

Abstract
Necrosis is a key factor in myocardial injury during cardiac pathological processes, such as myocardial infarction (MI), ischemia/reperfusion (I/R) injury and heart failure. Increasing evidence suggests that several aspects of necrosis are programmed and tightly regulated, so targeting the necrosis process has become a new trend for myocardial protection. Multiple cellular signaling pathways have been implicated in necrotic cell death, such as the death receptor-mediated extrinsic and mitochondrial intrinsic pathways. However, the precise mechanisms underlying myocardial necrosis remain unclear. In this study, we showed that apoptosis repressor with caspase recruitment domain (ARC) participated in the mitochondrial intrinsic pathway and inhibited myocardial necrosis by preventing the opening of the mitochondrial permeability transition pore (mPTP). ARC attenuated necrotic cell death triggered by exposure to 500 μM hydrogen peroxide (H2O2) in the cardiomyocyte cell line H9c2. In mice, ARC ameliorated myocardial necrosis, reduced the myocardial infarct size and improved long-term heart function during I/R injury. Mechanistically, it has been shown that the inhibition of necrosis by ARC was dependent on its mitochondrial localization and that ARC prevented the opening of mPTP by targeting CypD, the main regulator of mPTP. In addition, ARC expression was negatively regulated by the transcription factor p53 at the transcriptional level during the necrosis process. These findings identified the novel role of ARC in myocardial necrosis and delineated the p53-ARC-CypD/mPTP necrosis pathway during ischemia- and oxidative stress-induced myocardial damage, which can provide a new strategy for cardiac protection.
Keywords: ARC, Myocardial necrosis, mPTP, CypD
Graphical abstract
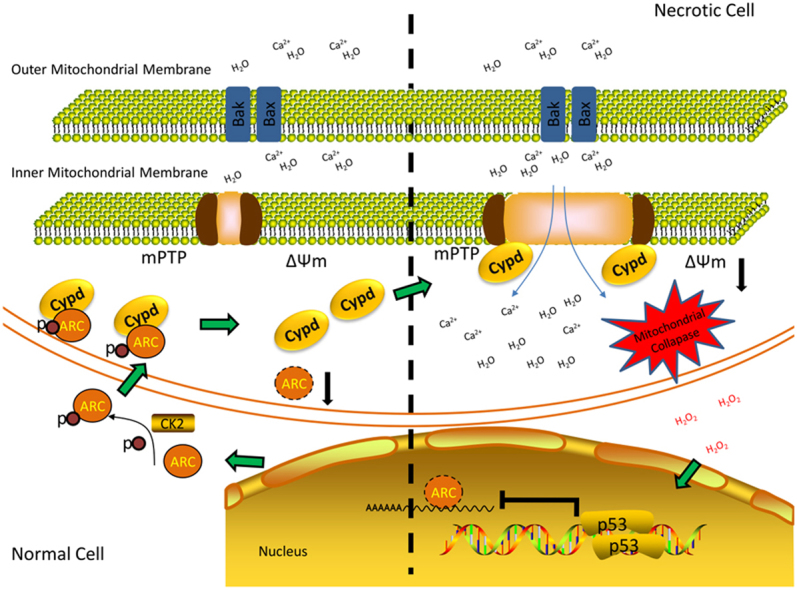
Highlights
-
•
ARC attenuates necrosis both in cardiomyocytes exposed to H2O2 and in mouse hearts with ischemia/reperfusion injury.
-
•
The attenuation of necrosis by ARC depends on its mitochondrial localization and its inhibition of mPTP opening.
-
•
ARC targets CypD and prevents mPTP opening.
-
•
p53 regulates necrosis by suppression of ARC expression at the transcriptional level.
1. Introduction
The death of terminally differentiated cardiomyocytes is the leading cause of various cardiac diseases, including heart failure, myocardial infarction (MI) and ischemia/reperfusion (I/R) injury. Apoptosis and necrosis both contribute to myocardial cell death during the pathogenesis of cardiac diseases [1], [2]. Apoptosis has received much attention over the past 30 years because it was considered the only form of programmed cell death, although necrotic cell death is the central feature of the failing and I/R heart [3], [4]. Myocardial necrosis is an irreversible cell death process, accompanied by rapid loss of cellular membrane potential, which leads to cell swelling, rupture, cytolysis and subsequent inflammation [5]. Increasing evidence suggests that several forms of necrosis are tightly regulated and programmed rather than merely an accidental form of cell death [6], [7]. Several signaling pathways have been shown to be involved in necrotic cell death [8], [9], [10]. However, knowledge of the regulation of necrosis remains limited and further studies are needed to explore the central factors involved in the process.
The mitochondrial permeability transition pore (mPTP), a multiple protein complex, is the key effector in the pathway to cell death whose molecular components remain elusive [11], [12], [13]. mPTP opening is the primary event of the mitochondrial intrinsic necrosis pathway, which is referred to as sudden mitochondrial permeability transition and loss of inner mitochondrial potential. The opening of mPTP leads to the interruption of mitochondrial respiration, matrix swelling and rupture of the mitochondrial membrane, which results in necrotic cell death [12], [13]. During cardiac I/R injury, necrosis induced by mPTP opening and the resulting mitochondrial damage is the main cause of myocardial cell death which contributes to more than 50% of the infarcted areas [14]. Understanding the regulation of necrosis induced by mPTP opening is essential for cardio-protection during I/R injury.
CypD has been reported to be a central factor in the mitochondrial intrinsic necrosis pathway, which promotes the opening of mPTP. CypD-deficient cardiomyocytes have been shown to have strong resistance to H2O2 or Ca2+-induced necrosis. CypD-knockout in mice was also found to play a cardio-protective role during I/R injury [15], [16], [17]. Cyclosporine A, a specific inhibitor of CypD, effectively prevents mPTP opening by binding to CypD and displacing it from the mPTP [18], [19]. Several lines of evidence suggest that CypD can also be regulated through protein-protein interaction or posttranslational modification [20], [21], [22], [23]. However, the precise mechanisms by which CypD/mPTP is regulated during necrosis remains largely unknown and further studies are required to understand these mechanisms.
Apoptosis repressor with caspase recruitment domain (ARC) is abundantly expressed in heart and muscle cells and has been reported to play a strong cardio-protective role during myocardial injury [24]. Previous studies mainly showed its anti-apoptotic effects by either directly binding to caspase-2 and −8 or indirectly decreasing the mitochondrial Ca2+ [25], [26]. However, whether ARC is involved in the regulation of myocardial necrosis is still unknown.
ARC has been reported to inhibit necrosis in mouse L929 fibrosarcoma cells by participating in the extrinsic death receptor-mediated necrosis pathway [27]. However, the cardio-protective effects of ARC depend on its mitochondrial localization during myocardial injury [28], [29]. The mitochondria is the convergence point for cell death signaling during the pathogenesis of cardiac diseases [30]. The work presented in this study focused on the mitochondrial intrinsic necrosis pathway and the potential role of ARC in the regulation of myocardial necrosis.
p53 is a tumor suppressor gene that has been reported to be a major factor influencing cell death processes. p53 promotes cell death by directly interacting with death-related proteins or by acting as a transcription factor in the regulation of vast gene expression programs [1]. It has been shown that p53 binds to the promoter region of ARC and decreases its expression at the transcriptional level at the initiation of apoptosis [31]. It was previously reported that p53 regulates necrosis by targeting CypD which promotes mPTP opening in MEF cells [23]. p53 is also a key mediator in myocardial cell death during I/R injury by influencing vast gene expression programs at the transcriptional level [1], [32]. However, whether p53 participates in myocardial necrosis remains largely unknown. Therefore, the specific role of the ARC and CypD/mPTP axis in the mitochondrial intrinsic necrosis pathway was explored in this study. Furthermore, we have also demonstrated the role of p53 in the ARC/CypD/mPTP axis at the transcriptional level during myocardial necrosis.
