

Abstract
The mechanisms underlying Ca2+/calmodulin-dependent protein kinase II (CaMKII)-induced arrhythmias in ischemia-reperfusion (I/R) are not fully understood. We tested the hypothesis that CaMKII increases late Na+ current (INa,L) via phosphorylation of Nav1.5 at Ser571 during I/R, thereby increasing arrhythmia susceptibility. To test our hypothesis, we studied isolated, Langendorff-perfused hearts from wild-type (WT) mice and mice expressing Nav channel variants Nav1.5-Ser571E (S571E) and Nav1.5-Ser571A (S571A). WT hearts showed a significant increase in the levels of phosphorylated CaMKII and Nav1.5 at Ser571 [p-Nav1.5(S571)] after 15 min of global ischemia (just before the onset of reperfusion). Optical mapping experiments revealed an increase in action potential duration (APD) and APD dispersion without changes in conduction velocity during I/R in WT and S571E compared with S571A hearts. At the same time, WT and S571E hearts showed an increase in spontaneous arrhythmia events (e.g., premature ventricular contractions) and an increase in the inducibility of reentrant arrhythmias during reperfusion. Pretreatment of WT hearts with the Na+ channel blocker mexiletine (10 μM) normalized APD dispersion and reduced arrhythmia susceptibility during I/R. We conclude that CaMKII-dependent phosphorylation of Nav1.5 is a crucial driver for increased INa,L, arrhythmia triggers, and substrate during I/R. Selective targeting of this CaMKII-dependent pathway may have therapeutic potential for reducing arrhythmias in the setting of I/R.
NEW & NOTEWORTHY Ca2+/calmodulin-dependent protein kinase II (CaMKII) phosphorylation of Nav1.5 at Ser571 leads to a prolongation of action potential duration (APD), increased APD dispersion, and increased arrhythmia susceptibility after ischemia-reperfusion in isolated mouse hearts. Genetic ablation of the CaMKII-dependent phosphorylation site Ser571 on Nav1.5 or low-dose mexiletine (to inhibit late Na+ current) reduced APD dispersion, arrhythmia triggers, and ventricular tachycardia inducibility.
Keywords: arrhythmia, Ca2+/calmodulin-dependent protein kinase II, ischemia-reperfusion, late sodium current
INTRODUCTION
Each year, ∼1,000,000 people suffer from myocardial infarction (MI) and an additional 700,000 patients experience cardioplegic arrest during completion of a cardiac procedure (38). While rapid return of normal coronary circulation is paramount to survival of these patients, the return of blood flow itself induces damage and arrhythmia (7, 14, 41). Therefore, it is of great importance to better understand the mechanisms underlying ischemia-reperfusion (I/R) injury. Intracellular Ca2+ overload secondary to reverse-mode Na+/Ca2+ exchanger activity plays a central role in tissue damage and arrhythmias after I/R (10). A critical step that leads to reverse-mode Na+/Ca2+ exchanger activity in I/R is an initial Na+ overload arising from a variety of sources, most notably the Na+/H+ exchanger (2, 17), although the precise sequence of events remains unknown. Normal cardiac function depends on voltage-gated Na+ (Nav) channels, essential for the rapid action potential (AP) upstroke and an important source of Na+ entry into the cell (34). Under normal conditions, Nav current (INa) rapidly activates and then inactivates to allow for AP repolarization. However, even under normal (nondiseased) conditions, a minor population of Nav channels fails to inactivate, giving rise to a small Na+ current that persists throughout the AP [late Na+ current (INa,L)]. Importantly, amplification of INa,L in the setting of disease has been shown to increase arrhythmia susceptibility (33), including in I/R, where increased INa,L has been linked to elevated intracellular Na+ (45). At the same time, Ca2+/calmodulin-dependent protein kinase II (CaMKII) activity has been found to be upregulated during I/R, with possible proarrhythmic effects on several downstream targets (47), including the dominant cardiac Na+ channel isoform Nav1.5 (54). Previous work from our group has shown that CaMKII phosphorylates Nav1.5 at Ser571 to increase INa,L and arrhythmia susceptibility (19, 22, 29).
Despite considerable work in this area, a potential role for direct CaMKII-dependent phosphorylation of Nav1.5 has not been studied in I/R. To explore the importance of this pathway for I/R-induced arrhythmia, we use two knockin mouse models where the Ser571 site on Nav1.5 was either ablated (S571A) or constitutively activated [phosphomimetic (S571E)] (19). An important aspect of these models is that they allow for specific targeting of INa,L without altering other critical channel properties. We anticipate that results from this study will advance our understanding of the role of INa,L in I/R injury and identify new therapeutic strategies to reduce arrhythmias after I/R without altering normal physiology.
MATERIALS AND METHODS
Mouse models.
The following mouse models were used: Scn5a knockin mice (C57BL/6 background) with a S571E or S571A point mutation in Nav1.5 or wild-type (WT) controls (19). Adult male mice were anesthetized by isoflurane; anesthesia was confirmed by the absence of response to probing of the lower extremities. Mice were intraperitoneally injected with heparin to avoid blood clots during heart extraction and cannulation. The whole heart was extracted and perfused via the aorta using a rolling pump at 2 ml/min and oxygenated Tyrode solution [containing (in mM) 140 NaCl, 1.0 MgCl2·6H2O, 1.2 NaH2PO4, 4.0 KCl, 1.8 CaCl2·2H2O, 5.6 glucose, and 10 HEPES sodium salt] maintained at 36.5°C and titrated to pH 7.4 using HCl. Experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals following protocols that were reviewed and approved by the Institutional Animal Care and Use Committee of The Ohio State University.
Optical mapping of ex vivo heart preparations.
