

Abstract
β-Adrenergic receptor (β-AR) activation produces the main positive inotropic response of the heart. During ischemia-reperfusion (I/R), however, β-AR activation can trigger life-threatening arrhythmias. Because I/R is frequently associated with oxidative stress, we investigated whether ryanodine receptor (RyR) oxidation contributes to proarrythmogenic Ca2+ waves during β-AR activation. Measurements of contractile and electrical activity from Langendorff-perfused rabbit hearts revealed that I/R produces tachyarrhythmias. Ventricular myocytes isolated from I/R hearts had an increased level of oxidized glutathione (i.e., oxidative stress) and a decreased level of free thiols in RyRs (i.e., RyR oxidation). Furthermore, myocytes from I/R hearts were characterized by increased sarcoplasmic reticulum (SR) Ca2+ leak and enhanced fractional SR Ca2+ release. In myocytes from nonischemic hearts, β-AR activation with isoproterenol (10 nM) produced only a positive inotropic effect, whereas in myocytes from ischemic hearts, isoproterenol at the same concentration triggered spontaneous Ca2+ waves. β-AR activation produced a similar effect on RyR phosphorylation in control and I/R myocytes. Treatment of myocytes from I/R hearts with the reducing agent mercaptopropionylglycine (100 μM) attenuated RyR oxidization and decreased Ca2+ wave frequency during β-AR activation. On the other hand, treatment of myocytes from nonischemic hearts with H2O2 (50 μM) increased SR Ca2+ leak and triggered Ca2+ waves during β-AR activation. Collectively, these results suggest that RyR oxidation after I/R plays a critical role in the transition from positive inotropic to arrhythmogenic effects during β-AR stimulation. Prevention of RyR oxidation can be a promising strategy to inhibit arrhythmias and preserve positive inotropic effect of β-AR activation during myocardial infarction.
NEW & NOTEWORTHY Oxidative stress induced by ischemia plays a critical role in triggering arrhythmias during adrenergic stimulation. The combined increase in sarcoplasmic reticulum Ca2+ leak (because of ryanodine receptor oxidation) and sarcoplasmic reticulum Ca2+ load (because of adrenergic stimulation) can trigger proarrythmogenic Ca2+ waves. Restoring normal ryanodine receptor redox status can be a promising strategy to prevent arrhythmias and preserve positive inotropic effect of adrenergic stimulation during myocardial infarction.
Keywords: Ca2+-induced Ca2+ release, calcium regulation, excitation-contraction coupling, oxidative stress, ryanodine receptor, sarcoplasmic reticulum
INTRODUCTION
Myocardial injuries induced by ischemia-reperfusion (I/R) are leading causes of morbidity and mortality in the modern world. Current medical interventions to restore blood flow to the ischemic region have proven to be successful in reducing the progression of necrosis and drastically improving mortality rates after ischemia. However, a major complication associated with reperfusing blood to the ischemic region is the increased risk of arrhythmias (42). Previous studies of postmyocardial infarction (post-MI) animal models have suggested that defective Ca2+ regulation plays an important role in cardiac arrhythmias (4, 5). It has been shown that excessive sarcoplasmic reticulum (SR) Ca2+ leak, particularly in the form of Ca2+ waves, can generate delayed afterdepolarizations (DADs) and cardiac arrhythmias. Despite its clinical significance, mechanisms of SR Ca2+ mishandling in ventricular myocytes subjected to I/R are not fully understood.
In the ischemic region, metabolites build up within the interstitium and intracellularly because of the energy consumption of the working myocardium and the lack of blood perfusion. Commonly associated with ischemia are complex cellular metabolic changes, including a decrease in ATP concentration and a subsequent increase in free Mg2+ concentration, K+ concentration, ADP concentration, and Pi concentration as well as a drop in intracellular pH (10). During ischemia, there are also important changes in the cytosolic redox potential. These changes can have broad impact on cell physiology, including the generation of reactive oxygen species (ROS) (24). Although the generation of ROS has been shown to play an important role in normal cell signaling (36), during periods of oxidative stress, excessive ROS production can have detrimental effects on normal protein function (34). Some proteins that are involved in intracellular Ca2+ regulation can be affected by ROS (43, 45). For example, the ryanodine receptor (RyR) Ca2+-release channel has ~360 cysteine residues per tetrameric channel, with an estimated 84 of those in a reduced free thiol state (41). Each free thiol residue can serve as a target for a number of oxidative modifications, including disulfide bond formation, S-nitrosylation, and S-glutathionylation. To date, a number of in vitro studies have shown that both ROS and other free radicals can induce changes in RyR channel activity. Lipid bilayer and single cell experiments have shown that ROS activates the single RyR channel function and SR Ca2+ release (43, 45). Moreover, an increase in overall oxidation of RyR2 with abnormal SR Ca2+ release has also been observed in post-MI dog hearts (5). Oxidation of RyR2 is also thought to play a role in myocardial preconditioning before an ischemic insult (15). It has been suggested that NADPH oxidase-dependent S-glutathionylation of RyRs can play a role in cardioprotection during ischemia (28).
MI causes life-threatening arrhythmias, particularly within several hours after the insult. During this time, an increased adrenergic tone manifests in the ischemic region because of elevated concentrations of catecholamines (21). Both ex vivo and in vivo I/R studies have shown that the main source of endogenous catecholamines is in fact from nonexocytotic release at sympathetic nerve endings that innervate the myocardium (20, 21). β-AR stimulation is considered to be an important contributor in I/R injury. Increased β-AR stimulation can further increase energy demand and intracellular ROS production in the ischemic region (12). Studies that have blocked PKA activation via β-blockers or direct inhibition of PKA have proven to be effective in reducing infarct size (22, 32) and preventing arrhythmias (25, 26). However, the cellular and molecular mechanisms of ischemia-induced arrhythmias are not completely understood. In the present study, we tested the hypothesis that oxidative stress during a period of I/R plays a critical role in triggering arrhythmias during β-AR stimulation. The obtained results revealed that RyR oxidation induced by I/R increases RyR activity and diastolic SR Ca2+ leak. During β-AR stimulation, the combined increase in SR Ca2+ leak (because of RyR oxidation) and SR Ca2+ load (because of SERCA stimulation) triggers proarrythmogenic Ca2+ waves.
MATERIALS AND METHODS
Ex vivo whole heart experiments.