2. Materials and methods
2.1. Cell culture and treatment
H9c2 cells (American Type Culture Collection) were cultured in Dulbecco's modified Eagle's medium (Invitrogen, 12100046) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 µg/L streptomycin and 110 mg/L sodium pyruvate in a humidified atmosphere containing 5% CO2 at 37 °C. Cells were treated with 100 µM or 500 µM H2O2 at the indicated times.
2.2. Cell death assays
The detailed procedures for PI staining are as follows: cardiomyocytes climbing to the carry sheet glass were washed with PBS three times after the H2O2 treatment. The concentration of working PI solution was 1.5 µM. Cells were then incubated on ice for 5 min. After washing with PBS three times, cells were fixed with 4% PFA on ice for 30 min. Following a quick wash, the cardiomyocytes were mounted with mounting medium containing DAPI and examined with a Nikon Eclipse Ti-S fluorescence microscope. The percentage of necrotic cell death was calculated by counting the total number of PI-positive nuclei divided by total DAPI-stained nuclei.
TUNEL assay was performed using TUNEL Apoptosis Detection Kit (Alexa Fluor 488, YEASEN, 40307ES20) according to the manufacturer's instructions. The percentage of apoptotic nuclei was calculated by counting the total number of TUNEL-positive nuclei divided by the total number of DAPI-stained nuclei.
2.3. Measurement of the lactate dehydrogenase (LDH) activity
Twenty microliters of supernatant was collected from each cell culture well after H2O2 treatment and LDH activity was measured using the Spectrophotometric kit (Nanjing Jiancheng, Jiangsu, China) following the manufacturer's instructions.
2.4. Measurement of the mPTP dynamics
2.4.1. TMRE staining
TMRE at a working concentration of 20 nM was loaded into the cell culture pore and cultured with the cells for 30 min, and then the supernatant was discarded. After washing with PBS three times, the cells were immediately treated with 500 μM H2O2 and sequential cellular fluorescence images were acquired with a Leica TCS SP8 MP laser scanning confocal microscope.
2.4.2. Calcein-AM staining
The cells were trypsinized and resuspended in PBS, then loaded with calcein AM and cytosolic calcein fluorescence quencher (CoCl2) for 15 min at 37 °C. The cells were centrifuged, washed, and the cell pellets were resuspended in PBS and characterized for mitochondrial calcein fluorescence by flow cytometry analysis.
The cells were loaded with calcein-AM and a cytosolic calcein fluorescence quencher (CoCl2) for 30 min at 37 °C after the H2O2 treatment. Mitochondrial calcein fluorescence images were acquired with a Leica TCS SP8 MP laser scanning confocal microscope.
The fluorescence intensity was calculated as a percentage by gating upon the positive and negative controls.
2.5. Adenoviruses and vectors for gene overexpression
Adenovirus ARC (Ad-ARC) and adenovirus h-galactosidase (Ad-β-gal) were as previously described [31]. Adenovirus ARCT149A (Ad-ARCT149A) was as previously described [33]. Adenovirus p53 (Ad-p53) was as previously described [31]. The CypD overexpression vector was constructed as follows: the CDS of CypD was cloned into the pKC-EF1α eukaryotic expression vector with an EF1α promoter.
The HA-ARC overexpression vector was constructed as follows: The CDS of ARC was cloned into the pKC-EF1α eukaryotic expression vector, the HA sequence was inserted into the C terminal of ARC; Myc-CypD was purchased from OriGene Company.
2.6. RNA interference (RNAi)
Adenovirus si-ARC (Ad-si-ARC) and the Adenovirus scr-ARC (Ad-scr-ARC) were as previously described [31]. Adenovirus si-p53 (Ad-si-p53) and the Adenovirus scr-p53 (Ad-scr-p53) were as previously described [31].
The CypD RNAi sense sequence was as follows:
si-CypD-1: 5′ GCGACUUCACCAAUCACAATT 3′
si-CypD-2: 5′ GCAUGUUGUGUUUGGCCAUTT 3′
The scramble sequence was as follows:
scr-CypD: 5′ UUCUCCGAACGUGUCACGUTT 3′
2.7. Quantitative real-time PCR (qRT-PCR)
The sequences of ARC primers were as follows:
Forward, 5′-CGGAAACGGCTGGTAGAAAC-3′;
Reverse, 5′-TGGCATGCTCACAGTTTGCT-3′
The sequences of IL-1β primers were as follows:
Forward, 5′-TGATGACGACCTGCTAGTGT-3′;
Reverse, 5′-CTTCTTTGGGTATTGTTTGG-3′;
The sequences of IL-6 primers were as follows:
Forward, 5′-AGTTGCCTTCTTGGGACTGA-3′;
Reverse, 5′-TGGGTGGTATCCTCTGTGAAG-3′;
The sequences of TNF-α primers were as follows:
Forward, 5′-CACTAAGAATTCAAACTGGGGC-3′;
Reverse, 5′-GAGGAAGGCCTAAGGTCCAC-3′.
2.8. Immunoblotting
The cells were collected and lysed for 1 h on ice in a strong RIPA buffer containing a protease inhibitor cocktail (Merk, 539134). The samples were subjected to 12% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred to PVDF (0.45 µm) membranes. The anti-ARC antibodies (1:2000) were from Abcam (ab126238) and Santa Cruz (sc-374177). The anti-CypD antibodies (1:2000) were from Abcam (ab110324). The anti-p53 antibody (1:1000) was from Cell Signaling Technology (mAb #2524). The anti-α-actin antibody was from Santa Cruz Biotechnology. The anti-myc antibody was from Biorbyt, China. The anti-HA antibody was from Santa Cruz Biotechnology. Primary antibody was incubated at 4 °C overnight. After washing with PBS-Tween 20 five times, HRP-conjugated secondary antibodies were incubated at room temperature for 1 h. Bands were quantitated using Image J and α-actin was used as the loading control. Fold change was normalized to the indicated control.
2.9. Immunoprecipitation
The cells were lysed on ice for 30 min in 500 μl NP-40 lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 10% glycerol, 0.2 mM PMSF and Protease inhibitor). The lysates were precleared by centrifugation, and 50 μl of the samples was aliquoted for input. The remainder of the samples were immunoprecipitated with 2 μg of antibody and 50 μl of Protein-A/G PLUS-Agarose (Santa Cruz Biotechnology). The samples were rotated at 4 °C overnight. The beads were washed three times with 1 ml of low-salt NP40 lysis buffer (300 mM NaCl) and twice with 1 ml of high-salt lysis buffer (500 mM NaCl). The beads were then boiled for 10 min in the presence of 25 μl 2× sample buffer and the released proteins were fractionated in 12% SDS-PAGE gels. Proteins were detected by immunoblotting as described above.
2.10. Immunofluorescence staining
The cells were fixed and permeabilized with 4% paraformaldehyde/0.1% Triton X-100 for 10 min at 4 °C, immediately prior to processing for immunofluorescence. Nonspecific antibody binding was blocked by a 30 min incubation period with 5% (v/v) fetal calf serum. The cells were then treated with ARC antibody (1:200) (Abcam, ab126238) for 2 h at room temperature. Following 3 washes, the cells were treated with Alexa Fluor 488 anti-rabbit secondary antibody (1:200) for 2 h at room temperature. The cells were then treated with CypD antibody (1:200) (Abcam, ab110324) for 2 h at room temperature. Following 3 washes, the cells were treated with Alexa Fluor 594 anti-rabbit secondary antibody (1:200) for 2 h at room temperature. Nuclei were identified by DAPI staining. Images were taken using a Nikon Eclipse Ti-S fluorescence microscope.
2.11. Mouse model of myocardial ischemia/reperfusion (I/R) injury
All procedures were in agreement with the standards for care of laboratory animals as outlined in the NIH Guide for the Care and Use of Laboratory Animals. All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of Qingdao University Medical College.
