outcomes Research . Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation 123: 933–944, 2011. doi: 10.1161/CIR.0b013e31820a55f5. [DOI] [PubMed] [Google Scholar]
- 45.Hiramatsu K, Yamada T, Ichikawa K, Izumiyama T, Nagata H. Changes in endocrine activities relative to obesity in patients with essential hypertension. J Am Geriatr Soc 29: 25–30, 1981. doi: 10.1111/j.1532-5415.1981.tb02389.x. [DOI] [PubMed] [Google Scholar]
- 46.Huby AC, Antonova G, Groenendyk J, Gomez-Sanchez CE, Bollag WB, Filosa JA, Belin de Chantemèle EJ. Adipocyte-derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. Circulation 132: 2134–2145, 2015. doi: 10.1161/CIRCULATIONAHA.115.018226. [DOI] [PubMed] [Google Scholar]
- 47.Huby A-C, Otvos L Jr, Belin de Chantemèle EJ. Leptin induces hypertension and endothelial dysfunction via aldosterone-dependent mechanisms in obese female mice. Hypertension 67: 1020–1028, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocorticoid receptors in human coronary artery smooth muscle cells. Circ Res 96: 643–650, 2005. doi: 10.1161/01.RES.0000159937.05502.d1. [DOI] [PubMed] [Google Scholar]
- 49.Jaffe IZ, Tintut Y, Newfell BG, Demer LL, Mendelsohn ME. Mineralocorticoid receptor activation promotes vascular cell calcification. Arterioscler Thromb Vasc Biol 27: 799–805, 2007. doi: 10.1161/01.ATV.0000258414.59393.89. [DOI] [PubMed] [Google Scholar]
- 50.Jaisser F, Farman N. Emerging roles of the mineralocorticoid receptor in pathology: toward new paradigms in clinical pharmacology. Pharmacol Rev 68: 49–75, 2016. doi: 10.1124/pr.115.011106. [DOI] [PubMed] [Google Scholar]
- 51.Jia G, Habibi J, Aroor AR, Hill MA, DeMarco VG, Lee LE, Ma L, Barron BJ, Whaley-Connell A, Sowers JR. Enhanced endothelium epithelial sodium channel signaling prompts left ventricular diastolic dysfunction in obese female mice. Metabolism 78: 69–79, 2018. doi: 10.1016/j.metabol.2017.08.008. [DOI] [PubMed] [Google Scholar]
- 52.Jia G, Habibi J, Aroor AR, Martinez-Lemus LA, DeMarco VG, Ramirez-Perez FI, Sun Z, Hayden MR, Meininger GA, Mueller KB, Jaffe IZ, Sowers JR. Endothelial mineralocorticoid receptor mediates diet-induced aortic stiffness in females. Circ Res 118: 935–943, 2016. doi: 10.1161/CIRCRESAHA.115.308269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jia G, Habibi J, DeMarco VG, Martinez-Lemus LA, Ma L, Whaley-Connell AT, Aroor AR, Domeier TL, Zhu Y, Meininger GA, Mueller KB, Jaffe IZ, Sowers JR. Endothelial mineralocorticoid receptor deletion prevents diet-induced cardiac diastolic dysfunction in females. Hypertension 66: 1159–1167, 2015. doi: 10.1161/HYPERTENSIONAHA.115.06015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Juncos LA, Juncos LI. Mineralocorticoid receptor antagonism in AKI: a new hope? J Am Soc Nephrol 27: 335–337, 2016. doi: 10.1681/ASN.2015080866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kadoya H, Satoh M, Sasaki T, Taniguchi S, Takahashi M, Kashihara N. Excess aldosterone is a critical danger signal for inflammasome activation in the development of renal fibrosis in mice. FASEB J 29: 3899–3910, 2015. doi: 10.1096/fj.15-271734. [DOI] [PubMed] [Google Scholar]
- 56.Khosla N, Kalaitzidis R, Bakris GL. Predictors of hyperkalemia risk following hypertension control with aldosterone blockade. Am J Nephrol 30: 418–424, 2009. doi: 10.1159/000237742. [DOI] [PubMed] [Google Scholar]
- 57.Kidambi S, Kotchen JM, Grim CE, Raff H, Mao J, Singh RJ, Kotchen TA. Association of adrenal steroids with hypertension and the metabolic syndrome in blacks. Hypertension 49: 704–711, 2007. doi: 10.1161/01.HYP.0000253258.36141.c7. [DOI] [PubMed] [Google Scholar]