Experiments were performed on hearts isolated from New Zealand White rabbits (27 animals, 2–2.5 kg, Myrtle’s Rabbitry). Rabbits were anesthetized with pentobarbital sodium (50 mg/kg iv). After a thoracotomy, hearts were quickly excised, mounted on a Langendorff apparatus, and retrogradely perfused with the Tyrode solution [containing (in mM) 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES; pH 7.4] at a flux of 25 ml/min for 10 min until stabilization of the ECG and left ventricular (LV) pressure (LVP). A low-flow ischemia model of the Langendorff-perfused rabbit heart was used in this study (13, 27, 29). The flux was reduced to 4 ml/min, subjecting the hearts to ischemic insult for 20 min. The initial flux was then restored for 15 min (reperfusion). The ECG was recorded with two electrodes placed across the ventricles and connected to an amplifier. LVP was measured with the aid of a plastic balloon inserted into the LV and connected to the same amplifier. ECG and LVP were measured simultaneously throughout the entire experiment. Sham (control) hearts were perfused at the same flux (25 ml/min) and for the same amount of time as I/R hearts. All whole heart experiments were performed at 37°C.
Myocyte isolation.
LV myocytes were isolated according to the previously described procedures (14). After ECG and LVP measurements in control conditions and during I/R, the same hearts were perfused with a solution containing Liberase Blendzyme (Roche Applied Science) for 16 min. The whole heart experiments and cell isolation procedure were approved by the Institutional Animal Care and Use Committee and complied with United States regulations on animal experimentation (16). Chemicals and reagents were purchased from Sigma-Aldrich unless otherwise stated.
Confocal microscopy.
Changes in cytosolic Ca2+ concentration ([Ca2+]i) and intra-SR free Ca2+ concentration ([Ca2+]SR) were measured with a laser scanning confocal microscopy [Radiance 2000 MP (Bio-Rad) and LSM 410 (Zeiss)] equipped with a ×40 oil-immersion objective lens (numerical aperture: 1.3). All confocal experiments were performed at 22–24°C.
Measurements of [Ca2+]i.
To record [Ca2+]i, we used the high-affinity Ca2+ indicator fluo 4 (Molecular Probes/Invitrogen). To load the cytosol with the Ca2+ indicator, cells were incubated at room temperature with 10 μM fluo 4-AM for 15 min in Tyrode solution followed by a 20-min wash. Fluo 4 was excited with the 488-nm line of an argon laser, and fluorescence was measured at >515 nm. Action potentials (APs) were induced by electrical field stimulation using a pair of platinum electrodes, which were connected to a Grass stimulator (Astro-Med). Fluo 4 images were acquired in line-scan mode (3 ms/scan). Ca2+ wave propensity was analyzed as numbers of waves per the cardiac cycle (in %).
Measurements of [Ca2+]SR and SR Ca2+ leak.
To record [Ca2+]SR, we used the low-affinity Ca2+ indicator fluo 5N (Molecular Probes/Invitrogen). To load the SR with the Ca2+ indicator, myocytes were incubated with 5 μM fluo 5N-AM for 2.5 h at 37°C. SR Ca2+ leak was measured as a function of [Ca2+]SR according to a previously described protocol (44). Briefly, fluo 5N was excited with the 488-nm line of an argon laser. Fluo 5N fluorescence was collected at >515 nm and averaged over the entire cellular width of the two-dimensional image (pixel size: 0.2 μm). Changes in [Ca2+]SR were calculated by the following formula (9): [Ca2+]SR = Kd × R/(Kd/[Ca2+]SR diast − R + 1), where Kd is the fluo 5N Ca2+ dissociation constant (390 μM based on in situ calibrations) (44), R is the normalized fluo 5N fluorescence [R = (F − Fmin)/(F0 − Fmin)], F0 and Fmin are the fluorescence levels at rest and after depletion of the SR with caffeine, respectively, and [Ca2+]SR diast is diastolic [Ca2+]SR at 0.75 Hz (900 μM) (14). SR Ca2+ leak was measured as the changes of total [Ca2+]SR ([Ca2+]SRT) over time (d[Ca2+]SRT/dt) after complete SERCA inhibition with thapsigargin. [Ca2+]SRT was calculated as follows: [Ca2+]SRT = Bmax/(1 + Kd/[Ca2+]SR) + [Ca2+]SR, where Bmax and Kd are 2,700 and 630 μM, respectively (31). The rate of SR Ca2+ leak (d[Ca2+]SRT/dt) was plotted as a function of [Ca2+]SR for each time point (15 s) during [Ca2+]SR decline. Changes in SR Ca2+ leak are presented as relative changes in the rate of leak (d[Ca2+]SRT/dt) for a given [Ca2+]SR beam. All two-dimensional and line-scan measurements for [Ca2+]SR were analyzed with ImageJ software (National Institutes of Health).
Western blot analysis of the RyR.
Equal amounts of myocyte suspensions (control and I/R) were treated with isoproterenol (ISO) under the same experimental conditions used for our Ca2+ concentration measurements. After treatment, cells were quickly settled down and lysed in Laemmli buffer (Sigma-Aldrich). The same amount of total lysate from each sample was subjected to 4–15% SDS-PAGE and transferred to nitrocellulose membranes. RyR phosphorylation at the PKA (Ser2809) and Ca2+/calmodulin-dependent protein kinase II (Ser2815) sites were quantified using phosphospecific antibodies. Western blot assay was performed with the primary antibody RyR-pS2809 (Badrilla) or RyR-pS2815 (kindly provided by Dr. Dmitry Terentyev, Brown University). Levels of phosphorylation were normalized to the maximum phosphorylation level (Fmax). Fmax was measured after treatment of myocytes with 10 μM ISO in the presence of phosphatase inhibitors okadaic acid (5 nM) and calyculin A (1 nM). The signal was normalized to total RyR level measured with the primary antibody C34 (DSHB). The secondary antibody was horseradish peroxidase conjugated; therefore, RyR bands were visualized using Luminata Forte Western HRP Substrate (Millipore), and signals were quantified using the UVP EpiChemi3 imaging system and ImageJ software.
Measurements of the GSSG-to-total glutathione ratio.