Mice were subjected to 30 min of LAD ligation followed by 3 h of reperfusion. The animals received PI injections of 10 mg/kg (PI; Sigma) to label necrotic cells after the termination of I/R surgery (n = 5/group). Frozen 5 µm sections were cut and counterstained with DAPI after which the quantification of necrotic cells was performed. Anti-a-actinin antibody was from Sigma, A7732. The immunofluorescence staining was performed as described above.
2.12. Detection of the pro-inflammatory factors
The I/R heart samples were harvested 24 h after I/R surgery and the pro-inflammatory factors were detected by Q-PCR analysis.
2.13. Masson trichrome staining for collagen
Masson trichrome staining was performed using the staining kit (Solarbio, G1340) following the manufacturer's instructions. Cardiac fibrosis stained blue with Masson's trichrome. The ratio of fibrotic area was quantified using software (Image J) by an observer blinded to the sample identity.
2.14. Cardiac functional assays
Left ventricular dimensions and cardiac function in mice exposed to I/R was analyzed by echocardiograph by an observer blinded to the sample identity two weeks after the I/R injury.
2.15. Statistical analysis
The results are expressed as the mean ± SEM of at least three independent experiments. The differences among experimental groups were evaluated by one-way ANOVA (analysis of variance). p < 0.05 was considered statistically significant.
3. Results
3.1. ARC attenuated H2O2-induced necrotic cell death in cardiomyocytes
Hydrogen peroxide (H2O2) induces both apoptosis and necrosis in cardiomyocytes and has been used to generate oxidative stress mimicking I/R injury [8], [17], [34], [35]. Our previous work, and the work of others, has shown that the response to oxidative stress induced by H2O2 in cardiomyocytes depends upon the concentration of H2O2 applied [8], [36]. Exposing the cardiomyocytes to lower concentrations of H2O2 (100 μM) mainly triggered apoptosis, while the higher concentration of H2O2 (500 μM) predominantly induced necrosis, as shown in Supplementary Fig. 1A and Supplementary Fig. 1B, which is consistent with previous results. Apoptosis and necrosis were respectively assessed by the terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) assay and propidium iodide (PI) staining [8], [37]. To investigate the potential role of ARC in necrosis, cardiomyocytes were exposed to the higher concentration of H2O2 (500 μM), and the protein levels of ARC were examined during necrosis induced by oxidative stress. The protein levels of ARC showed a significant downregulation (Fig. 1A and Supplementary Fig. 4A). We then forced the expression of ARC in cardiomyocytes by adenoviral infection (Fig. 1B). Our results showed that the overexpression of ARC attenuated necrosis in cardiomyocytes exposed to H2O2 compared to the negative control group (Fig. 1C, D and Supplementary Fig. 1C). To mimic the H2O2-induced ARC downregulation, we knocked down the endogenous ARC in cardiomyocytes (Fig. 1E). Our results showed that the knockdown of ARC sensitized the cardiomyocytes to undergo necrosis after H2O2 exposure even at the lower concentration (100 μM) compared to the negative control (Fig. 1F and Supplementary Fig. 1D). Taken together, these results indicated that ARC could attenuate myocardial necrosis induced by oxidative stress as a result of H2O2 exposure.
Fig. 1.

Apoptosis repressor with CARD (ARC) inhibited H2O2-induced necrosis. (A) ARC protein levels in cardiomyocytes treated with 500 μM H2O2 for the indicated time were detected by Western blotting. (B) Adenovirus could efficiently force the expression of ARC in cardiomyocytes. (C) Forced expression of ARC prevented H2O2-induced necrosis in cardiomyocytes exposed to 500 μM H2O2 for 12 h compared to the control. Necrosis was detected by PI assay; scale bar, 30 µm. (D) Statistical analysis of the proportion of the PI positive cells in each group. Error bars represent SEM. *P < 0.05 vs control. (E) Adenovirus could efficiently knockdown the endogenous ARC. (F) Knockdown of ARC increased H2O2-induced necrosis in cardiomyocytes exposed to 100 μM H2O2 for 12 h compared with the control. Necrosis was detected by PI assay; Error bars represent SEM. *P < 0.05 vs control.
3.2. ARC inhibited mPTP opening induced by H2O2 in cardiomyocytes
We next sought to understand the mechanism by which ARC inhibited myocardial necrosis. The mitochondrial permeability transition is the primary component of oxidative damage-induced necrosis during cardiac I/R injury. We then examined whether ARC-based attenuation of H2O2-induced myocardial necrosis was due to the prevention of the mitochondrial permeability transition pore (mPTP) opening. The opening of the mPTP is determined by the loss of the inner-membrane potential (ΔΨm). TMRE (tetramethylrhodamine ethyl ester) is a specific mitochondrial inner-membrane potential indicator and is widely used in the detection of ΔΨm during the induction of necrosis [17], [20], [38]. The cardiomyocytes were loaded with TMRE (20 nM) and the opening of the mPTP was visualized as a rapid dissipation of TMRE fluorescence. Our results showed that the rapid loss of TMRE fluorescence in a time dependent manner was indicating the opening of mPTP in cardiomyocytes that were exposed to H2O2 (Fig. 2A). Conversely, treating cells with CsA (5 μM), the most specific inhibitor of mPTP opening [39], could delay the loss of TMRE fluorescence (Supplementary Fig. 2B). These results demonstrated that the dissipation of fluorescence in cells exposed to H2O2 was the result of the pores opening. Next, we explored whether ARC was involved in the regulation of the mPTP opening in cardiomyocytes upon H2O2 exposure. We overexpressed ARC in cardiomyocytes successfully, as shown in Fig. 1B. The overexpression of ARC significantly delayed the TMRE loss in cardiomyocytes exposed to H2O2 compared to the negative control group indicating that ARC significantly inhibited mPTP opening during necrosis (Fig. 2B and C). To further confirm the specificity of these events on the pore dynamics, we utilized another established method to detect mPTP opening in the intact cells [40], [41]. We incubated cardiomyocytes with calcein-AM (1 μM) and cobalt-chloride (CoCl2, 1 mM), which localized calcein fluorescence in the mitochondria. The mPTP opening was determined by the reduction in calcein fluorescence in the mitochondria. Our results showed that H2O2-induced necrosis in cardiomyocytes could significantly induce loss of calcein fluorescence in the mitochondria compared to the negative control. Moreover, ARC overexpression in cardiomyocytes delayed the calcein fluorescence loss upon H2O2 exposure (Fig. 2D and E). Taken together, these results demonstrated that ARC could significantly inhibit mPTP opening in H2O2-induced necrosis in cardiomyocytes.
Fig. 2.

ARC prevented mPTP opening induced by 500 μM H2O2. (A) Inner mitochondrial membrane potential (ΔΨm) was detected by TMRE staining at the indicated time in cardiomyocytes exposed to 500 μM H2O2 or without any treatment. The intensity of TMRE fluorescence was measured by confocal microscopy. The relative TMRE fluorescence intensity was the ratio to the initial value. Error bars represent SEM. (B) Forced expression of ARC delayed the loss of TMRE fluorescence in cardiomyocytes exposed to 500 μM H2O2 compared to the control; Scale bar, 20 µm. (C) Statistical data showed the intensity of the TMRE fluorescence in each group. Error bars represent SEM. *P < 0.05 vs control. (D) Forced expression of ARC delayed the loss of calcein fluorescence intensity in cardiomyocytes exposed to 500 μM H2O2 compared to the control; scale bar, 10 µm. (E) Statistical data showed the intensity of the calcein fluorescence in each group. Error bars represent SEM. *P < 0.05 vs control.