- 58.Kim SK, McCurley A, DuPont JJ, Aronovitz M, Moss ME, Stillman IE, Karumanchi SA, Christou DD, Jaffe IZ. Smooth muscle cell mineralocorticoid receptor as a mediator of and therapeutic target for cardiovascular aging. Hypertension 71: 609–621, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koenig JB, Jaffe IZ. Direct role for smooth muscle cell mineralocorticoid receptors in vascular remodeling: novel mechanisms and clinical implications. Curr Hypertens Rep 16: 427, 2014. doi: 10.1007/s11906-014-0427-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kolkhof P, Bärfacker L. 30 Years of the mineralocorticoid receptor: mineralocorticoid receptor antagonists: 60 years of research and development. J Endocrinol 234: T125–T140, 2017. doi: 10.1530/JOE-16-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kornel L, Kanamarlapudi N, Travers T, Taff DJ, Patel N, Chen C, Baum RM, Raynor WJ. Studies on high affinity binding of mineralo- and glucocorticoids in rabbit aorta cytosol. J Steroid Biochem 16: 245–264, 1982. doi: 10.1016/0022-4731(82)90173-X. [DOI] [PubMed] [Google Scholar]
- 62.Kornel L, Kanamarlapudi N, Von Dreele MM. The role of arterial mineralocorticoid receptors in the mechanism of hypertension: findings and hypothesis. Clin Biochem 20: 113–120, 1987. doi: 10.1016/S0009-9120(87)80109-1. [DOI] [PubMed] [Google Scholar]
- 63.Krug AW, Allenhöfer L, Monticone R, Spinetti G, Gekle M, Wang M, Lakatta EG. Elevated mineralocorticoid receptor activity in aged rat vascular smooth muscle cells promotes a proinflammatory phenotype via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase and epidermal growth factor receptor-dependent pathways. Hypertension 55: 1476–1483, 2010. doi: 10.1161/HYPERTENSIONAHA.109.148783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krysiak R, Okopien B. The effect of treatment on monocyte and lymphocyte cytokine release in patients with aldosteronoma. Hypertens Res 35: 123–125, 2012. doi: 10.1038/hr.2011.142. [DOI] [PubMed] [Google Scholar]
- 65.Kusche-Vihrog K, Tarjus A, Fels J, Jaisser F. The epithelial Na+ channel: a new player in the vasculature. Curr Opin Nephrol Hypertens 23: 143–148, 2014. doi: 10.1097/01.mnh.0000441054.88962.2c. [DOI] [PubMed] [Google Scholar]
- 66.Lakatta EG, Wang M, Najjar SS. Arterial aging and subclinical arterial disease are fundamentally intertwined at macroscopic and molecular levels. Med Clin North Am 93: 583–604, 2009. doi: 10.1016/j.mcna.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lastra G, Manrique C, Jia G, Aroor AR, Hayden MR, Barron BJ, Niles B, Padilla J, Sowers JR. Xanthine oxidase inhibition protects against Western diet-induced aortic stiffness and impaired vasorelaxation in female mice. Am J Physiol Regul Integr Comp Physiol 313: R67–R77, 2017. doi: 10.1152/ajpregu.00483.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lattenist L, Lechner SM, Messaoudi S, Le Mercier A, El Moghrabi S, Prince S, Bobadilla NA, Kolkhof P, Jaisser F, Barrera-Chimal J. Nonsteroidal mineralocorticoid receptor antagonist finerenone protects against acute kidney injury-mediated chronic kidney disease: role of oxidative stress. Hypertension 69: 870–878, 2017. doi: 10.1161/HYPERTENSIONAHA.116.08526. [DOI] [PubMed] [Google Scholar]