Equal amounts of myocyte suspensions (control and I/R) were quickly settled down and homogenized using a bead-beater homogenizer. The addition of 5-sulfo-salicylic acid dihydrate [5% (wt/vol)] was used to precipitate protein from homogenate samples. Samples were centrifuged at 14,000 rpm at 4°C for 10 min. The resulting supernatant was used for the analysis of the ratio of GSSG to total glutathione. The concentration of reduced glutathione (GSH) was defined using fluorometric glutathione detection assay DetectX (Arbor Assays), using a fluorescent label that covalently binds to GSH. The sample fluorescence was measured using a fluorometer (OLIS DM 45) at an excitation/emission of 390/510 nm. A standard plot of known GSH concentrations was developed before each set of experiments. After GSH fluorescence was determined for each sample, the subsequent reduction of GSSG by glutathione reductase yielded total glutathione fluorescence. Absolute concentrations were determined based on the linear fit of the standard fluorescence. The GSSG concentration was quantified as follows: GSSG concentration = (total glutathione − free GSH)/2. Results are presented as changes in the GSSG-to-total glutathione ratio.
Measurements of free thiol content of RyRs.
The content of free thiols in RyRs was measured with the monobromobimane (mBB) fluorescence method as previously described (6, 19). The maximal free thiol content (maximal reduction) was determined after treatment of cells with a strong reducing agent, DTT (5 mM). The minimal free thiol content (maximal oxidation) was measured after incubation of cells with a strong oxidant, 2.2′-dithiodipyridine (0.5 mM). Afterward, cells were permeabilized with saponin, incubated with mBB (400 μM) for 1 h, and then washed three times to eliminate any extra unbound mBB. Cells were then lysed in Laemmli buffer, and an equal amount of lysate was loaded on two separate 4–15% SDS-PAGEs. One gel was used to measure mBB fluorescence with the UVP EpiChemi3 imaging system. The second gel was stained with Coomassie blue or used for the Western blot analysis. The nitrocellulose was probed with the primary antibody against RyR C34. These gels were used to normalize the mBB signal to the total RyR level. the level of RyR oxidation was measured as a decrease of the mBB signal at the 560-kDa band compared with the maximal free thiol level of RyR.
Statistics.
Data are presented as means ± SE of n measurements or averaged values from each myocyte isolation. Statistical comparisons between groups were performed with a Student’s t-test for paired or unpaired data sets. Differences were considered statistically significant at P < 0.05. Comparisons among multiple sample groups were performed with one-way ANOVA. Statistical analysis and graphical representation of averaged data were carried out on OriginPro7.5 software (OriginLab).
RESULTS
Effects of I/R on whole heart function.
In these experiments, a low-flow ischemia model of the Langendorff-perfused rabbit heart was used to ensure that all ventricular myocytes isolated for biochemical and functional experiments had undergone I/R. LVP and the ECG were monitored throughout the entire experiment. Figure 1A shows the experimental protocol and corresponding changes in LVP. Figure 1B shows representative recordings of the ECG and LVP under control conditions (Fig. 1Bi), during low-low ischemia (Fig. 1B, ii and iii), and during reperfusion (Fig. 1Biv). Because the ECG electrodes were placed across the ventricles, no P waves can be discerned on the recording. However, QRS complexes and T waves were clearly observed so that the rate of pacemaker automaticity and regular conduction pathway could be defined. Based on the polarity and the duration and intrinsic frequency of the QRS complex, ventricular ectopic beats (not originating nor following the normal conduction pathway) were recorded and analyzed. Changes in LVP were calculated during the ischemia and reperfusion phases relative to the average pressure recorded under control conditions (~100 mmHg). With low-flow ischemia, the relative change in LVP (Fig. 2A) and pacemaker automaticity were markedly decreased (Fig. 2B), whereas the propensity of arrhythmogenic events increased substantially (Fig. 2C). With reperfusion, the relative heart rate was significantly increased, and arrhythmias were observed in all hearts used for single myocyte isolation and experimentation. Although LVP was significantly decreased during ischemia, no significant difference was observed in LVP during reperfusion compared with the control steady state.
Fig. 1.

Recordings of left ventricular pressure (LVP) and electrocardiogram (ECG) during low-flow ischemia and reperfusion in Langendorff-perfused rabbit hearts. A: LVP recording under control (Ctrl) conditions and during low-flow ischemia and reperfusion. The experimental protocol to induce ischemia-reperfusion is shown above the LVP recording. To induce low-low ischemia, flux was reduced to 4 ml/min for 20 min. Flux was then restored to the control value (25 ml/min) for 15 min (reperfusion). Simultaneous ECG recording is not shown in this recording. B: representative recordings of ECG and LVP under control conditions (i), during low-flow ischemia (ii and iii), and during reperfusion (iv). The recordings were made at points depicted by the arrows in A. Episodes of arrhythmias are marked by gray boxes.
Fig. 2.

Effect of low-flow ischemia and reperfusion on left ventricular pressure (LVP), electrocardiogram (ECG), and arrhythmia in Langendorff-perfused rabbit hearts. A−C: average effects of low-flow ischemia (Isch) and reperfusion (Rep) on LVP (A), ECG (B), and the propensity of arrhythmia (C). Results were obtained from 13 animals. N/D, not defined. #P < 0.05 vs. control (Ctrl).
Effects of I/R on the cytosolic redox status and RyR2 oxidation.
To examine the effect of low-flow I/R on the intracellular redox environment, the GSSG-to-total glutathione ratio was measured from cardiomyocytes isolated from control and I/R hearts. GSH is the main cytosolic redox buffer, and an increase in GSSG concentration is indicative of oxidative stress (18). Analysis of GSH and total glutathione fluorescence were used to determine the GSSG concentration (GSSG concentration = (total glutathione concentration – GSH concentration)/2. We found that the GSSG-to-total glutathione ratio was significantly elevated after I/R (Fig. 3A), confirming that low-flow I/R causes oxidative stress in ventricular myocytes. To determine if oxidation of RyRs took place as a result of I/R, changes in the relative free thiol content of RyRs were measured in isolated myocytes from control and I/R hearts. Using the mBB assay to label protein free thiols, we found that RyR free thiol content was decreased almost twice in myocytes isolated from I/R hearts (Fig. 3B), indicating a significant level of RyR oxidation. Treatment of myocytes from ischemic hearts with the reducing agent mercaptopropionylglycine (MPG; 100 μM) abolished RyR oxidization induced by I/R.
Fig. 3.