3.3. Mitochondrial localization of ARC was essential for its role in the inhibition of H2O2-induced necrotic cell death
It has been reported that ARC suppresses TNF-α-induced necrosis in mouse L929 fibrosarcoma cells by interacting with TNFR and disrupting RIP1 recruitment in the cytoplasm [27]. However, it is well known that ARC is mainly localized in the mitochondria [42]. Our results also showed that ARC significantly inhibited mPTP opening in H2O2-induced necrosis in cardiomyocytes. Therefore, we hypothesized that ARC was involved in the mitochondrial intrinsic necrosis pathway. Our previous results showed that the phosphorylation of ARC at the T149 residue by CK2 is involved in the localization to mitochondria in cardiomyocytes, whereas the nonphosphorylated ARC is unable to migrate to the mitochondria and remains distributed in the cytoplasm [28], [43]. We forced the expression of the mutated form of ARC with T149 converted to a nonphosphorylatable alanine residue (A149) by the adenoviral infection. As shown in Fig. 3A and Supplementary Fig. 4B, the forced expression of ARCT149A was mainly distributed in the cytoplasm which was consistent with the previous results [28], [43]. The normal ARC inhibited necrosis significantly compared to the control group, while mutated ARCT149A could not inhibit necrosis in H2O2-exposed cardiomyocytes (Fig. 3B and C). Furthermore, the mutated ARCT149A could not inhibit the opening of mPTP in H2O2-exposed cardiomyocytes (Fig. 3D). These results demonstrated that the role of ARC on the inhibition of H2O2-induced necrotic cell death was dependent on its mitochondrial localization.
Fig. 3.

Mitochondrial localization of ARC was essential for inhibition of H2O2-induced necrotic cell death. (A) Different distributions of ARCT149A in the cytosol and mitochondria. (B) Adenovirus could efficiently force the expression of ARC and ARCT149A in cardiomyocytes. The forced expression of ARC prevented necrosis, while forced expression of ARCT149A could not prevent necrosis in cardiomyocytes exposed to 500 μM H2O2 for 12 h. Necrosis was detected by PI assay (C) and indicated by LDH release assay (D). Error bars represent SEM. *P < 0.05 vs control. (E) Inner mitochondrial membrane potential (ΔΨm) was detected by TMRE staining at the indicated time. Forced expression of ARCT149A could not delay the loss of TMRE fluorescence intensity in cardiomyocytes exposed to 500 μM H2O2 compared with the control. Error bars represent SEM.
3.4. ARC inhibited H2O2-induced necrotic cell death by targeting CypD in cardiomyocytes
Our results showed that ARC was localized to mitochondria and significantly inhibited mPTP opening during H2O2-induced myocardial necrosis. Next, we explored the ARC mechanism for the inhibition of mPTP opening. CypD is the main regulator of mPTP, which can promote the opening of mPTP and induce necrosis in cardiomyocytes [44], [45]. First, we forced the expression of CypD in cardiomyocytes using the eukaryon expression plasmid with the EF1α promoter to confirm its role in the induction of necrosis in cardiomyocytes (Fig. 4A). The overexpression of CypD sensitized cardiomyocytes to undergo necrosis when exposed to H2O2 (100 μM) (Fig. 4B and C). Then, we used siRNA to knockdown the expression of endogenous CypD in cardiomyocytes (Fig. 4D). Our results showed that the knockdown of CypD protected cardiomyocytes from necrosis induced by H2O2 exposure (Fig. 4E and F). CsA is the specific inhibitor which binds with CypD and antagonizes its activity [16]. We found that treatment with CsA (5 μM) could also protect cardiomyocytes from necrosis induced by H2O2 (Fig. 4G and H). These results confirmed that CypD was involved in H2O2-induced myocardial necrosis.
Fig. 4.

CypD regulated H2O2-induced myocardial necrosis (A) Forced expression of CypD using the eukaryon expression plasmid with EF1α promoter in cardiomyocytes. Forced expression of CypD sensitized cardiomyocytes to undergo necrosis when exposed to 100 μM H2O2 for 12 h compared with the control. Necrosis was detected by PI assay (B) and indicated by LDH release assay (C); error bars represent SEM. *P < 0.05 vs control. (D) CypD was knocked down by using the small interfering RNA. Knockdown of CypD prevented H2O2-induced necrosis in cardiomyocytes exposed to 500 μM H2O2 for 12 h compared with the control. Necrosis was detected by PI assay (E) and indicated by LDH release assay (F); error bars represent SEM. *P < 0.05 vs control. CsA (5 μM) treatment prevented H2O2-induced necrosis in cardiomyocytes exposed to 500 μM H2O2 for 12 h compared with the control. Necrosis was detected by PI assay (G) and indicated by LDH release assay (H). Error bars represent SEM. *P < 0.05 vs control.
Then, we sought to investigate whether ARC inhibited mPTP opening and necrosis by targeting the CypD protein. It has been reported that the interacting proteins regulate the activity of CypD [20]. Our results showed that ARC colocalized with CypD in cardiomyocytes (Fig. 5A). We performed ectopic expression of HA-ARC and Myc-CypD in 293T cells and demonstrated that ARC could interact with CypD (Fig. 5B). We further confirmed the physical interaction between endogenous ARC and CypD by immunoprecipitation assay (Fig. 5C). Next, we explored whether ARC regulated myocardial necrosis by targeting CypD. Our results showed that the overexpression of ARC inhibited H2O2-induced necrosis compared to the control, whereas this inhibition was abolished by simultaneous overexpression of CypD (Fig. 5D). Moreover, the knockdown of ARC sensitized cardiomyocytes to undergo necrosis when exposed to H2O2 (100 μM) and this effect was also antagonized by the simultaneous knockdown of CypD (Fig. 5E and F). Furthermore, ARC could also inhibit necrosis induced by CypD overexpression in cardiomyocytes exposed to H2O2 (100 μM) as shown in Fig. 5G and H. Taken together, these results demonstrated that ARC inhibited necrosis by targeting CypD in cardiomyocytes.
Fig. 5.

ARC prevented mPTP opening and necrotic cell death by inhibiting the activity of CypD. (A) Colocalization of ARC with CypD in cardiomyocytes was detected by immunofluorescence staining. ARC, green; CypD, red; nuclei, blue; scale bar, 10 µm. (B) Interaction of ARC with CypD was detected by immunoprecipitation in 293 T cells with ectopic expression of HA-ARC and Myc-CypD. (C) Interaction of endogenous ARC with CypD in cardiomyocytes was detected by immunoprecipitation. (D) Overexpression of ARC inhibited necrosis in cardiomyocytes, which was abolished by simultaneous overexpression of CypD in cardiomyocytes exposed to 500 μM H2O2 for 12 h. Necrosis was detected by PI assay. Error bars represent SEM; *P < 0.05 vs control. Knockdown of ARC sensitized cardiomyocytes to undergo necrosis which was inhibited by simultaneous knockdown of CypD in cardiomyocytes exposed to 100 μM H2O2 for 12 h. Necrosis was detected by PI assay (E) and indicated by LDH release assay (F). Error bars represent SEM; *P < 0.05 vs control. Forced expression of CypD sensitized cardiomyocytes to undergo necrosis which was abolished by simultaneous overexpression of ARC. Necrosis was detected by PI assay (G) and indicated by LDH release assay (H). Error bars represent SEM; *P < 0.05 vs control.