Optical mapping techniques were used to measure the membrane electrical potential on the anterior portion of the isolated heart. The heart was perfused for 15 min with blebbistatin (from a stock solution of 5 mg/ml in DMSO) to decrease motion for clearer signal production without significantly altering the electrophysiology (51). Perfusion continued with the voltage dye di-4-ANEPPS (4 μM) for 15 min (4, 56). Images were taken to ensure that the dye reached the extremity tissues of the organ. The fast voltage-sensitive dye was chosen based on previous work showing a good signal-to-noise ratio, negligible light interference, and reduced autofluorescence signals (which contribute to inaccurate readings) (5). Global ischemia was induced by stopping perfusion for 15 min; the heart was then reperfused with Tyrode solution for the remainder of the experiment.
To excite the dye, the LEX2-LZ4-G light source (SciMedia) was used in conjunction with a 531/40 filter. Signals were collected at 1,000 Hz (1 ms/frame) using MiCAM05 complementary metal oxide semiconductor cameras and filtered with a 600-nm long-pass filter. The area of the resulting images was 1.06 ×1.06 cm2, with 100 × 100 pixels. A custom-designed MATLAB code (provided by Dr. Steven Poelzing) was used to obtain values from the signals measured during experimentation. AP duration at 80% repolarization (APD) was measured from optical signals and reported as an average value throughout the mapping area. APD spatial heterogeneity was estimated as the standard deviation of the mean value over the mapping area. Conduction velocity was also measured in the transverse and longitudinal directions, and activation maps were created using RHYTHM (18).
Electrocardiogram activity was measured using three leads placed around the temperature-controlled dish, adjacent to the heart, and was analyzed for spontaneous arrhythmia events [e.g., premature ventricular contractions (PVCs)] throughout the I/R protocol. Arrhythmia susceptibility was also assessed after 5 min of reperfusion using a S1S2 pacing protocol. Briefly, after 6 s of S1 pacing (150-ms cycle length), a S2 beat was delivered at an initial S1S2 interval of 100 ms and then decreased by 10 ms until 1:1 capture was not possible or ventricular tachycardia (VT) was observed. Pacing was performed using a unipolar lead at the center of the anterior heart wall and a stimulus amplitude equal to twice threshold at baseline (with amplitude held constant for the entire I/R period). VT in response to S1S2 pacing was defined as three or more nonpaced beats in rapid succession.
Immunoblot analysis.
Ventricular lysates were analyzed by SDS-PAGE and immunoblotting, as described elsewhere (19, 22, 23, 29). Equal protein loading was achieved using standard BCA protocols and verified by Ponceau staining of immunoblots. The following antibodies were used: total CaMKII (Badrilla), phosphorylated CaMKIIδ antibody (Santa Cruz Biotechnology), and total and phosphorylated Nav1.5 (custom antibodies) (22).
Data and statistical analysis.
SigmaPlot 13.0 was used for statistical analysis. One-way ANOVA was used for multiple comparisons with Tukey’s test for post hoc testing. Values are means ± SE. Contingency data were analyzed using a χ2-test. The null hypothesis was rejected for P < 0.05.
RESULTS
Phosphorylation of Nav1.5 at Ser571 contributes to the generation of arrhythmias during I/R.
To test our hypothesis that increased CaMKII-dependent phosphorylation of Nav1.5 promotes increased INa,L, Na+/Ca2+ overload, and arrhythmias during I/R, we subjected isolated Langendorff-perfused hearts to global ischemia followed by reperfusion (Fig. 1). We first measured levels of total/phosphorylated CaMKII and Nav1.5 at baseline and during the I/R protocol in WT hearts but not in S571A and S571E hearts because of loss of the phosphorylated Nav epitope in these models (19). Consistent with previous findings (47), autophosphorylated (activated) CaMKII was significantly increased after 15 min of ischemia. Nav1.5 also showed a significant increase in phosphorylation at Ser571 (CaMKII site) after ischemia (Fig. 2). Surprisingly, CaMKII and Nav1.5 phosphorylation levels rapidly returned to baseline with reperfusion; normal values were restored by 5 min (Fig. 2). These results demonstrate that global I/R induces a transient increase in CaMKII-dependent phosphorylation of Nav1.5.
Fig. 1.

Top: proposed hypothesis linking Ca2+/calmodulin-dependent protein kinase II (CaMKII)-dependent phosphorylation of dominant cardiac Na+ channel isoform Nav1.5 and arrhythmia susceptibility. p-CaMKII, phosphorylated CaMKII; p-Nav1.5, phosphorylated Nav1.5; INa, Na+ current; APD, action potential duration at 80% repolarization. Bottom: experimental protocol to study arrhythmia susceptibility in isolated, Langendorff-perfused mouse hearts.
Fig. 2.
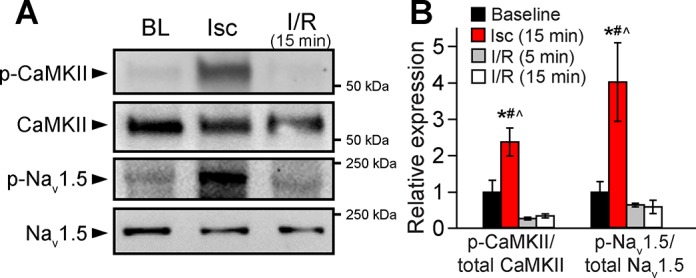
A and B: representative immunoblots and densitometric measurements for phosphorylated Ca2+/calmodulin-dependent protein kinase II (p-CaMKII) and phosphorylated Na+ channel isoform Nav1.5 (p-Nav1.5) expressed as ratio to total CaMKII and Nav1.5, respectively, in wild-type (WT) mice at baseline, after 15 min of ischemia (Isc), and after reperfusion (I/R) for 5 and 15 min. Values are means ± SE; n = 3 in each group. *P < 0.05 vs. baseline; #P < 0.05 vs. reperfusion for 5 min; ^P < 0.05 vs. reperfusion for 15 min.
Phosphorylation of Nav1.5 at Ser571 prolongs APD and increases APD heterogeneity during I/R.