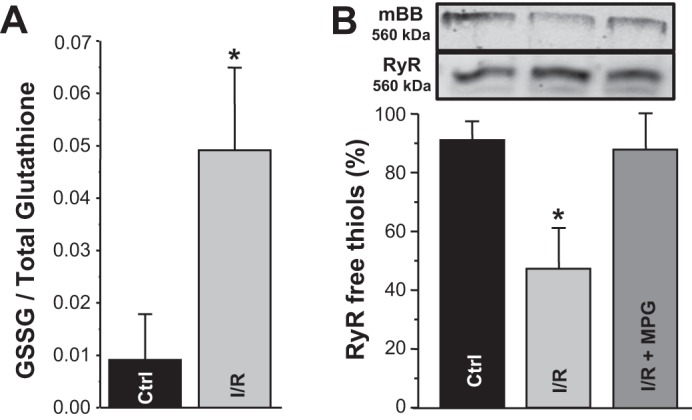
Changes in the GSH redox potential and ryanodine receptor (RyR) oxidation induced by ischemia-reperfusion (I/R). A: GSSG-to-total glutathione ratio in control (Ctrl) and I/R ventricular myocytes. The ratio was measured using the fluorometric detection assay in myocytes isolated from 4 control hearts and 4 I/R hearts. B: RyR free thiol content in control and I/R ventricular myocytes. Top: original gels of mBB and anti-RyR at 560 kDa. Bottom: changes in the free thiol content (measured with the monobromobimane assay) in control myocytes, I/R myocytes, and I/R myocytes treated with mercaptopropionylglycine (MPG; 100 μM). Myocytes were isolated from 4 control hearts and 4 I/R hearts. Maximal and minimal free thiol content were determined after treatment of cells with DTT (5 mM) and 2.2′-dithiodipyridine (0.5 mM), respectively. #P < 0.05 vs. control.
Effects of I/R on SR Ca2+ fractional release and SR Ca2+ leak.
To assess the effect of I/R on SR Ca2+ handling, AP- and caffeine-induced Ca2+ transients were measured in ventricular myocytes isolated from control and low-flow I/R hearts (Fig. 4A). Myocytes were constantly paced at 0.75 Hz except during caffeine application. The amplitude of AP-induced Ca2+ transients in I/R myocytes was relatively similar to the amplitude recorded in control myocytes (Fig. 4, A and B). However, SR Ca2+ load was significantly lower in I/R myocytes (Fig. 4, A and C). As a result, SR Ca2+ fractional release was significantly higher in I/R myocytes compared with control myocytes (Fig. 4D). In these experiments, SR Ca2+ load was measured as the amplitude of the cytosolic Ca2+ transient induced by caffeine (10 mM) application. Fractional release was calculated as the ratio between the amplitude of AP-induced and caffeine-induced Ca2+ transients. The decay of AP-induced Ca2+ transients was 2.8 times slower in I/R myocytes than in control myocytes. Treatment of I/R myocytes with MPG (100 μM) partially normalized the decay rate to the control level.
Fig. 4.

Changes in Ca2+ transient amplitude, sarcoplasmic reticulum (SR) Ca2+ load, and fractional intra-SR free Ca2+ concentration ([Ca2+]SR) release induced by ischemia-reperfuion (I/R). A: representative traces of cytosolic Ca2+ transients at a pacing frequency of 0.75 Hz followed by application of caffeine (10 mM) in ventricular myocytes isolated from control (Ctrl) and I/R hearts. [Ca2+]i, cytosolic Ca2+ concentration; F, fluorescence level; F0, florescence level at rest. B−D: changes in Ca2+ transient amplitude (B), SR Ca2+ load (C), and fractional SR Ca2+ release in control (n = 20 myocytes, 4 animals) and I/R myocytes (n = 25 myocytes, 5 animals) (D). #P < 0.05 vs. control.
We also studied how I/R affects RyR-mediated SR Ca2+ leak in ventricular myocytes. The low-affinity Ca2+ dye fluo 5N trapped into the SR was used to track changes in [Ca2+]SR. SR Ca2+ leak was measured as the rate of [Ca2+]SR decline after SERCA inhibition with thapsigargin (Fig. 5A) (44). Because the SR Ca2+ leak rate was analyzed as a function of [Ca2+]SR, changes in SR Ca2+ leak between control and I/R myocytes were compared at the same [Ca2+]SR (Fig. 5B). We found that SR Ca2+ leak was higher in I/R myocytes almost at all measured [Ca2+]SR. As a result, diastolic SR Ca2+ load was significantly lower in I/R myocytes. All together, these results demonstrate that systolic SR Ca2+ release and diastolic SR Ca2+ leak are enhanced in myocytes exposed to I/R. Despite the increased RyR activity, spontaneous Ca2+ waves were rarely observed in I/R myocytes.
Fig. 5.

Changes in sarcoplasmic reticulum (SR) Ca2+ leak induced by ischemia-reperfusion (I/R). A: changes of intra-SR free Ca2+ concentration ([Ca2+]SR) during electrical stimulation at a pacing frequency of 0.75 Hz and during rest in the presence of thapsigargin (TG) in control (Ctrl; black dashed line) and I/R myocytes (light gray circles). B: SR Ca2+ leak rate as a function of [Ca2+]SR in control (n = 18 myocytes, 4 animals) and I/R myocytes (n = 12 myocytes, 3 animals). SR Ca2+ leak was measured as the rate of decline of [Ca2+]SR after SERCA inhibition with TG. SR Ca2+ leak in control and I/R myocytes was compared at the same level of [Ca2+]SR. #P < 0.05 vs. control.
Effect of I/R on SR Ca2+ release during β-AR activation.
According to previous publications (20, 21, 25, 26), β-AR activation is responsible for the occurrence of arrhythmias during I/R in whole heart experiments (Fig. 1). Therefore, in the next set of experiments, we examined how ex vivo stimulation of β-ARs with ISO affects SR Ca2+ handling in control and I/R myocytes. Figure 6A, top, shows that ISO (10 nM) increased systolic Ca2+ transient amplitude, accelerated the decay of Ca2+ transients, and decreased diastolic [Ca2+]i in control myocytes. Spontaneous Ca2+ waves were rarely observed during β-AR activation with 10 nM ISO in control myocytes (Fig. 6B). However, in I/R myocytes, the same concentration of ISO produced spontaneous Ca2+ waves almost in every studied cell (Fig. 6A, middle). Ca2+ wave frequency reached a maximal level (~35% or 1 wave/3 transients) after 6 min of β-AR stimulation (Fig. 6B). Incubation of I/R myocytes with the reducing agent MPG (100 μM) slightly decreased Ca2+ transient amplitude but did not change SR Ca2+ load (results are not shown). The subsequent application of ISO (10 nM) in the presence of MPG increased SR Ca2+ release during APs, without producing Ca2+ waves (Fig. 6A, bottom). Average results of ISO effect on Ca2+ wave frequency in control and I/R myocytes are shown in Fig. 6B. These results suggest that β-AR activation does not trigger spontaneous Ca2+ waves unless myocytes are previously subjected to I/R-induced oxidation stress.