3.5. p53 promoted myocardial necrosis by transcriptionally repressing ARC expression
As ARC was downregulated during the induction of necrosis, we explored the upstream regulator of ARC. In our previous work, we revealed that p53 could transcriptionally repress the expression of ARC during apoptosis induction [31]. Another study showed that p53 mediated necrosis by targeting CypD in MEF cells [23]. However, whether p53 is involved in myocardial necrosis remains largely unknown. The protein levels of p53 were significantly increased, which was conversely related to the decreased protein levels of ARC during necrosis induced by H2O2 in cardiomyocytes (Fig. 6A and Fig. 1A). Therefore, we hypothesized that p53 might participate in H2O2-induced myocardial necrosis by repressing the expression of ARC. To assess the role of p53 in myocardial necrosis, p53 was stably overexpressed in cardiomyocytes by adenoviral infection (Fig. 6B). Our result showed that overexpression of p53 sensitized cardiomyocytes to undergo necrosis upon H2O2 exposure (100 μM) (Supplementary Fig. 3A and Supplementary Fig. 3B). Conversely, the knockdown of p53 inhibited H2O2-induced necrosis in cardiomyocytes (Fig. 6F and Supplementary Fig. 3C). These results showed that p53 participated in H2O2-induced myocardial necrosis. Next, we examined the link between p53 and ARC in the regulation of necrosis. Our results showed that the overexpression of p53 could significantly decrease the expression levels of ARC in cardiomyocytes at both the mRNA and protein levels (Fig. 6B and D). Conversely, the knockdown of p53 could rescue the expression of ARC at both the mRNA and protein levels in cardiomyocytes exposed to H2O2 (500 μM) (Fig. 6C and E). The overexpression of p53 sensitized cardiomyocytes to undergo necrosis when exposed to H2O2 (100 μM), but either the simultaneous overexpression of ARC or the knockdown of CypD could attenuate the sensitivity of cardiomyocytes to oxidative stress (Supplementary Fig. 3D-3G). Furthermore, the knockdown of p53 inhibited necrosis induced by H2O2 exposure and this inhibition was abolished either by the simultaneous knockdown of ARC or by the overexpression of CypD (Fig. 6G and H). Taken together, these results demonstrated that p53 promoted myocardial necrosis by repressing ARC expression at the transcriptional level.
Fig. 6.

p53 promoted myocardial necrosis by transcriptionally repressing ARC expression. (A) p53 protein levels in cardiomyocytes treated with 500 μM H2O2 for the indicated time were detected by Western blotting. (B) Overexpression of p53 by adenovirus decreased the protein levels of ARC in cardiomyocytes. (C) Knockdown of p53 partially recovered the protein levels of ARC in cardiomyocytes exposed to 500 μM H2O2. (D) Overexpression of p53 decreased the mRNA levels of ARC. (E) Knockdown of p53 partially recovered the mRNA levels of ARC in cardiomyocytes exposed to 500 μM H2O2. (F) Knockdown of p53 inhibited necrosis in cardiomyocytes exposed to 500 μM H2O2. Necrosis was detected by PI assay. Error bars represent SEM; *P < 0.05 vs control. (G) Knockdown of p53 inhibited necrosis which was abolished by simultaneous knockdown of ARC. Necrosis was detected by PI assay. Error bars represent SEM; *P < 0.05 vs control. (H) Knockdown of p53 inhibited necrosis which was abolished by simultaneous overexpression of CypD. Necrosis was detected by PI assay. Error bars represent SEM; *P < 0.05 vs control.
3.6. ARC protected cardiomyocytes from necrotic cell death in the heart
After validating the role of ARC in H2O2-induced necrosis in vitro, we further investigated the role of ARC in the pathogenesis of cardiac infarction in a mouse model of I/R14. First, we examined the protein levels of ARC and p53 in the mice ischemic heart tissues. Our results showed that the protein levels of ARC were decreased but the protein levels of p53 were increased significantly in a time dependent manner in the ischemic heart tissues (Fig. 7A). Next, we examined whether ARC inhibited myocardial necrosis in the mouse I/R model. Our results showed that the adenovirus harboring, ARC overexpressing mice group had significantly reduced myocardial necrosis compared to the negative control group, as indicated by the PI staining (Fig. 7B and C) and by the LDH activity in the serum (Fig. 7D) after the I/R surgery. Moreover, the myocardial infarct size was also reduced in the ARC overexpressing mice group compared to the negative control group (Fig. 7E) during cardiac I/R damage. These results demonstrated that ARC protected cardiomyocytes from necrosis during the cardiac I/R injury.
Fig. 7.

ARC protected cardiomyocytes from necrotic cell death in the heart. (A) The protein levels of ARC and p53 in the ischemic heart tissues for the indicated ischemia time were detected by Western blotting. (B) Overexpression of ARC by adenovirus infection attenuated myocardial necrosis during cardiac ischemia/reperfusion (I/R). Mice were subjected to 30 min of LAD ligation followed by 3 h of reperfusion (n = 5/group). Propidium iodide (PI) was injected into the mice to label necrotic cells after the I/R. Representative images of ventricular myocardium sections are shown. Red, PI-positive cardiomyocyte nuclei; blue, 4′,6-diamidino-2-phenylindole–stained nuclei; green, α-actinin; scale bar, 40 µm. (C) Quantitative analysis of PI-positive cells is shown. Error bars represent SEM; *P < 0.05 vs control. (D) Serum LDH was measured 3 h after reperfusion. Error bars represent SEM; *P < 0.05 vs control. (E) The ratios of area at risk (AAR) to left ventricular (LV) area and infarct (INF) area to AAR are shown (n = 5/group). Error bars represent SEM; *P < 0.05 vs control; Scale bar, 1 mm.
It is well known that myocardial necrosis induces robust inflammation which further damages the heart during I/R injury [46], [47]. To assess the role of ARC in the inflammatory response after I/R injury, we quantified the expression levels of pro-inflammatory markers. Our results showed that pro-inflammatory markers, such as IL-1β, TNF-α and IL-6, were upregulated significantly during the I/R injury, but expression of these markers was attenuated significantly in the ARC overexpressing mice group compared to the negative control group (Fig. 8A, B and C).
Fig. 8.

ARC alleviated the inflammation caused by I/R injury and improved the long-term function of the heart after I/R injury. The levels of (A) interleukin (IL)-1β, (B) tumor necrosis factor-α (TNF-α) and (C) interleukin (IL)–6 were analyzed in mice I/R heart tissues by Q-PCR. Error bars represent SEM; *P < 0.05 vs control. Mice were treated as described in Fig. 7C (n = 5/group). (D) Representative photomicrographs show Masson trichrome staining for collagen. (E) The ratios of collagen area were shown. Error bars represent SEM; *P < 0.05 vs control; Scale bar, 60 µm. Echocardiographic analysis of left ventricular dimensions and cardiac function in mice (n = 5/group) 2 weeks after surgery; (F) EF, ejection fraction of left ventricular diameter; (G) FS, fractional shortening of left ventricular diameter; (H) LVIDd, diastolic left ventricular internal diameters. Error bars represent SEM; *P < 0.05 vs control.
Long-term cardiac function and remodeling has been considered an important criterion for cardio-protective strategies. Fibrosis of the myocardium is closely associated with cardiac remodeling and exacerbates the deterioration of heart function after I/R injury [48]. Our results showed that the collagen areas were reduced two weeks after I/R injury in the ARC overexpressing mice compared to the negative control group (Fig. 8D and E). Moreover, the ARC overexpressing mice group had improved cardiac function as shown by preservation of the ejection fraction (EF) and fractional shortening (FS) and by a decrease in heart chamber dimensions after myocardial I/R injury. (Fig. 8F, G and H). Taken together, these results demonstrated that ARC prevented myocardial necrosis and reduced the infarcted areas of the heart exposed to I/R injury in mice. Moreover, ARC also attenuated the inflammation induced in response to necrosis and improved long-term heart function after I/R injury in mice hearts. Our findings revealed a significant role for ARC in regulating the myocardial necrosis that suggests ARC as a potential therapeutic target for cardio-protection during ischemia and oxidative stress-related cardiac diseases.