To assess the role of CaMKII-dependent phosphorylation of Nav1.5 in creating a substrate for I/R-induced arrhythmias, we used knockin mouse models with the CaMKII phosphorylation site at Ser571 replaced with glutamine (S571E) to mimic constitutive phosphorylation or alanine (S571A) to prevent phosphorylation (19). Optical recordings revealed significant APD prolongation in S571E hearts at baseline, after 15 min of ischemia, and after 5 min of reperfusion compared with WT or S571A hearts (Fig. 3). WT and S571A hearts showed similar APD except immediately after ischemia, when APD was significantly shorter in S571A than WT hearts (Fig. 3, A and B) Consistent with the known action of ATP-sensitive K+ channels, APD was significantly shorter in all groups after ischemia than at respective baselines (P < 0.05 for all groups) (16). Interestingly, although WT and S571A hearts had a similar mean APD after 5 min of reperfusion, WT hearts showed a significant increase in APD dispersion compared with S571A hearts (measured as standard deviation of APD across the mapping area) (Fig. 3, C and D). APD dispersion was also high in S571E compared with S571A hearts during reperfusion (similar to the level in WT hearts). In contrast, conduction velocity was not significantly different between the three groups at baseline or after 5 min of reperfusion (Table 1). These data suggest that CaMKII-dependent phosphorylation of Nav1.5 affects APD and APD dispersion to potentially alter the triggers and substrate for arrhythmia during I/R.
Fig. 3.

A and B: optically recorded action potentials (A) and summary data (B) for action potential duration at 80% repolarization (APD) in wild-type (WT), S571E, and S571A hearts at baseline and during the ischemia (Isch)-reperfusion (I/R) protocol. Data are also shown for a subset of WT hearts pretreated with mexiletine (WT + mex). Scale bars = 50 ms. C and D: APD maps and summary data showing APD standard deviation as an index of spatial dispersion during the I/R protocol. Maps span a change in APD of 30 ms for all groups, with a shift in the absolute range to facilitate comparison of dispersion independent of differences in average APD. Approximate locations of right ventricle (RV), left ventricle (LV), base, and apex are labeled on APD maps. A pacing cycle length of 150 ms was used for all measurements. Values are means ± SE; n = 5 independent preparations for all groups. *P < 0.05 vs. WT; # P < 0.05 vs. S571A; ^P < 0.05 vs. WT + mex.
Table 1.
Ca2+/calmodulin-dependent protein kinase II-dependent phosphorylation of Nav1.5 does not alter conduction velocity during ischemia-reperfusion
| WT |
S571E |
S571A |
WT + Mexiletine |
|||||
|---|---|---|---|---|---|---|---|---|
| Longitudinal | Transverse | Longitudinal | Transverse | Longitudinal | Transverse | Longitudinal | Transverse | |
| Baseline | 57.6 ± 1.7 | 35.6 ± 1.5 | 53.8 ± 3.0 | 27.4 ± 2.8 | 57.6 ± 1.7 | 38.2 ± 1.6 | 56.2 ± 1.8 | 36.2 ± 0.14 |
| Ischemia-reperfusion (5 min) | 56.8 ± 2.0 | 36.4 ± 2.1 | 53.2 ± 3.6 | 26.2 ± 3.0 | 57.6 ± 1.4 | 38.0 ± 0.7 | 54.0 ± 1.1 | 35.0 ± 1.3 |
Values (in cm/s) are means ± SE; n = 5 independent preparations for all groups. WT, wild-type. P = not significant.
To determine whether CaMKII-dependent phosphorylation of Nav1.5 affected arrhythmia susceptibility during reperfusion, electrocardiogram measurements were analyzed during spontaneous (unpaced) activity. Although spontaneous VT was not observed in any strain during reperfusion, the incidence of arrhythmia triggers (PVCs) was significantly higher in S571E and WT than S571A hearts (Fig. 4). Arrhythmia inducibility was also assessed in WT, S571E, and S571A hearts after 5 min of reperfusion using a S1S2 pacing protocol. Interestingly, reentry giving rise to VT was readily induced in WT and S571E, but not S571A, hearts (Fig. 5). Notably, arrhythmias were not inducible in S571A hearts for a wide range of S1S2 intervals (decreased until the tissue could no longer be captured). Together, these findings indicate that CaMKII-dependent phosphorylation of Nav1.5 alters excitability to promote both triggers and substrate for arrhythmia during I/R.
Fig. 4.

A and B: representative electrocardiogram recordings after 5 min of reperfusion (scale bars = 500 ms) and summary data for spontaneous arrhythmia events over the entire reperfusion period for wild-type (WT), S571E, and S571A hearts. Data are also shown from a subset of WT and S571E hearts pretreated with mexiletine (WT + mex). Representative arrhythmia events (premature ventricular contractions) are identified with red arrowheads. Values are means ± SE; n = 5 independent preparations for all groups. *P < 0.05 vs. WT; #P < 0.05 vs. S571E.
Fig. 5.

A and B: summary data and representative action potentials during the S1S2 protocol to induce arrhythmia following 5 min of reperfusion for wild-type (WT), S571 mutant (SE), and S571 mutant (SA) hearts. Data are also shown for a subset of WT and S571E hearts pretreated with mexiletine (WT + mex and S571E + mex). Cycle length of 150 ms was used for S1 pacing. The red arrow denotes the initiation of S2 protocol. VT, ventricular tachycardia. Values are means ± SE; n = 5 independent preparations for all groups. *P < 0.05 vs. WT; #P < 0.05 vs. SA. C: representative activation maps showing the spatial spread of activation after the last S1 beat, subsequent S2 beat, and first unpaced beat. WT and S571E maps demonstrate reentrant activation driving ventricular tachycardia after S2 (reentry path indicated by white arrows). Map for S571A shows conduction block near site of S2 (no unpaced activity after S2). The WT + mex map displays a single unpaced beat (no unpaced activity after the first unpaced beat). The location of pacing for S1 and S2 is indicated by a white asterisk.