Fig. 6.

Effects of β-adrenergic activation on Ca2+ transients and Ca2+ waves in control (Ctrl) and ischemia-reperfused (I/R) myocytes. A: representative traces of cytosolic Ca2+ transients (at a pacing frequency of 0.75 Hz) recorded at different times of isoproterenol (ISO; 10 nM) application in control myocytes, I/R myocytes, and I/R myocytes treated with mercaptopropionylglycine (I/R + MPG). The arrows indicate spontaneous Ca2+ waves. B: ISO effect on Ca2+ wave propensity in control myocytes, I/R myocytes, and I/R myocytes in the presence of MPG. Ca2+ wave propensity is presented as the number of waves per pacing interval (1.33 s). The analysis was based on results obtained from 12 control and 18 I/R myocytes. [Ca2+]i, cytosolic Ca2+ concentration; F, fluorescence level; F0, florescence level at rest. #P < 0.05 vs. control.
Effect of I/R on RyR phosphorylation.
In whole hearts, I/R can increase RyR phosphorylation at the PKA site (Ser2809) as a result of the local adrenergic stimulation. Although β-AR activation is no longer present in isolated myocytes, an elevated level of RyR phosphorylation might persist after myocyte isolation. Thus, ex vivo β-AR activation in I/R myocytes would have a higher chance to reach the critical level of RyR phosphorylation at which spontaneous Ca2+ waves become inevitable. In this set of experiments, we studied whether the increased propensity of spontaneous Ca2+ waves during ISO application is associated with increased RyR phosphorylation in I/R myocytes. Figure 7A shows images of representative Western blots with the phosphospecific antibody (RyR-pS2809) against the PKA site in control and I/R myocytes. In both groups, myocytes were paced at 0.75 Hz under control conditions (pacing) or during β-AR activation (+ISO). Western blot analysis did not detect any significant difference in RyR phosphorylation between control and I/R myocytes under control conditions (Fig. 7B, pacing). However, RyR phosphorylation after β-AR activation was slightly decreased in I/R myocytes compared with control myocytes. RyR phosphorylation at the Ca2+/calmodulin-dependent protein kinase II site was not significantly different between control and I/R myocytes (data not shown). Because samples for the Western blot analysis were collected 30 min after reperfusion, these results suggest that any changes in RyR phosphorylation induced by I/R (presumably because of high adrenergic tone) return to its control level during this time. Thus, an increase of RyR phosphorylation by itself cannot be the key factor that triggers Ca2+ waves and arrhythmias during I/R, because β-AR activation produced a similar effect on RyR phosphorylation in control and I/R myocytes.
Fig. 7.
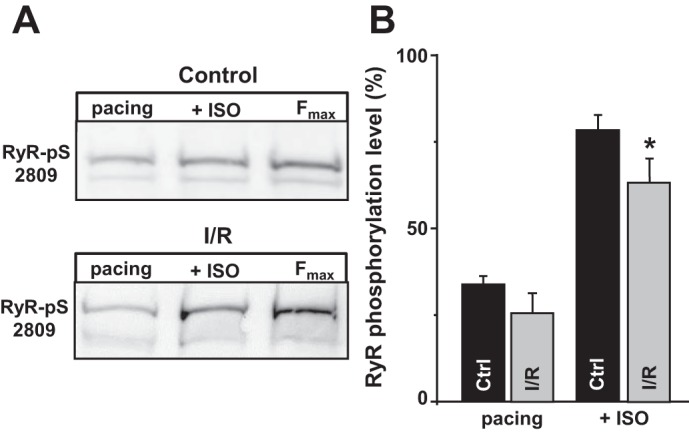
Effects of β-adrenergic activation on ryanodine receptor (RyR) phosphorylation at the PKA site in control (Ctrl) and ischemia-reperfused (I/R) myocytes. A: representative Western blots showing RyR phosphorylation at the PKA site (Ser2809) in control and I/R myocytes during electrical pacing (pacing) and electrical pacing in the presence of isoproterenol (+ISO). RyR phosphorylation was normalized to the maximum phosphorylation level (ISO + phosphatase inhibitors). B: changes in RyR phosphorylation level at the PKA site in control (n = 4 animals) and I/R myocytes (n = 4 animals) during electrical pacing (pacing) and during ISO application. Fmax, maximum phosphorylation level. #P < 0.05 vs. control.
Effect of RyR oxidation on SR Ca2+ release during β-AR activation.
In the following experiments, we studied whether oxidative stress can trigger proarrhythmogenic Ca2+ waves during β-AR activation in control myocytes. To induce oxidative stress, control myocytes were treated with H2O2 (50 μM) for 5 min. We found that treatment of control myocytes with H2O2 significantly increased SR Ca2+ leak over a whole range of SR Ca2+ loads (Fig. 8A). Despite the decreased SR Ca2+ load (Fig. 8A), H2O2 did not significantly change Ca2+ transient amplitude (Fig. 8B). These results suggest that SR Ca2+ fractional release is increased during RyR oxidation (similar to I/R; Fig. 4D). Initially, ISO (10 nM) increased Ca2+ transient amplitude (Fig. 8B) without triggering frequent Ca2+ weaves (only 1 wave/10 transients). However, 4 min after ISO application, Ca2+ wave frequency substantially increased, reaching a level of 60% (6 waves/10 transients). Because ISO alone does not trigger Ca2+ waves in control myocytes (Fig. 6), these results suggest that oxidative stress plays the key role in the transition from positive inotropic to arrhythmogenic effect during β-AR activation in I/R myocytes.
Fig. 8.

Effects of ryanodine receptor (RyR) oxidation on sarcoplasmic reticulum (SR) Ca2+ leak, Ca2+ transients, and Ca2+ waves in control (Ctrl) myocytes during β-adrenergic receptor (β-AR) activation. A: SR Ca2+ leak rate as a function of intra-SR free Ca2+ concentration ([Ca2+]SR) under control conditions and during H2O2 (50 μM) application (n = 12 myocytes, 3 animals). SR Ca2+ leak was measured as the rate of decline of [Ca2+]SR after SERCA inhibition. SR Ca2+ leak was compared at the same level of [Ca2+]SR. B: Ca2+ transients (pacing frequency of 0.75 Hz) recorded at different time of isoproterenol (ISO; 10 nM) application in control myocytes treated with H2O2 (50 μM). The arrows indicate spontaneous Ca2+ waves. [Ca2+]i, cytosolic Ca2+ concentration; F, fluorescence level; F0, florescence level at rest. C: ISO effect on Ca2+ wave propensity under control conditions and during H2O2 application (n = 16 myocytes, 4 animals). #P < 0.05 vs. control.