4. Discussion
Necrosis is the major form of cell death during myocardial I/R injury [1], [3]. Understanding the underlying molecular mechanisms of necrosis will benefit clinical practice in protecting cardiomyocytes from I/R injury. In this study, we reported the pivotal role of ARC in regulating myocardial necrosis induced by oxidative stress. Protein levels of ARC significantly decreased in H2O2-induced necrosis and overexpression of ARC attenuated necrosis both in vitro and in vivo utilizing cardiomyocytes and the mouse model of I/R injury. Moreover, the inhibition of necrosis by ARC was critically dependent upon ARC localization to mitochondria. Mechanistically, ARC inhibited the opening of mPTP by targeting CypD in H2O2-induced necrosis in cardiomyocytes. Furthermore, we also confirmed that p53 was the upstream regulator of ARC in H2O2-induced necrosis and promoted myocardial necrosis by transcriptional suppression of ARC expression.
ARC has been reported to exert its strong cardio-protective properties through the inhibition of apoptosis. ARC binds to caspase-8 and caspase-2 through its CARD domain and inhibits apoptosis initiation [24], [25], [26]. ARC can also preserve mitochondrial integrity and prevents cytochrome c release by inhibiting Bax in cardiomyocytes [49]. Our data also showed that ARC significantly inhibited H2O2-induced apoptosis in cardiomyocytes and confirmed previously published results [31] (Supplementary Fig. 2A). Although the cardio-protective role of ARC has been revealed in apoptosis, the function of ARC remains unknown in necrosis. Necrotic cell death has been shown to be involved in human cardiac diseases and contributes several-fold more to disease pathogenesis than apoptosis [50]. The well-established concept of programmed necrosis has drawn more attention toward targeting necrosis in cardiac pathologies [51], [52]. Our present work has demonstrated the central role of ARC in the inhibition of oxidative stress-induced necrosis.
mPTP is a nonspecific pore in the inner mitochondrial membrane. The prolonged opening of mPTP usually converts the mitochondria from organelles that support cell survival to those that actively induce apoptotic and necrotic cell death [53]. There is increasing evidence that mPTP opening is of critical importance during cardiac I/R injury [16], [53]. Therefore, understanding the regulation of mPTP opening is crucial for clinical cardio-protection strategies. It has been reported that CypD is localized in the mitochondrial matrix but under oxidative stress it trans-locates to the inner mitochondrial membrane, allowing CypD to bind to ANT, the major pore-formation element that induces the opening of mPTP [54], [55]. CypD exhibits peptidyl prolyl cis/trans isomerase (PPIase) activity, which causes a conformational change in ANT that converts it into a nonspecific pore [53]. This activity of CypD is regulated by either posttranslational modification or protein-protein interactions. For instance, it has been reported that acetylation of CypD at lysine 166 promotes age-related cardiac hypertrophy by regulating the mPTP opening, which can be reversed by SIRT3-mediated deacetylation of CypD [21]. HAX-1 has been reported to regulate the activity of CypD through interference with CypD binding to a chaperon protein in mitochondria, leaving CypD prone to degradation [20]. However, we could not detect significant changes in the protein levels of CypD in both in vitro and in vivo. Therefore, our results suggested that ARC could possibly prevent CypD translocation to the mPTP complex from the mitochondrial matrix, keeping the mPTP pore inactive. The activation of JNK has been reported to promote the activity of CypD and mPTP opening [56]. It has also been reported that ARC inhibits JNK activation by specific interaction with JNK1 and JNK2 in hepatic cells [57]. Additionally, ARC has also been reported as an inhibitor of TNF-α-mediated necrosis in which ARC interferes with recruitment of RIP1, a critical mediator of TNF-α-induced necrosis [27]. RIP1 has been reported as a central molecule for the initiation of multiple pathways that can contribute in necrotic cell death. For instance, RIP1 can disrupt the interaction between ANT and CypD, and impairs the function of ANT and increases ROS production [58], [59]. However, there needs to be further exploration into whether ARC inhibits CypD through the JNK pathway or through interference with recruitment of RIP1 and/or RIP1 disruption of CypD in cardiomyocytes during oxidative stress.
The transcription factor p53 has been reported as a master regulator of the cardiac transcriptome and an important modulator during I/R injury [32], [60]. In our previous work, we found that the ARC promoter region contains two p53 binding sites, and p53 represses the expression of ARC at the transcriptional level during apoptosis induction [31]. p53 has also been shown to induce necrosis in MEF cells [23]. However, little is known about the role of p53 in myocardial necrosis. Our results highlighted that p53 promoted myocardial necrosis by the transcriptional suppression of ARC in cardiomyocytes.
It has been reported that p53 could directly interact with CypD in MEF cells [23]. In our work, we found that ARC could regulate necrosis by targeting CypD, and p53 suppressed the expression of ARC at the transcriptional level to initiate necrosis. It is well known that ARC is abundant exclusively in terminally differentiated cells, for instance, cardiomyocytes and skeletal muscle cells, as well as some cancer cell lines, and is an important protein for maintaining cell survival [24], [61]. The protein levels of ARC in cardiomyocytes determined the switching point from cell survival to cell death. Therefore, we reasoned that the transcriptional suppression of ARC by p53 was an important step for necrosis initiation in cardiomyocytes. However, whether p53 could directly target CypD in cardiomyocytes needs to be explored further.
5. Conclusion
In summary, our data revealed the pivotal role of ARC in myocardial necrosis and delineated the p53-ARC-CypD/mPTP central necrosis pathway, which can provide a potential therapeutic avenue for ischemia and oxidative stress-related cardiac diseases.
Acknowledgements
Thanks very much to Dr. AKRAM for the language help.
Acknowledgments
Funding
This work was supported by National Natural Science Foundation of China (81622005 and 81770232); Natural Science Foundation of Shandong Province (JQ201815); China Postdoctoral Science Foundation (2016M602095). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Declarations of interest
None.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.10.023
Appendix A. Supplementary material
Supplementary Fig. 1.

(A) Necrosis and apoptosis were detected simultaneously in cardiomyocytes exposed to 500 μM H2O2 or 100 μM H2O2 at the indicated time by the PI and TUNEL assays. Representative photomicrographs show the PI and TUNEL double staining in the cardiomyocytes. (B) Statistical data showed the ratio of TUNEL positive cells and PI positive cells to the total cardiomyocytes exposed to 500 μM H2O2 or 100 μM H2O2. Error bars represent SEM; *P < 0.05. (C) Forced expression of ARC prevented H2O2-induced necrosis in cardiomyocytes exposed to 500 μM H2O2 for 12 h compared to the control. (D) Knockdown of ARC increased H2O2-induced necrosis in cardiomyocytes exposed to 100 μM H2O2 for 12 h compared with the control. Necrosis was indicated by LDH release assay. Error bars represent SEM; *P < 0.05.
Supplementary Fig. 2.

(A) ARC overexpression inhibited apoptosis in cardiomyocytes exposed to 100 μM H2O2 for 24 h. Apoptosis was detected by TUNEL assay. Error bars represent SEM; *P < 0.05 vs control. (B) CsA (5 μM) delayed the loss of TMRE fluorescence in cardiomyocytes exposed to 500 μM H2O2 compared with the control. Error bars represent SEM; *P < 0.05 vs control.
Supplementary Fig. 3.

(A) Overexpression of p53 sensitized cardiomyocytes to undergo necrosis when exposed to 100 μM H2O2. Necrosis was indicated by LDH release assay. Error bars represent SEM; *P < 0.05 vs control. (B) Overexpression of p53 sensitized cardiomyocytes to undergo necrosis when exposed to 100 μM H2O2; Necrosis was detected by PI assay; Error bars represent SEM; *P < 0.05 vs control. (C) Knockdown of p53 inhibited necrosis in cardiomyocytes exposed to 500 μM H2O2. Necrosis was indicated by LDH release assay; Error bars represent SEM; *P < 0.05 vs control. (D) Overexpression of p53 sensitized cardiomyocytes to undergo necrosis which was prevented by simultaneous overexpression of ARC; Necrosis was detected by PI assay; Error bars represent SEM; *P < 0.05 vs control. (E) Overexpression of p53 sensitized cardiomyocytes to undergo necrosis which was prevented by simultaneous knockdown of CypD; Necrosis was detected by PI assay; Error bars represent SEM; *P < 0.05 vs control. (F) Overexpression of p53 sensitized cardiomyocytes to undergo necrosis which was prevented by simultaneous overexpression of ARC; Necrosis was indicated by LDH release assay; Error bars represent SEM; *P < 0.05 vs control. (G) Overexpression of p53 sensitized cardiomyocytes to undergo necrosis which was prevented by simultaneous knockdown of CypD; Necrosis was indicated by LDH release assay; Error bars represent SEM; *P < 0.05 vs control.