Treatment with mexiletine reduces APD dispersion and arrhythmia susceptibility after I/R.
To test the hypothesis that CaMKII-dependent phosphorylation promotes arrhythmia after I/R via increased INa,L, we examined the effects of the selective Na+ channel blocker mexiletine on WT hearts subjected to our I/R protocol (Figs. 3–5). We used 10 μM mexiletine on the basis of previous reports showing preferential block of INa,L over peak at this dose (49). Interestingly, WT hearts treated with mexiletine showed APD changes with I/R similar to those observed in S571A hearts: mexiletine resulted in a shorter APD after ischemia without significant differences at baseline or after reperfusion compared with untreated hearts (Fig. 3, A and B). Mexiletine also reduced APD dispersion after reperfusion similar to that measured in S571A hearts (Fig. 3, C and D). At the same time, mexiletine reduced spontaneous arrhythmia events during reperfusion in both WT and S571E hearts (38 ± 9% and 49 ± 7% reduction, respectively, although differences were not significant; Fig. 4) while completely eliminating inducibility of VT (Fig. 5). These results support our hypothesis that CaMKII-dependent phosphorylation of Nav1.5 enhances INa,L to alter arrhythmia triggers and substrate during I/R.
DISCUSSION
In the United States, ∼1,000,000 patients suffer MI each year, yet the mechanisms contributing to arrhythmogenesis in the setting of I/R remain incompletely understood (38, 52). While efforts have been made to limit Na+/Ca2+ overload after I/R, effective targeting of related pathways without altering normal physiology remains a challenge for the field. In this study we define a novel mechanism to limit arrhythmias after I/R by specifically targeting CaMKII-induced INa,L. Consistent with previous studies (28, 47), we found that I/R promotes CaMKII activation (autophosphorylation). For the first time, we report a parallel increase in phosphorylation of Nav1.5 at Ser571. Through the use of Scn5a knockin mouse models (S571E and S571A), we show that Ser571 on Nav1.5 is critical for CaMKII-dependent regulation of INa,L, tissue excitability, and arrhythmias during I/R. Our findings demonstrate that ablation of CaMKII-dependent phosphorylation of Nav1.5 or pharmacological inhibition of INa,L with mexiletine reduces APD heterogeneity, arrhythmia triggers, and inducibility in response to I/R. Our data support the hypothesis that CaMKII-dependent hyperphosphorylation of Nav1.5 promotes both triggers and the substrate for arrhythmia during I/R. We propose that Nav1.5 hyperphosphorylation promotes spontaneous arrhythmia events via increased INa,L and consequent Na+/Ca2+ overload.
Earlier work identified CaMKII as an important contributor to cardiac arrhythmias in heart failure and chronic disease states (3, 12, 13, 52a, 57). These previous studies showed that, through various mechanisms (e.g., β-adrenergic stimulation and oxidative stress), CaMKII activity is increased, leading to abnormal cardiac excitability and function. While CaMKII has received considerable attention for its role in chronic disease, it also has the ability to promote arrhythmogenesis under acute stress relevant to acute MI (6, 28, 37, 46, 47). Studies of acute CaMKII activation in I/R showed an increase in phosphorylated CaMKII after as little as 15 min of I/R, with measurable effects on the downstream targets phospholamban and ryanodine receptor 2 (9, 47, 48). Other work pointed to Na+/Ca2+ overload as a source of arrhythmias in I/R (6, 10). Consistent with these studies, we have shown an increase in CaMKII autophosphorylation after 15 min of ischemia, with a concomitant increase in arrhythmia susceptibility after reperfusion. Beyond this previous work focusing heavily on CaMKII and its regulation of Ca2+ cycling after I/R, our study sought to elucidate Nav1.5 as a potential upstream source of Na+ overload (and subsequent Ca2+ overload during I/R). In line with this objective, we found that phosphorylated Nav1.5 is upregulated, together with CaMKII autophosphorylation, after 15 min of ischemia. Interestingly, we found that CaMKII and Nav1.5 phosphorylation levels are rapidly normalized during reperfusion (by 5 min), although the proarrhythmogenic effects on AP repolarization (and, presumably, ion homeostasis) remain. Related to these findings, previous work in rats showed that intracellular Na+ concentration doubles within 5 min of the start of global ischemia and continues to steadily increase for ≥30 min (25). Furthermore, intracellular Na+ concentrations return to normal within 10 min of reperfusion. In part, these rapid changes in intracellular Na+ are due to alterations in Na+/H+ exchanger activity, but INa,L also likely plays a role (27). Investigation of the upregulation of phosphorylated Nav1.5 at shorter and multiple time points for I/R would help elucidate the role of INa,L in mechanisms of Na+ overload and how it relates to changes in phosphorylated CaMKII and other Ca2+-handling proteins. It is important to note that, in addition to changes in phosphorylation status, CaMKII and Nav1.5 are likely subject to posttranslational modification downstream of a myriad of stress signals during I/R. While our experiments focused on CaMKII phosphorylation of Ser571 on Nav1.5, a host of phosphorylation sites have been identified for multiple signaling pathways (including additional sites for CaMKII) with potential to regulate Nav1.5 function in I/R (35). Furthermore, reactive oxygen species overproduction during reperfusion likely contributes to CaMKII activation and downstream effects but may also directly affect Nav1.5 (and other channel) behavior (30, 55). In this vein, it would be interesting to examine whether reactive oxygen species scavenging could reduce CaMKII activity, restore normal Nav1.5 activity, and mitigate arrhythmias produced by their interaction during I/R.