DISCUSSION
Ventricular tachyarrhythmias remain a common cause of sudden death during MI (17). The underlying arrhythmogenic mechanism is frequently attributed to spontaneous APs because of triggered activity (40). It has been shown that spontaneous SR Ca2+ release during diastole plays a key role in the triggered activity. During spontaneous Ca2+ waves, Ca2+ extrusion by the electrogenic Na+/Ca2+ exchanger can generate DADs with amplitudes that are large enough to trigger premature APs (30, 33). Although the defective Ca2+ regulation is well recognized as an important proarrhythmogenic factor during I/R, specific mechanisms of SR Ca2+ mishandling are not fully understood.
To gain a better understanding of cellular and molecular mechanisms of SR Ca2+ regulation during I/R, a majority of experiments were conducted in isolated ventricular myocytes. To ensure that all myocytes used for single cell experiments were exposed to I/R, the low-flow ischemia model (13, 27, 29) was applied to Langendorff-perfused rabbit hearts. We found that during low-flow ischemia, heart rate and LV contraction were significantly decreased. During reperfusion, however, the increased heart rate was accompanied by frequent arrhythmias (Fig. 1). Based on the polarity, the amplitude, the duration, and the intrinsic frequency of the QRS complex, we concluded that the observed arrhythmias originated within the ventricles. It has been shown that I/R is also associated with complex changes in the cellular metabolism, including intracellular acidification, decrease in ATP concentration, and increase of Mg2+ concentration, ADP concentration, and Pi concentration (10). All these metabolic changes can affect [Ca2+]i regulation and, therefore, may cause SR Ca2+ mishandling and arrhythmias during ischemia. Previous studies of a similar I/R model revealed that all these factors are restored back to a normal level within 2–3 min of reperfusion (13, 29). Because tachyarrhythmias persisted during the entire period of reperfusion (>15 min; Fig. 1), we suggest that other proarrhythmogenic mechanisms remain active for an extensive period time after reperfusion.
Oxidative stress, because of an increased supply of oxygen to the ischemic myocardium, has been implicated as the underlying factor that promotes I/R injury (38, 46). For many cells, including cardiomyocytes, glutathione is considered the major cytosolic redox buffer (18). Under normal physiological conditions, the cytosolic glutathione pool is mainly maintained at the reduced state (GSH). High GSH concentration contributes significantly to the cytosolic antioxidant defense and an increase in GSSG is indicative of oxidative stress (7, 11, 35, 39). We found that in myocytes isolated from I/R hearts, the GSSG-to-total glutathione ratio was significantly increased (Fig. 3). Although a mechanism of oxidative stress has not been identified in this study, it has been shown that several ROS sources are involved in oxidative stress during I/R. In the ischemic myocardium, the electron transport chain in the mitochondria becomes uncoupled and incompletely reduces O2, forming superoxide radicals (37). This sudden burst of mitochondrial ROS overwhelms the intrinsic antioxidant system. The decreased GSH concentration during I/R (11, 39) can also contribute to mitochondrial ROS spillover (2, 8). Other sources of ROS, including NADPH oxidase, uncoupled nitric oxide synthase, and xanthine oxidase, are also believed to play a role in I/R injury (1, 3, 47).
As a result of oxidative stress, RyR cysteines become partially oxidized during I/R (Fig. 3). It is well established that changes in the cysteine redox potential affect the activity of many channels, including RyR (43, 45). There are three major types of redox modification of RyR: S-nitrosylation, S-glutathionylation, and disulfide bond formation. These modifications can be reversed by reducing agents and specific enzymes. Oxidation of cysteines can also cause formation of sulfinic and sulfonic acids. Sulfinic acids can be reduced to free thiol by DTT or other reducing agents. Sulfinic acids can be further oxidized to produce sulfonic acids. Although these redox modifications are irreversible, sulfonic acids can only be induced by very strong oxidants. In this study, we did not identify the specific mechanism of RyR redox modification. It is possible that several different mechanisms are involved in RyR modification during I/R. More important is, however, that MPG reversed RyR oxidation (Fig. 3B) and prevented Ca2+ waves during adrenergic stimulation (Fig. 6), suggesting that these two mechanisms are causally related.
Consistent with previous studies (5, 7, 23), we found that RyR oxidation during I/R contributes to the increased SR Ca2+ leak and decreased SR Ca2+ load (Fig. 5). However, AP-induced Ca2+ transients in I/R myocytes had relatively similar amplitude to control myocytes (Fig. 4). It appears that the increased RyR activity can partially normalize Ca2+ transient amplitude at depleted SR Ca2+ load by increasing the fractional SR Ca2+ release. Surprisingly, no spontaneous Ca2+ waves were recorded during diastole in I/R myocytes. These results seem to be paradoxical in comparison with the data obtained in the whole heart experiments (Fig. 1), where arrhythmias were very frequent during reperfusion. The difference could be explained by β-AR activation in the intact myocardium. The heart is equipped with sympathetic nerve endings that release norepinephrine to produce positive inotropic and dromotropic effects. It has been shown in both in vivo and ex vivo studies that this effect is particularly exacerbated during I/R to compensate negative inotropic and dromotropic effects of metabolites (e.g., adenosine and lactate) (20, 21). Therefore, in the whole heart, high adrenergic tone during reperfusion would increase [Ca2+]SR sufficiently to produce spontaneous Ca2+ waves and arrhythmias. Indeed, β-blockers can effectively suppress arrhythmias during I/R in whole heart experiments (25, 26). In isolated I/R myocytes, however, β-AR activation is no longer present (Fig. 7). Consequently, SR Ca2+ load is too low to trigger Ca2+ waves, despite RyRs being more active because of its oxidation. However, ex vivo β-AR activation with ISO (10 nM) in I/R myocytes triggered Ca2+ waves in every studied cell (Fig. 6). The same dose of ISO produced only a positive inotropic effect in control myocytes. Treatment of I/R myocytes with the reducing agent attenuated RyR oxidization and decreased Ca2+ wave frequency during β-AR activation (Fig. 6). On the other hand, treatment of myocytes from nonischemic hearts with H2O2 to induce oxidative stress triggered Ca2+ waves during β-AR activation (Fig. 8). These results suggest that RyR oxidation during I/R plays an important role in the transition from positive inotropic to proarrhythmogenic effects during β-AR activation. Our findings are in agreement with recent studies of Ca2+ regulation and arrhythmias in a post-MI dog model (4, 5). The authors showed that DADs and arrhythmogenesis induced by MI are associated with RyR oxidation. Furthermore, we have previously shown that RyR oxidation plays a decisive role in the generation of spontaneous Ca2+ waves during prolonged and excessive β-AR stimulation (6).