- 69.Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, Stanton RC, Pitt B, Loscalzo J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med 13: 189–197, 2007. [Erratum in Nat Med 15:1093, 2009]. doi: 10.1038/nm1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lind L, Berglund L, Larsson A, Sundström J. Endothelial function in resistance and conduit arteries and 5-year risk of cardiovascular disease. Circulation 123: 1545–1551, 2011. doi: 10.1161/CIRCULATIONAHA.110.984047. [DOI] [PubMed] [Google Scholar]
- 71.Liu SL, Schmuck S, Chorazcyzewski JZ, Gros R, Feldman RD. Aldosterone regulates vascular reactivity: short-term effects mediated by phosphatidylinositol 3-kinase-dependent nitric oxide synthase activation. Circulation 108: 2400–2406, 2003. doi: 10.1161/01.CIR.0000093188.53554.44. [DOI] [PubMed] [Google Scholar]
- 72.Lurbe E, Torro I, Aguilar F, Alvarez J, Alcon J, Pascual JM, Redon J. Added impact of obesity and insulin resistance in nocturnal blood pressure elevation in children and adolescents. Hypertension 51: 635–641, 2008. doi: 10.1161/HYPERTENSIONAHA.107.099234. [DOI] [PubMed] [Google Scholar]
- 73.Marzolla V, Armani A, Zennaro MC, Cinti F, Mammi C, Fabbri A, Rosano GM, Caprio M. The role of the mineralocorticoid receptor in adipocyte biology and fat metabolism. Mol Cell Endocrinol 350: 281–288, 2012. doi: 10.1016/j.mce.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 74.McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, Chambon P, Hill MA, Dorrance AM, Mendelsohn ME, Jaffe IZ. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med 18: 1429–1433, 2012. doi: 10.1038/nm.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mejía-Vilet JM, Ramírez V, Cruz C, Uribe N, Gamba G, Bobadilla NA. Renal ischemia-reperfusion injury is prevented by the mineralocorticoid receptor blocker spironolactone. Am J Physiol Renal Physiol 293: F78–F86, 2007. doi: 10.1152/ajprenal.00077.2007. [DOI] [PubMed] [Google Scholar]
- 76.Muñoz-Durango N, Vecchiola A, Gonzalez-Gomez LM, Simon F, Riedel CA, Fardella CE, Kalergis AM. Modulation of immunity and inflammation by the mineralocorticoid receptor and aldosterone. BioMed Res Int 2015: 652738, 2015. doi: 10.1155/2015/652738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mutoh A, Isshiki M, Fujita T. Aldosterone enhances ligand-stimulated nitric oxide production in endothelial cells. Hypertens Res 31: 1811–1820, 2008. doi: 10.1291/hypres.31.1811. [DOI] [PubMed] [Google Scholar]
- 78.Newfell BG, Iyer LK, Mohammad NN, McGraw AP, Ehsan A, Rosano G, Huang PL, Mendelsohn ME, Jaffe IZ. Aldosterone regulates vascular gene transcription via oxidative stress-dependent and -independent pathways. Arterioscler Thromb Vasc Biol 31: 1871–1880, 2011. doi: 10.1161/ATVBAHA.111.229070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Noren Hooten N, Abdelmohsen K, Gorospe M, Ejiogu N, Zonderman AB, Evans MK. microRNA expression patterns reveal differential expression of target genes with age. PLoS One 5: e10724, 2010. doi: 10.1371/journal.pone.0010724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ojeda-Cervantes M, Barrera-Chimal J, Alberú J, Pérez-Villalva R, Morales-Buenrostro LE, Bobadilla NA. Mineralocorticoid receptor blockade reduced oxidative stress in renal transplant recipients: a double-blind, randomized pilot study. Am J Nephrol 37: 481–490, 2013. doi: 10.1159/000350539. [DOI] [PubMed] [Google Scholar]
- 81.Olivier A, Pitt B, Girerd N, Lamiral Z, Machu JL, McMurray JJV, Swedberg K, van Veldhuisen DJ, Collier TJ, Pocock SJ, Rossignol P, Zannad F, Pizard A. Effect of eplerenone in patients with heart failure and reduced ejection fraction: potential effect modification by abdominal obesity. Insight from the EMPHASIS-HF trial. Eur J Heart Fail 19: 1186–1197, 2017. doi: 10.1002/ejhf.792. [DOI] [PubMed] [Google Scholar]