Beyond direct effects on ion homeostasis, it is interesting to consider the multiple ways that INa,L alters excitability and arrhythmogenesis during I/R. We provide interesting data that alteration of Nav1.5 phosphorylation impacts both the triggers (spontaneous PVCs) and substrate (inducibility of tachycardia) for reperfusion arrhythmias. The effect of Nav1.5 phosphorylation on arrhythmia triggers is likely mediated by changes in ion homeostasis, although this remains to be tested directly. It is less clear how changes in Nav1.5 phosphorylation and INa,L impact the substrate for arrhythmia. Our data showing decreased epicardial APD dispersion (base to apex) during reperfusion in S571A compared with WT or S571E hearts hint at a potential mechanism (Fig. 3, C and D). Nav1.5 has variable regional expression in the mouse heart, although base-to-apex changes are less well characterized than transmural or across chamber (e.g., right to left ventricle) changes (44, 53). At the same time, others observed that interventricular electrical heterogeneities are amplified by ischemia, exacerbating the arrhythmia substrate (36, 50). It is possible that the increase in APD dispersion and arrhythmia susceptibility observed with I/R-induced augmentation of phosphorylated Nav1.5 reflects the inherent heterogeneous expression of Nav1.5. It will be interesting in the future to determine the expression pattern of total and phosphorylated Nav1.5 throughout the heart at baseline and during I/R.
Previous efforts to prevent arrhythmogenesis associated with Na+/Ca2+ overload have focused primarily on inhibiting the Na+/Ca2+ exchanger (10, 24, 32). Much less attention has been given to INa,L. as a source of Na+ overload upstream of the Na+/Ca2+ exchanger. Recently, it was shown that INa,L activity increases to promote intracellular Na+ accumulation during I/R, providing a potential link between dysregulation of INa,L and arrhythmogenesis (45). At the same time, it was shown that CaMKII exerts specific regulation over Nav1.5 through phosphorylation, and when CaMKII activity is increased, this leads to a higher amount of INa,L (54). Our study builds on this previous work by identifying CaMKII-dependent phosphorylation of Nav1.5 at Ser571 upstream of increased INa,L, Na+ overload, and arrhythmias in I/R. Our investigation depended on the use of novel S571E and S571A knockin mice, which allowed for targeted perturbation of CaMKII-dependent regulation of Nav1.5 and INa,L. Interestingly, we were able to alter repolarization and arrhythmogenesis during I/R by manipulating the Ser571 site. These data support the notion that CaMKII-dependent phosphorylation of Nav1.5 is likely an important upstream event in I/R-induced arrhythmias.
Clinical relevance.
Despite efforts to reduce arrhythmogenesis after MI, there remains a great need for new and improved therapies (15). High-profile examples of past failures in this area include the use of Nav channel blockers, such as flecainide and encainide, which were successful in reducing PVCs but increased the risk of fatal reentrant arrhythmias in high-risk patients (8a). While improved outcomes have been observed for a Na+ channel inhibitor, such as ranolazine (more specific for late than for peak Na+ current), concerns remain about efficacy and off-target effects that can affect normal physiology (1, 8, 11, 20). Although implantable devices have shown greater efficacy in post-MI patients, they have several limitations, including cost, durability, and invasiveness (39). There has also been an effort after MI to treat patients with nonspecific anti-inflammatory steroids to assist recovery, but these drugs are linked with worsened ventricular and clinical outcomes (21, 42). As such, there is certainly a need for more specific targeting of pathogenic pathways in post-MI patients to combat these life-threatening arrhythmias (14). Our study attempts to explore this need by investigating a novel arrhythmogenic pathway involving CaMKII and Nav1.5 in the setting of I/R. Our findings suggest that targeting of INa,L may be an effective way to reduce arrhythmia triggers without increasing risk for reentrant arrhythmias.
Limitations.
While our findings provide novel insights into the mechanism linking CaMKII-dependent phosphorylation of Nav1.5 with increased INa,L and arrhythmias in I/R, we acknowledge multiple study limitations. First, our study used a global ischemia model in which perfusion to the entire heart is stopped for a period of time. In reality, ischemia is a much more heterogeneous condition, with regions of poor perfusion surrounded by nonischemic tissue. We anticipate that the role of the CaMKII-dependent pathway identified here will be just as important in the more heterogeneous case as in the global ischemia model. In vivo studies using temporary ligation of a coronary artery would be important for extending these findings to more a physiological situation. The use of blebbistatin is an additional limitation, particularly in the ischemia model, because of its mechanical uncoupling action, which would be expected to conserve ATP and aid in recovery during the reperfusion period (49). Another limitation is the difference in electrophysiology (especially repolarization) between mice and humans (40). However, the CaMKII-dependent pathway under investigation in this study also is highly conserved across species (31, 33).
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-135096, HL-134824, and HL-114893 (to T. J. Hund) and HL-135754, HL-134824, and HL-114383 (to P. J. Mohler) and by an American Heart Association Postdoctoral Fellowship (to A. Greer-Short).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.H., A.G.-S., T.S., N.P., D.N., P.J.M., and T.J.H. conceived and designed research; T.H., A.G.-S., T.S., N.P., and D.N. performed experiments; T.H., A.G.-S., T.S., N.P., D.N., P.J.M., and T.J.H. analyzed data; T.H., A.G.-S., T.S., N.P., D.N., P.J.M., and T.J.H. interpreted results of experiments; T.H., A.G.-S., T.S., and T.J.H. prepared figures; T.H., A.G.-S., T.S., and T.J.H. drafted manuscript; T.H., A.G.-S., T.S., N.P., D.N., P.J.M., and T.J.H. edited and revised manuscript; T.H., A.G.-S., T.S., N.P., D.N., P.J.M., and T.J.H. approved final version of manuscript.