In conclusion, the results of the present study describe mechanisms underlying the generation of proarrhythmogenic Ca2+ waves in ventricular myocytes exposed to I/R. It appears that the redox status of the RyR plays an important role in a response of the SR Ca2+-handling machinery to β-AR activation. At elevated SR Ca2+ load (because of β-AR-mediated SERCA activation), RyR oxidation during I/R can increase SR Ca2+ leak to a critical level that can trigger propagating Ca2+ waves. Thus, preventing RyR oxidation can be a promising therapeutic strategy to inhibit arrhythmias and preserve positive inotropic effect of β-AR activation during MI.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant HL-130231 (to A. V. Zima) and an American Heart Association postdoctoral award (to E. Bovo).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.B., S.R.M., and A.V.Z. conceived and designed research; E.B., S.R.M., and A.V.Z. performed experiments; E.B., S.R.M., and A.V.Z. analyzed data; E.B., S.R.M., and A.V.Z. interpreted results of experiments; E.B., S.R.M., and A.V.Z. prepared figures; E.B., S.R.M., and A.V.Z. drafted manuscript; E.B., S.R.M., and A.V.Z. edited and revised manuscript; E.B., S.R.M., and A.V.Z. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Dmitry Terentyev (Brown University) for providing the phospho-specific antibody agonist Ser2815 RyR2.
REFERENCES
- 1.Angelos MG, Kutala VK, Torres CA, He G, Stoner JD, Mohammad M, Kuppusamy P. Hypoxic reperfusion of the ischemic heart and oxygen radical generation. Am J Physiol Heart Circ Physiol 290: H341–H347, 2006. doi: 10.1152/ajpheart.00223.2005. [DOI] [PubMed] [Google Scholar]
- 2.Aon MA, Cortassa S, O’Rourke B. Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta 1797: 865–877, 2010. doi: 10.1016/j.bbabio.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res 61: 461–470, 2004. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 4.Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, Carnes CA, Billman GE, Györke S. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res 110: 569–577, 2012. doi: 10.1161/CIRCRESAHA.111.260455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belevych AE, Terentyev D, Viatchenko-Karpinski S, Terentyeva R, Sridhar A, Nishijima Y, Wilson LD, Cardounel AJ, Laurita KR, Carnes CA, Billman GE, Gyorke S. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc Res 84: 387–395, 2009. doi: 10.1093/cvr/cvp246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bovo E, Lipsius SL, Zima AV. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during β-adrenergic receptor stimulation in rabbit cardiomyocytes. J Physiol 590: 3291–3304, 2012. doi: 10.1113/jphysiol.2012.230748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bovo E, Mazurek SR, Zima AV. The role of RyR2 oxidation in the blunted frequency-dependent facilitation of Ca2+ transient amplitude in rabbit failing myocytes. Pflugers Arch 470: 959–968, 2018. doi: 10.1007/s00424-018-2122-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown DA, Aon MA, Frasier CR, Sloan RC, Maloney AH, Anderson EJ, O’Rourke B. Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J Mol Cell Cardiol 48: 673–679, 2010. doi: 10.1016/j.yjmcc.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cannell MB, Cheng H, Lederer WJ. Spatial non-uniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Biophys J 67: 1942–1956, 1994. doi: 10.1016/S0006-3495(94)80677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev 79: 917–1017, 1999. doi: 10.1152/physrev.1999.79.3.917. [DOI] [PubMed] [Google Scholar]
- 11.Ceconi C, Curello S, Cargnoni A, Ferrari R, Albertini A, Visioli O. The role of glutathione status in the protection against ischaemic and reperfusion damage: effects of N-acetyl cysteine. J Mol Cell Cardiol 20: 5–13, 1988. doi: 10.1016/S0022-2828(88)80174-3. [DOI] [PubMed] [Google Scholar]
- 12.Christensen NJ, Videbaek J. Plasma catecholamines and carbohydrate metabolism in patients with acute myocardial infarction. J Clin Invest 54: 278–286, 1974. doi: 10.1172/JCI107763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cross HR, Clarke K, Opie LH, Radda GK. Is lactate-induced myocardial ischaemic injury mediated by decreased pH or increased intracellular lactate? J Mol Cell Cardiol 27: 1369–1381, 1995. doi: 10.1006/jmcc.1995.0130. [DOI] [PubMed] [Google Scholar]
- 14.Domeier TL, Blatter LA, Zima AV. Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. J Physiol 587: 5197–5209, 2009. doi: 10.1113/jphysiol.2009.177576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Domenech RJ, Macho P, Vélez D, Sánchez G, Liu X, Dhalla N. Tachycardia preconditions infarct size in dogs: role of adenosine and protein kinase C. Circulation 97: 786–794, 1998. doi: 10.1161/01.CIR.97.8.786. [DOI] [PubMed] [Google Scholar]
- 16.Drummond GB. Reporting ethical matters in the Journal of Physiology: standards and advice. J Physiol 587: 713–719, 2009. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gheeraert PJ, De Buyzere ML, Taeymans YM, Gillebert TC, Henriques JP, De Backer G, De Bacquer D. Risk factors for primary ventricular fibrillation during acute myocardial infarction: a systematic review and meta-analysis. Eur Heart J 27: 2499–2510, 2006. doi: 10.1093/eurheartj/ehl218. [DOI] [PubMed] [Google Scholar]
- 18.Griffith OW. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic Biol Med 27: 922–935, 1999. doi: 10.1016/S0891-5849(99)00176-8. [DOI] [PubMed] [Google Scholar]
- 19.Kosower NS, Kosower EM. Thiol labeling with bromobimanes. Methods Enzymol 143: 76–84, 1987. doi: 10.1016/0076-6879(87)43015-2. [DOI] [PubMed] [Google Scholar]
- 20.Kurz T, Offner B, Schreieck J, Richardt G, Tölg R, Schömig A. Nonexocytotic noradrenaline release and ventricular fibrillation in ischemic rat hearts. Naunyn Schmiedebergs Arch Pharmacol 352: 491–496, 1995. doi: 10.1007/BF00169382. [DOI] [PubMed] [Google Scholar]