- 82.Ozata M, Ozdemir IC, Licinio J. Human leptin deficiency caused by a missense mutation: multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J Clin Endocrinol Metab 84: 3686–3695, 1999. doi: 10.1210/jcem.84.10.5999. [DOI] [PubMed] [Google Scholar]
- 83.Paz-Filho G, Mastronardi C, Delibasi T, Wong ML, Licinio J. Congenital leptin deficiency: diagnosis and effects of leptin replacement therapy. Arq Bras Endocrinol Metabol 54: 690–697, 2010. doi: 10.1590/S0004-27302010000800005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pojoga LH, Adamová Z, Kumar A, Stennett AK, Romero JR, Adler GK, Williams GH, Khalil RA. Sensitivity of NOS-dependent vascular relaxation pathway to mineralocorticoid receptor blockade in caveolin-1-deficient mice. Am J Physiol Heart Circ Physiol 298: H1776–H1788, 2010. doi: 10.1152/ajpheart.00661.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pojoga LH, Yao TM, Opsasnick LA, Siddiqui WT, Reslan OM, Adler GK, Williams GH, Khalil RA. Cooperative role of mineralocorticoid receptor and caveolin-1 in regulating the vascular response to low nitric oxide-high angiotensin ii-induced cardiovascular injury. J Pharmacol Exp Ther 355: 32–47, 2015. doi: 10.1124/jpet.115.226043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pruthi D, McCurley A, Aronovitz M, Galayda C, Karumanchi SA, Jaffe IZ. Aldosterone promotes vascular remodeling by direct effects on smooth muscle cell mineralocorticoid receptors. Arterioscler Thromb Vasc Biol 34: 355–364, 2014. doi: 10.1161/ATVBAHA.113.302854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rickard AJ, Morgan J, Tesch G, Funder JW, Fuller PJ, Young MJ. Deletion of mineralocorticoid receptors from macrophages protects against deoxycorticosterone/salt-induced cardiac fibrosis and increased blood pressure. Hypertension 54: 537–543, 2009. doi: 10.1161/HYPERTENSIONAHA.109.131110. [DOI] [PubMed] [Google Scholar]
- 88.Rocha R, Rudolph AE, Frierdich GE, Nachowiak DA, Kekec BK, Blomme EA, McMahon EG, Delyani JA. Aldosterone induces a vascular inflammatory phenotype in the rat heart. Am J Physiol Heart Circ Physiol 283: H1802–H1810, 2002. doi: 10.1152/ajpheart.01096.2001. [DOI] [PubMed] [Google Scholar]
- 89.Sánchez-Pozos K, Barrera-Chimal J, Garzón-Muvdi J, Pérez-Villalva R, Rodríguez-Romo R, Cruz C, Gamba G, Bobadilla NA. Recovery from ischemic acute kidney injury by spironolactone administration. Nephrol Dial Transplant 27: 3160–3169, 2012. doi: 10.1093/ndt/gfs014. [DOI] [PubMed] [Google Scholar]
- 90.Schäfer N, Lohmann C, Winnik S, van Tits LJ, Miranda MX, Vergopoulos A, Ruschitzka F, Nussberger J, Berger S, Lüscher TF, Verrey F, Matter CM. Endothelial mineralocorticoid receptor activation mediates endothelial dysfunction in diet-induced obesity. Eur Heart J 34: 3515–3524, 2013. doi: 10.1093/eurheartj/eht095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schiffrin EL. Immune mechanisms in hypertension and vascular injury. Clin Sci (Lond) 126: 267–274, 2014. doi: 10.1042/CS20130407. [DOI] [PubMed] [Google Scholar]
- 92.Sorof J, Daniels S. Obesity hypertension in children: a problem of epidemic proportions. Hypertension 40: 441–447, 2002. doi: 10.1161/01.HYP.0000032940.33466.12. [DOI] [PubMed] [Google Scholar]