Supplementary Fig. 4.

Full gel exposure of the Western blot results indicating the expression of the ARC during necrosis induction. (B) Full gel exposure of the Western blot results indicating the different distribution of ARCT149A in the cytosol and mitochondria. (C) Full gel exposure of the Western blot results indicating the expression of ARC in the ischemic heart tissues.
References
- 1.Whelan R.S., Kaplinskiy V., Kitsis R.N. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu. Rev. Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PubMed] [Google Scholar]
- 2.Konstantinidis K., Whelan R.S., Kitsis R.N. Mechanisms of cell death in heart disease. Arterioscler. Thromb. Vasc. Biol. 2012;32(7):1552–1562. doi: 10.1161/ATVBAHA.111.224915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu H., Sun A. Programmed necrosis in heart disease: molecular mechanisms and clinical implications. J. Mol. Cell. Cardiol. 2018;116:125–134. doi: 10.1016/j.yjmcc.2018.01.018. [DOI] [PubMed] [Google Scholar]
- 4.Kung G., Konstantinidis K., Kitsis R.N. Programmed necrosis, not apoptosis, in the heart. Circ. Res. 2011;108(8):1017–1036. doi: 10.1161/CIRCRESAHA.110.225730. [DOI] [PubMed] [Google Scholar]
- 5.Zong W.X., Thompson C.B. Necrotic death as a cell fate. Genes Dev. 2006;20(1):1–15. doi: 10.1101/gad.1376506. [DOI] [PubMed] [Google Scholar]
- 6.He S., Wang L., Miao L., Wang T., Du F., Zhao L. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 7.Zhang D.W., Shao J., Lin J., Zhang N., Lu B.J., Lin S.C. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 8.Wang J.X., Zhang X.J., Li Q., Wang K., Wang Y., Jiao J.Q. MicroRNA-103/107 regulate programmed necrosis and myocardial ischemia/reperfusion injury through targeting FADD. Circ. Res. 2015;117(4):352–363. doi: 10.1161/CIRCRESAHA.117.305781. [DOI] [PubMed] [Google Scholar]
- 9.Zhang T., Zhang Y., Cui M., Jin L., Wang Y., Lv F. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016;22(2):175–182. doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]
- 10.Yang Z., Wang Y., Zhang Y., He X., Zhong C.Q., Ni H. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell Biol. 2018;20(2):186–197. doi: 10.1038/s41556-017-0022-y. [DOI] [PubMed] [Google Scholar]
- 11.J. Karch, J.D. Molkentin, Identifying the components of the elusive mitochondrial permeability transition pore, in: Proceedings of the National Academy of Sciences of the United States of America, vol. 111(29), 2014, pp. 10396–10397. [DOI] [PMC free article] [PubMed]
- 12.Baines C.P., Song C.X., Zheng Y.T., Wang G.W., Zhang J., Wang O.L. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ. Res. 2003;92(8):873–880. doi: 10.1161/01.RES.0000069215.36389.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karch J., Kwong J.Q., Burr A.R., Sargent M.A., Elrod J.W., Peixoto P.M. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife. 2013;2:e00772. doi: 10.7554/eLife.00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Z., Alloush J., Beck E., Weisleder N. A murine model of myocardial ischemia-reperfusion injury through ligation of the left anterior descending artery. J. Vis. Exp.: JoVE. 2014;(86) doi: 10.3791/51329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakayama H., Chen X., Baines C.P., Klevitsky R., Zhang X., Zhang H. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J. Clin. Investig. 2007;117(9):2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakagawa T., Shimizu S., Watanabe T., Yamaguchi O., Otsu K., Yamagata H. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434(7033):652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 17.Baines C.P., Kaiser R.A., Purcell N.H., Blair N.S., Osinska H., Hambleton M.A. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434(7033):658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 18.Kajitani K., Fujihashi M., Kobayashi Y., Shimizu S., Tsujimoto Y., Miki K. Crystal structure of human cyclophilin D in complex with its inhibitor, cyclosporin A at 0.96-A resolution. Proteins. 2008;70(4):1635–1639. doi: 10.1002/prot.21855. [DOI] [PubMed] [Google Scholar]
- 19.Kroemer G., Galluzzi L., Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007;87(1):99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 20.C.K. Lam, W. Zhao, G.S. Liu, W.F. Cai, G. Gardner, G. Adly, , et al., HAX-1 regulates cyclophilin-D levels and mitochondria permeability transition pore in the heart, in: Proceedings of the National Academy of Sciences of the United States of America, vol. 112(47), 2015, pp. E6466–E6475. [DOI] [PMC free article] [PubMed]
- 21.Hafner A.V., Dai J., Gomes A.P., Xiao C.Y., Palmeira C.M., Rosenzweig A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging. 2010;2(12):914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boengler K., Hilfiker-Kleiner D., Heusch G., Schulz R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 2010;105(6):771–785. doi: 10.1007/s00395-010-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaseva A.V., Marchenko N.D., Ji K., Tsirka S.E., Holzmann S., Moll U.M. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149(7):1536–1548. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.T. Koseki, N. Inohara, S. Chen, Nunez G. , ARC, an inhibitor of apoptosis expressed in skeletal muscle and heart that interacts selectively with caspases, in: Proceedings of the National Academy of Sciences of the United States of America, vol. 95(9), 1998, pp. 5156–5160. [DOI] [PMC free article] [PubMed]
- 25.Zhang Y.Q., Herman B. ARC protects rat cardiomyocytes against oxidative stress through inhibition of caspase-2 mediated mitochondrial pathway. J. Cell. Biochem. 2006;99(2):575–588. doi: 10.1002/jcb.20946. [DOI] [PubMed] [Google Scholar]
- 26.Jo D.G., Jun J.I., Chang J.W., Hong Y.M., Song S., Cho D.H. Calcium binding of ARC mediates regulation of caspase 8 and cell death. Mol. Cell. Biol. 2004;24(22):9763–9770. doi: 10.1128/MCB.24.22.9763-9770.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kung G., Dai P., Deng L., Kitsis R.N. A novel role for the apoptosis inhibitor ARC in suppressing TNFalpha-induced regulated necrosis. Cell Death Differ. 2014;21(4):634–644. doi: 10.1038/cdd.2013.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li P.F., Li J., Muller E.C., Otto A., Dietz R., von Harsdorf R. Phosphorylation by protein kinase CK2: a signaling switch for the caspase-inhibiting protein ARC. Mol. Cell. 2002;10(2):247–258. doi: 10.1016/s1097-2765(02)00600-7. [DOI] [PubMed] [Google Scholar]
- 29.Tan W.Q., Wang J.X., Lin Z.Q., Li Y.R., Lin Y., Li P.F. Novel cardiac apoptotic pathway: the dephosphorylation of apoptosis repressor with caspase recruitment domain by calcineurin. Circulation. 2008;118(22):2268–2276. doi: 10.1161/CIRCULATIONAHA.107.750869. [DOI] [PubMed] [Google Scholar]