REFERENCES
- 1.Aldakkak M, Camara AKS, Heisner JS, Yang M, Stowe DF. Ranolazine reduces Ca2+ overload and oxidative stress and improves mitochondrial integrity to protect against ischemia reperfusion injury in isolated hearts. Pharmacol Res 64: 381–392, 2011. doi: 10.1016/j.phrs.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen DG, Xiao XH. Role of the cardiac Na+/H+ exchanger during ischemia and reperfusion. Cardiovasc Res 57: 934–941, 2003. doi: 10.1016/S0008-6363(02)00836-2. [DOI] [PubMed] [Google Scholar]
- 3.Anderson ME. Multiple downstream proarrhythmic targets for calmodulin kinase II: moving beyond an ion channel-centric focus. Cardiovasc Res 73: 657–666, 2007. doi: 10.1016/j.cardiores.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 4.Arora R, Das MK, Zipes DP, Wu J. Optical mapping of cardiac arrhythmias. Indian Pacing Electrophysiol J 3: 187–196, 2003. [PMC free article] [PubMed] [Google Scholar]
- 5.Attin M, Clusin WT. Basic concepts of optical mapping techniques in cardiac electrophysiology. Biol Res Nurs 11: 195–207, 2009. doi: 10.1177/1099800409338516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bell JR, Vila-Petroff M, Delbridge LMD. CaMKII-dependent responses to ischemia and reperfusion challenges in the heart. Front Pharmacol 5: 96, 2014. doi: 10.3389/fphar.2014.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braunwald E, Kloner RA. Myocardial reperfusion: a double-edged sword? J Clin Invest 76: 1713–1719, 1985. doi: 10.1172/JCI112160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calderón-Sánchez EM, Domínguez-Rodríguez A, López-Haldón J, Jiménez-Navarro MF, Gómez AM, Smani T, Ordóñez A. Cardioprotective effect of ranolazine in the process of ischemia-reperfusion in adult rat cardiomyocytes. Rev Esp Cardiol (Engl Ed) 69: 45–53, 2016. doi: 10.1016/j.recesp.2015.02.027. [DOI] [PubMed] [Google Scholar]
- 8a.Cardiac Arrhythmia Suppression Trial (CAST) Investigators Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med 321: 406–412, 1989. doi: 10.1056/NEJM198908103210629. [DOI] [PubMed] [Google Scholar]
- 9.Di Carlo MN, Said M, Ling H, Valverde CA, De Giusti VC, Sommese L, Palomeque J, Aiello EA, Skapura DG, Rinaldi G, Respress JL, Brown JH, Wehrens XHT, Salas MA, Mattiazzi A. CaMKII-dependent phosphorylation of cardiac ryanodine receptors regulates cell death in cardiac ischemia/reperfusion injury. J Mol Cell Cardiol 74: 274–283, 2014. doi: 10.1016/j.yjmcc.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen S, Li S. The Na+/Ca2+ exchanger in cardiac ischemia/reperfusion injury. Med Sci Monit 18: RA161–RA165, 2012. doi: 10.12659/MSM.883533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dhalla AK, Wang W-Q, Dow J, Shryock JC, Belardinelli L, Bhandari A, Kloner RA. Ranolazine, an antianginal agent, markedly reduces ventricular arrhythmias induced by ischemia and ischemia-reperfusion. Am J Physiol Heart Circ Physiol 297: H1923–H1929, 2009. doi: 10.1152/ajpheart.00173.2009. [DOI] [PubMed] [Google Scholar]
- 12.Erickson JR, Anderson ME. CaMKII and its role in cardiac arrhythmia. J Cardiovasc Electrophysiol 19: 1332–1336, 2008. doi: 10.1111/j.1540-8167.2008.01295.x. [DOI] [PubMed] [Google Scholar]
- 13.Feng Y, Cheng J, Wei B, Wang Y. CaMKII inhibition reduces isoproterenol-induced ischemia and arrhythmias in hypertrophic mice. Oncotarget 8: 17504–17509, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Francis Stuart SD, De Jesus NM, Lindsey ML, Ripplinger CM. The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J Mol Cell Cardiol 91: 114–122, 2016. doi: 10.1016/j.yjmcc.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frommeyer G, Milberg P, Maier LS, Eckardt L. Late sodium current inhibition: the most promising antiarrhythmic principle in the near future? Curr Med Chem 21: 1271–1280, 2014. doi: 10.2174/09298673113209990220. [DOI] [PubMed] [Google Scholar]
- 16.Furukawa T, Kimura S, Furukawa N, Bassett AL, Myerburg RJ. Role of cardiac ATP-regulated potassium channels in differential responses of endocardial and epicardial cells to ischemia. Circ Res 68: 1693–1702, 1991. doi: 10.1161/01.RES.68.6.1693. [DOI] [PubMed] [Google Scholar]
- 17.Garciarena CD, Boum J, Swietach P, Vaughan-jones RD. H+-activated Na+ influx in the ventricular myocyte couples Ca2+-signalling to intracellular pH. J Mol Cell Cardiol 61: 51–59, 2013. doi: 10.1016/j.yjmcc.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Gloschat C, Aras K, Gupta S, Faye NR, Zhang H, Syunyaev RA, Pryamonosov RA, Rogers J, Kay MW, Efimov IR. RHYTHM: an open source imaging toolkit for cardiac panoramic optical mapping. Sci Rep 8: 2921, 2018. doi: 10.1038/s41598-018-21333-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glynn P, Musa H, Wu X, Unudurthi SD, Little S, Qian L, Wright PJ, Radwanski PB, Gyorke S, Mohler PJ, Hund TJ. Voltage-gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation 132: 567–577, 2015. doi: 10.1161/CIRCULATIONAHA.114.015218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hale SL, Leeka JA, Kloner RA. Improved left ventricular function and reduced necrosis after myocardial ischemia/reperfusion in rabbits treated with ranolazine, an inhibitor of the late sodium channel. J Pharmacol Exp Ther 318: 418–423, 2006. doi: 10.1124/jpet.106.103242. [DOI] [PubMed] [Google Scholar]