- 21.Lameris TW, de Zeeuw S, Alberts G, Boomsma F, Duncker DJ, Verdouw PD, Veld AJ, van Den Meiracker AH. Time course and mechanism of myocardial catecholamine release during transient ischemia in vivo. Circulation 101: 2645–2650, 2000. doi: 10.1161/01.CIR.101.22.2645. [DOI] [PubMed] [Google Scholar]
- 22.Makaula S, Lochner A, Genade S, Sack MN, Awan MM, Opie LH. H-89, a non-specific inhibitor of protein kinase A, promotes post-ischemic cardiac contractile recovery and reduces infarct size. J Cardiovasc Pharmacol 45: 341–347, 2005. doi: 10.1097/01.fjc.0000156825.80951.14. [DOI] [PubMed] [Google Scholar]
- 23.Mazurek SR, Bovo E, Zima AV. Regulation of sarcoplasmic reticulum Ca2+ release by cytosolic glutathione in rabbit ventricular myocytes. Free Radic Biol Med 68: 159–167, 2014. doi: 10.1016/j.freeradbiomed.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 24.Misra MK, Sarwat M, Bhakuni P, Tuteja R, Tuteja N. Oxidative stress and ischemic myocardial syndromes. Med Sci Monit 15: RA209–RA219, 2009. [PubMed] [Google Scholar]
- 25.Murphy SR, Wang L, Wang Z, Domondon P, Lang D, Habecker BA, Myles RC, Ripplinger CM. β-Adrenergic inhibition prevents action potential and calcium handling changes during regional myocardial ischemia. Front Physiol 8: 630, 2017. doi: 10.3389/fphys.2017.00630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Norris RM, Barnaby PF, Brown MA, Geary GG, Clarke ED, Logan RL, Sharpe DN. Prevention of ventricular fibrillation during acute myocardial infarction by intravenous propranolol. Lancet 324: 883–886, 1984. doi: 10.1016/S0140-6736(84)90651-2. [DOI] [PubMed] [Google Scholar]
- 27.Owen P, Dennis S, Opie LH. Glucose flux rate regulates onset of ischemic contracture in globally underperfused rat hearts. Circ Res 66: 344–354, 1990. doi: 10.1161/01.RES.66.2.344. [DOI] [PubMed] [Google Scholar]
- 28.Sánchez G, Pedrozo Z, Domenech RJ, Hidalgo C, Donoso P. Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J Mol Cell Cardiol 39: 982–991, 2005. doi: 10.1016/j.yjmcc.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 29.Schaefer S, Prussel E, Carr LJ. Requirement of glycolytic substrate for metabolic recovery during moderate low flow ischemia. J Mol Cell Cardiol 27: 2167–2176, 1995. doi: 10.1016/S0022-2828(95)91407-2. [DOI] [PubMed] [Google Scholar]
- 30.Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ Res 87: 774–780, 2000. doi: 10.1161/01.RES.87.9.774. [DOI] [PubMed] [Google Scholar]
- 31.Shannon TR, Ginsburg KS, Bers DM. Reverse mode of the sarcoplasmic reticulum calcium pump and load-dependent cytosolic calcium decline in voltage-clamped cardiac ventricular myocytes. Biophys J 78: 322–333, 2000. doi: 10.1016/S0006-3495(00)76595-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spear JF, Prabu SK, Galati D, Raza H, Anandatheerthavarada HK, Avadhani NG. β1-Adrenoreceptor activation contributes to ischemia-reperfusion damage as well as playing a role in ischemic preconditioning. Am J Physiol Heart Circ Physiol 292: H2459–H2466, 2007. doi: 10.1152/ajpheart.00459.2006. [DOI] [PubMed] [Google Scholar]
- 33.Spencer CI, Sham JS. Effects of Na+/Ca2+ exchange induced by SR Ca2+ release on action potentials and afterdepolarizations in guinea pig ventricular myocytes. Am J Physiol Heart Circ Physiol 285: H2552–H2562, 2003. doi: 10.1152/ajpheart.00274.2003. [DOI] [PubMed] [Google Scholar]
- 34.Stadtman ER, Berlett BS. Reactive oxygen-mediated protein oxidation in aging and disease. Drug Metab Rev 30: 225–243, 1998. doi: 10.3109/03602539808996310. [DOI] [PubMed] [Google Scholar]
- 35.Terentyev D, Györke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Györke S. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res 103: 1466–1472, 2008. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol 279: L1005–L1028, 2000. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 37.Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep 17: 3–8, 1997. doi: 10.1023/A:1027374931887. [DOI] [PubMed] [Google Scholar]
- 38.Vanden Hoek TL, Shao Z, Li C, Zak R, Schumacker PT, Becker LB. Reperfusion injury on cardiac myocytes after simulated ischemia. Am J Physiol Heart Circ Physiol 270: H1334–H1341, 1996. doi: 10.1152/ajpheart.1996.270.4.H1334. [DOI] [PubMed] [Google Scholar]
- 39.Werns SW, Fantone JC, Ventura A, Lucchesi BR. Myocardial glutathione depletion impairs recovery of isolated blood-perfused hearts after global ischaemia. J Mol Cell Cardiol 24: 1215–1220, 1992. doi: 10.1016/0022-2828(92)93088-2. [DOI] [PubMed] [Google Scholar]
- 40.Xing D, Martins JB. Triggered activity due to delayed afterdepolarizations in sites of focal origin of ischemic ventricular tachycardia. Am J Physiol Heart Circ Physiol 287: H2078–H2084, 2004. doi: 10.1152/ajpheart.00027.2004. [DOI] [PubMed] [Google Scholar]
- 41.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279: 234–237, 1998. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 42.Yavuz S. Surgery as early revascularization after acute myocardial infarction. Anadolu Kardiyol Derg 8, Suppl 2: 84–92, 2008. [PubMed] [Google Scholar]
- 43.Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res 71: 310–321, 2006. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 44.Zima AV, Bovo E, Bers DM, Blatter LA. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol 588: 4743–4757, 2010. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zima AV, Mazurek SR. Functional impact of ryanodine receptor oxidation on intracellular calcium regulation in the heart. Rev Physiol Biochem Pharmacol 171: 39–62, 2016. doi: 10.1007/112_2016_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci USA 84: 1404–1407, 1987. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res 70: 181–190, 2006. doi: 10.1016/j.cardiores.2006.02.025. [DOI] [PubMed] [Google Scholar]