- 93.Sowers JR. Diabetes mellitus and cardiovascular disease in women. Arch Intern Med 158: 617–621, 1998. doi: 10.1001/archinte.158.6.617. [DOI] [PubMed] [Google Scholar]
- 94.Terada Y, Ueda S, Hamada K, Shimamura Y, Ogata K, Inoue K, Taniguchi Y, Kagawa T, Horino T, Takao T. Aldosterone stimulates nuclear factor-kappa B activity and transcription of intercellular adhesion molecule-1 and connective tissue growth factor in rat mesangial cells via serum- and glucocorticoid-inducible protein kinase-1. Clin Exp Nephrol 16: 81–88, 2012. doi: 10.1007/s10157-011-0498-x. [DOI] [PubMed] [Google Scholar]
- 95.Toblli J, Cao G, Rivas C, Munoz M, Giani J, Dominici F, Angerosa M. Cardiovascular protective effects of nebivolol in Zucker diabetic fatty rats. J Hypertens 28: 1007–1019, 2010. doi: 10.1097/HJH.0b013e328337598c. [DOI] [PubMed] [Google Scholar]
- 96.Tostes RC, Touyz RM, He G, Chen X, Schiffrin EL. Contribution of endothelin-1 to renal activator protein-1 activation and macrophage infiltration in aldosterone-induced hypertension. Clin Sci (Lond) 103, Suppl 48: 25S–30S, 2002. doi: 10.1042/CS103S025S. [DOI] [PubMed] [Google Scholar]
- 97.Towfighi A, Zheng L, Ovbiagele B. Weight of the obesity epidemic: rising stroke rates among middle-aged women in the United States. Stroke 41: 1371–1375, 2010. doi: 10.1161/STROKEAHA.109.577510. [DOI] [PubMed] [Google Scholar]
- 98.Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci 65: 1028–1041, 2010. doi: 10.1093/gerona/glq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wada T, Ishikawa A, Watanabe E, Nakamura Y, Aruga Y, Hasegawa H, Onogi Y, Honda H, Nagai Y, Takatsu K, Ishii Y, Sasahara M, Koya D, Tsuneki H, Sasaoka T. Eplerenone prevented obesity-induced inflammasome activation and glucose intolerance. J Endocrinol 235: 179–191, 2017. doi: 10.1530/JOE-17-0351. [DOI] [PubMed] [Google Scholar]
- 100.Wang M, Monticone RE, Lakatta EG. Arterial aging: a journey into subclinical arterial disease. Curr Opin Nephrol Hypertens 19: 201–207, 2010. doi: 10.1097/MNH.0b013e3283361c0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wenzel U, Turner JE, Krebs C, Kurts C, Harrison DG, Ehmke H. Immune mechanisms in arterial hypertension. J Am Soc Nephrol 27: 677–686, 2016. doi: 10.1681/ASN.2015050562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Willum-Hansen T, Staessen JA, Torp-Pedersen C, Rasmussen S, Thijs L, Ibsen H, Jeppesen J. Prognostic value of aortic pulse wave velocity as index of arterial stiffness in the general population. Circulation 113: 664–670, 2006. doi: 10.1161/CIRCULATIONAHA.105.579342. [DOI] [PubMed] [Google Scholar]
- 103.Wilsgaard T, Schirmer H, Arnesen E. Impact of body weight on blood pressure with a focus on sex differences: the Tromso Study, 1986-1995. Arch Intern Med 160: 2847–2853, 2000. doi: 10.1001/archinte.160.18.2847. [DOI] [PubMed] [Google Scholar]
- 104.Xu X, Wang B, Ren C, Hu J, Greenberg DA, Chen T, Xie L, Jin K. Age-related impairment of vascular structure and functions. Aging Dis 8: 590–610, 2017. doi: 10.14336/AD.2017.0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yang L, Humphreys BD, Bonventre JV. Pathophysiology of acute kidney injury to chronic kidney disease: maladaptive repair. Contrib Nephrol 174: 149–155, 2011. doi: 10.1159/000329385. [DOI] [PubMed] [Google Scholar]
- 106.Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, Kim YB, Elmquist JK, Tartaglia LA, Kahn BB, Neel BG. PTP1B regulates leptin signal transduction in vivo. Dev Cell 2: 489–495, 2002. doi: 10.1016/S1534-5807(02)00148-X. [DOI] [PubMed] [Google Scholar]