- 21.Hammerman H, Kloner RA, Schoen FJ, Brown EJ Jr, Hale S, Braunwald E. Indomethacin-induced scar thinning after experimental myocardial infarction. Circulation 67: 1290–1295, 1983. doi: 10.1161/01.CIR.67.6.1290. [DOI] [PubMed] [Google Scholar]
- 22.Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, Gudmundsson H, Kline CF, Davidson NP, Cardona N, Rasband MN, Anderson ME, Mohler PJ. A βIV-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest 120: 3508–3519, 2010. doi: 10.1172/JCI43621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hund TJ, Snyder JS, Wu X, Glynn P, Koval OM, Onal B, Leymaster ND, Unudurthi SD, Curran J, Camardo C, Wright PJ, Binkley PF, Anderson ME, Mohler PJ. βIV-spectrin regulates TREK-1 membrane targeting in the heart. Cardiovasc Res 102: 166–175, 2014. doi: 10.1093/cvr/cvu008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imahashi K, Pott C, Goldhaber JI, Steenbergen C, Philipson KD, Murphy E. Cardiac-specific ablation of the Na+-Ca2+ exchanger confers protection against ischemia/reperfusion injury. Circ Res 97: 916–921, 2005. doi: 10.1161/01.RES.0000187456.06162.cb. [DOI] [PubMed] [Google Scholar]
- 25.Imahashi K, Kusuoka H, Hashimoto K, Yoshioka J, Yamaguchi H, Nishimura T. Intracellular sodium accumulation during ischemia as the substrate for reperfusion injury. Circ Res 84: 1401–1406, 1999. doi: 10.1161/01.RES.84.12.1401. [DOI] [PubMed] [Google Scholar]
- 27.Jung IS, Lee SH, Yang MK, Park JW, Yi KY, Yoo SE, Kwon SH, Chung H-J, Choi W-S, Shin H-S. Cardioprotective effects of the novel Na+/H+ exchanger-1 inhibitor KR-32560 in a perfused rat heart model of global ischemia and reperfusion: involvement of the Akt-GSK-3β cell survival pathway and antioxidant enzyme. Arch Pharm Res 33: 1241–1251, 2010. doi: 10.1007/s12272-010-0815-z. [DOI] [PubMed] [Google Scholar]
- 28.Kong L-H, Gu X-M, Wu F, Jin Z-X, Zhou J-J. CaMKII inhibition mitigates ischemia/reperfusion-elicited calpain activation and the damage to membrane skeleton proteins in isolated rat hearts. Biochem Biophys Res Commun 491: 687–692, 2017. doi: 10.1016/j.bbrc.2017.07.128. [DOI] [PubMed] [Google Scholar]
- 29.Koval OM, Snyder JS, Wolf RM, Pavlovicz RE, Glynn P, Curran J, Leymaster ND, Dun W, Wright PJ, Cardona N, Qian L, Mitchell CC, Boyden PA, Binkley PF, Li C, Anderson ME, Mohler PJ, Hund TJ. Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation 126: 2084–2094, 2012. doi: 10.1161/CIRCULATIONAHA.112.105320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu M, Liu H, Dudley SCJ Jr. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res 107: 967–974, 2010. doi: 10.1161/CIRCRESAHA.110.220673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma J, Luo A, Wu L, Wan W, Zhang P, Ren Z, Zhang S, Qian C, Shryock JC, Belardinelli L. Calmodulin kinase II and protein kinase C mediate the effect of increased intracellular calcium to augment late sodium current in rabbit ventricular myocytes. Am J Physiol Cell Physiol 302: C1141–C1151, 2012. doi: 10.1152/ajpcell.00374.2011. [DOI] [PubMed] [Google Scholar]
- 32.MacDonald AC, Howlett SE. Differential effects of the sodium calcium exchange inhibitor, KB-R7943, on ischemia and reperfusion injury in isolated guinea pig ventricular myocytes. Eur J Pharmacol 580: 214–223, 2008. doi: 10.1016/j.ejphar.2007.10.055. [DOI] [PubMed] [Google Scholar]
- 33.Maltsev VA, Sabbah HN, Higgins RSD, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 98: 2545–2552, 1998. doi: 10.1161/01.CIR.98.23.2545. [DOI] [PubMed] [Google Scholar]
- 34.Marban E, Yamagishi T, Tomaselli GF. Structure and function of voltage-gated sodium channels. J Physiol 508: 647–657, 1998. doi: 10.1111/j.1469-7793.1998.647bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marionneau C, Abriel H. Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications. J Mol Cell Cardiol 82: 36–47, 2015. doi: 10.1016/j.yjmcc.2015.02.013. [DOI] [PubMed] [Google Scholar]
- 36.Matiukas A, Pertsov AM, Kothari P, Cram A, Tolkacheva EG. Optical mapping of electrical heterogeneities in the heart during global ischemia. Conf Proc IEEE Eng Med Biol Soc 2009: 6321–6324, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitsuyama H, Yokoshiki H, Watanabe M, Mizukami K, Shimokawa J, Tsutsui H. Ca2+/calmodulin-dependent protein kinase II increases the susceptibility to the arrhythmogenic action potential alternans in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 307: H199–H206, 2014. doi: 10.1152/ajpheart.00387.2012. [DOI] [PubMed] [Google Scholar]
- 38.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, De Ferranti S, Després J, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jiménez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, Mcguire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB; Writing Group Members; American Heart Association Statistics Committee; Stroke Statistics Subcommittee . Heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation 133: e38–e360, 2016. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 39.Nattel S, Andrade J, Macle L, Rivard L, Dyrda K, Mondesert B, Khairy P. New directions in cardiac arrhythmia management: present challenges and future solutions. Can J Cardiol 30, Suppl: S420–S430, 2014. doi: 10.1016/j.cjca.2014.09.027. [DOI] [PubMed] [Google Scholar]
- 40.Nerbonne JM. Studying cardiac arrhythmias in the mouse−a reasonable model for probing mechanisms? Trends Cardiovasc Med 14: 83–93, 2004. doi: 10.1016/j.tcm.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 41.Neri M, Riezzo I, Pascale N, Pomara C, Turillazzi E. Ischemia/reperfusion injury following acute myocardial infarction: a critical issue for clinicians and forensic pathologists. Mediators Inflamm 2017: 7018393, 2017. doi: 10.1155/2017/7018393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Gara PT, Kushner FG, Ascheim DD, Casey DE Jr, Chung MK, de Lemos JA, Ettinger SM, Fang JC, Fesmire FM, Franklin BA, Granger CB, Krumholz HM, Linderbaum JA, Morrow DA, Newby LK, Ornato JP, Ou N, Radford MJ, Tamis-Holland JE, Tommaso CL, Tracy CM, Woo YJ, Zhao DX, Anderson JL, Jacobs AK, Halperin JL, Albert NM, Brindis RG, Creager MA, DeMets D, Guyton RA, Hochman JS, Kovacs RJ, Kushner FG, Ohman EM, Stevenson WG, Yancy CW; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines . 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 127: e362–e425, 2013. doi: 10.1161/CIR.0b013e3182742c84. [DOI] [PubMed] [Google Scholar]
- 44.Remme CA, Verkerk AO, Hoogaars WMH, Aanhaanen WTJ, Scicluna BP, Annink C, van den Hoff MJB, Wilde AAM, van Veen TAB, Veldkamp MW, de Bakker JMT, Christoffels VM, Bezzina CR. The cardiac sodium channel displays differential distribution in the conduction system and transmural heterogeneity in the murine ventricular myocardium. Basic Res Cardiol 104: 511–522, 2009. doi: 10.1007/s00395-009-0012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ronchi C, Torre E, Rizzetto R, Bernardi J, Rocchetti M, Zaza A. Late sodium current and intracellular ionic homeostasis in acute ischemia. Basic Res Cardiol 112: 12, 2017. doi: 10.1007/s00395-017-0602-9. [DOI] [PubMed] [Google Scholar]
- 46.Said M, Becerra R, Palomeque J, Rinaldi G, Kaetzel MA, Diaz-Sylvester PL, Copello JA, Dedman JR, Mundiña-Weilenmann C, Vittone L, Mattiazzi A. Increased intracellular Ca2+ and SR Ca2+ load contribute to arrhythmias after acidosis in rat heart. Role of Ca2+/calmodulin-dependent protein kinase II. Am J Physiol Heart Circ Physiol 295: H1669–H1683, 2008. doi: 10.1152/ajpheart.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Said M, Becerra R, Valverde CA, Kaetzel MA, Dedman JR, Mundiña-Weilenmann C, Wehrens XH, Vittone L, Mattiazzi A. Calcium-calmodulin dependent protein kinase II (CaMKII): a main signal responsible for early reperfusion arrhythmias. J Mol Cell Cardiol 51: 936–944, 2011. doi: 10.1016/j.yjmcc.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salas MA, Valverde CA, Sánchez G, Said M, Rodriguez JS, Portiansky EL, Kaetzel MA, Dedman JR, Donoso P, Kranias EG, Mattiazzi A. The signalling pathway of CaMKII-mediated apoptosis and necrosis in the ischemia/reperfusion injury. J Mol Cell Cardiol 48: 1298–1306, 2010. doi: 10.1016/j.yjmcc.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Signore S, Sorrentino A, Borghetti G, Cannata A, Meo M, Zhou Y, Kannappan R, Pasqualini F, O’Malley H, Sundman M, Tsigkas N, Zhang E, Arranto C, Mangiaracina C, Isobe K, Sena BF, Kim J, Goichberg P, Nahrendorf M, Isom LL, Leri A, Anversa P, Rota M. Late Na+ current and protracted electrical recovery are critical determinants of the aging myopathy. Nat Commun 6: 8803, 2015. doi: 10.1038/ncomms9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith RM, Velamakanni SS, Tolkacheva EG. Interventricular heterogeneity as a substrate for arrhythmogenesis of decoupled mitochondria during ischemia in the whole heart. Am J Physiol Heart Circ Physiol 303: H224–H233, 2012. doi: 10.1152/ajpheart.00017.2012. [DOI] [PubMed] [Google Scholar]
- 51.Swift LM, Asfour H, Posnack NG, Arutunyan A, Kay MW, Sarvazyan N. Properties of blebbistatin for cardiac optical mapping and other imaging applications. Pflugers Arch 464: 503–512, 2012. doi: 10.1007/s00424-012-1147-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am J Cardiol 106: 360–368, 2010. doi: 10.1016/j.amjcard.2010.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52a.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XHT. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation 122: 2669–2679, 2010. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Veerman CC, Podliesna S, Tadros R, Lodder EM, Mengarelli I, de Jonge B, Beekman L, Barc J, Wilders R, Wilde AAM, Boukens BJ, Coronel R, Verkerk AO, Remme CA, Bezzina CR. The Brugada syndrome susceptibility gene HEY2 modulates cardiac transmural ion channel patterning and electrical heterogeneity. Circ Res 121: 537–548, 2017. doi: 10.1161/CIRCRESAHA.117.310959. [DOI] [PubMed] [Google Scholar]
- 54.Wagner S, Dybkova N, Rasenack ECL, Jacobshagen C, Fabritz L, Kirchhof P, Maier SKG, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest 116: 3127–3138, 2006. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J, Belardinelli L, Maier LS. Reactive oxygen species-activated Ca/calmodulin kinase IIδ is required for late INa augmentation leading to cellular Na and Ca overload. Circ Res 108: 555–565, 2011. doi: 10.1161/CIRCRESAHA.110.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang L, Myles RC, De Jesus NM, Ohlendorf AKP, Bers DM, Ripplinger CM. Optical mapping of sarcoplasmic reticulum Ca2+ in the intact heart: ryanodine receptor refractoriness during alternans and fibrillation. Circ Res 114: 1410–1421, 2014. doi: 10.1161/CIRCRESAHA.114.302505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, Roden DM, Passier R, Olson EN, Colbran RJ, Anderson ME. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation 106: 1288–1293, 2002. doi: 10.1161/01.CIR.0000027583.73268.E7. [DOI] [PubMed] [Google Scholar]