- 30.Chiang S., Kalinowski D.S., Jansson P.J., Richardson D.R., Huang M.L. Mitochondrial dysfunction in the neuro-degenerative and cardio-degenerative disease, Friedreich's ataxia. Neurochem. Int. 2017 doi: 10.1016/j.neuint.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 31.Li Y.Z., Lu D.Y., Tan W.Q., Wang J.X., Li P.F. p53 initiates apoptosis by transcriptionally targeting the antiapoptotic protein ARC. Mol. Cell. Biol. 2008;28(2):564–574. doi: 10.1128/MCB.00738-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J.X., Jiao J.Q., Li Q., Long B., Wang K., Liu J.P. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 2011;17(1):71–78. doi: 10.1038/nm.2282. [DOI] [PubMed] [Google Scholar]
- 33.Wang J.X., Li Q., Li P.F. Apoptosis repressor with caspase recruitment domain contributes to chemotherapy resistance by abolishing mitochondrial fission mediated by dynamin-related protein-1. Cancer Res. 2009;69(2):492–500. doi: 10.1158/0008-5472.CAN-08-2962. [DOI] [PubMed] [Google Scholar]
- 34.Choi K., Kim J., Kim G.W., Choi C. Oxidative stress-induced necrotic cell death via mitochondira-dependent burst of reactive oxygen species. Curr. Neurovasc. Res. 2009;6(4):213–222. doi: 10.2174/156720209789630375. [DOI] [PubMed] [Google Scholar]
- 35.Saberi B., Shinohara M., Ybanez M.D., Hanawa N., Gaarde W.A., Kaplowitz N. Regulation of H(2)O(2)-induced necrosis by PKC and AMP-activated kinase signaling in primary cultured hepatocytes. Am. J. Physiol. Cell Physiol. 2008;295(1):C50–C63. doi: 10.1152/ajpcell.90654.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saito Y., Nishio K., Ogawa Y., Kimata J., Kinumi T., Yoshida Y. Turning point in apoptosis/necrosis induced by hydrogen peroxide. Free Radic. Res. 2006;40(6):619–630. doi: 10.1080/10715760600632552. [DOI] [PubMed] [Google Scholar]
- 37.Sawai H., Domae N. Discrimination between primary necrosis and apoptosis by necrostatin-1 in Annexin V-positive/propidium iodide-negative cells. Biochem. Biophys. Res. Commun. 2011;411(3):569–573. doi: 10.1016/j.bbrc.2011.06.186. [DOI] [PubMed] [Google Scholar]
- 38.R.S. Whelan, K. Konstantinidis, A.C. Wei, Y. Chen, D.E. Reyna, S. Jha, , et al., Bax regulates primary necrosis through mitochondrial dynamics, in: Proceedings of the National Academy of Sciences of the United States of America, vol. 109(17), 2012, pp. 6566–6571. [DOI] [PMC free article] [PubMed]
- 39.Crompton M., Ellinger H., Costi A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 1988;255(1):357–360. [PMC free article] [PubMed] [Google Scholar]
- 40.Katoh H., Nishigaki N., Hayashi H. Diazoxide opens the mitochondrial permeability transition pore and alters Ca2+ transients in rat ventricular myocytes. Circulation. 2002;105(22):2666–2671. doi: 10.1161/01.cir.0000016831.41648.04. [DOI] [PubMed] [Google Scholar]
- 41.Petronilli V., Miotto G., Canton M., Brini M., Colonna R., Bernardi P. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys. J. 1999;76(2):725–734. doi: 10.1016/S0006-3495(99)77239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ekhterae D., Lin Z., Lundberg M.S., Crow M.T., Brosius F.C., 3rd, Nunez G. ARC inhibits cytochrome c release from mitochondria and protects against hypoxia-induced apoptosis in heart-derived H9c2 cells. Circ. Res. 1999;85(12):e70–e77. doi: 10.1161/01.res.85.12.e70. [DOI] [PubMed] [Google Scholar]
- 43.Murtaza I., Wang H.X., Feng X., Alenina N., Bader M., Prabhakar B.S. Down-regulation of catalase and oxidative modification of protein kinase CK2 lead to the failure of apoptosis repressor with caspase recruitment domain to inhibit cardiomyocyte hypertrophy. J. Biol. Chem. 2008;283(10):5996–6004. doi: 10.1074/jbc.M706466200. [DOI] [PubMed] [Google Scholar]
- 44.Matas J., Young N.T., Bourcier-Lucas C., Ascah A., Marcil M., Deschepper C.F. Increased expression and intramitochondrial translocation of cyclophilin-D associates with increased vulnerability of the permeability transition pore to stress-induced opening during compensated ventricular hypertrophy. J. Mol. Cell. Cardiol. 2009;46(3):420–430. doi: 10.1016/j.yjmcc.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 45.A.C. Schinzel, O. Takeuchi, Z. Huang, J.K. Fisher, Z. Zhou, J. Rubens, , et al., Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia, in: Proceedings of the National Academy of Sciences of the United States of America, vol. 102(34), 2005, pp. 12005–12010. [DOI] [PMC free article] [PubMed]
- 46.Frangogiannis N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014;11(5):255–265. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cusack M.R., Marber M.S., Lambiase P.D., Bucknall C.A., Redwood S.R. Systemic inflammation in unstable angina is the result of myocardial necrosis. J. Am. Coll. Cardiol. 2002;39(12):1917–1923. doi: 10.1016/s0735-1097(02)01899-5. [DOI] [PubMed] [Google Scholar]
- 48.Janicki J.S., Brower G.L. The role of myocardial fibrillar collagen in ventricular remodeling and function. J. Card. Fail. 2002;8(6 Suppl.):S319–S325. doi: 10.1054/jcaf.2002.129260. [DOI] [PubMed] [Google Scholar]
- 49.Gustafsson A.B., Tsai J.G., Logue S.E., Crow M.T., Gottlieb R.A. Apoptosis repressor with caspase recruitment domain protects against cell death by interfering with Bax activation. J. Biol. Chem. 2004;279(20):21233–21238. doi: 10.1074/jbc.M400695200. [DOI] [PubMed] [Google Scholar]
- 50.Chiong M., Wang Z.V., Pedrozo Z., Cao D.J., Troncoso R., Ibacache M. Cardiomyocyte death: mechanisms and translational implications. Cell Death Dis. 2011;2:e244. doi: 10.1038/cddis.2011.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou W., Yuan J. Necroptosis in health and diseases. Semin. Cell Dev. Biol. 2014;35:14–23. doi: 10.1016/j.semcdb.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 52.Pavlosky A., Lau A., Su Y., Lian D., Huang X., Yin Z. RIPK3-mediated necroptosis regulates cardiac allograft rejection. Am. J. Transplant.: Off. J. Am. Soc. Transplant. Am. Soc. Transplant. Surg. 2014;14(8):1778–1790. doi: 10.1111/ajt.12779. [DOI] [PubMed] [Google Scholar]
- 53.Halestrap A.P., Clarke S.J., Javadov S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc. Res. 2004;61(3):372–385. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 54.Halestrap A.P., Woodfield K.Y., Connern C.P. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J. Biol. Chem. 1997;272(6):3346–3354. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- 55.Connern C.P., Halestrap A.P. Recruitment of mitochondrial cyclophilin to the mitochondrial inner membrane under conditions of oxidative stress that enhance the opening of a calcium-sensitive non-specific channel. Biochem. J. 1994;302(Pt 2):321–324. doi: 10.1042/bj3020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou C., Chen Z., Lu X., Wu H., Yang Q., Xu D. Icaritin activates JNK-dependent mPTP necrosis pathway in colorectal cancer cells. Tumour Biol.: J. Int. Soc. Oncodev. Biol. Med. 2016;37(3):3135–3144. doi: 10.1007/s13277-015-4134-3. [DOI] [PubMed] [Google Scholar]
- 57.An J., Mehrhof F., Harms C., Lattig-Tunnemann G., Lee S.L., Endres M. ARC is a novel therapeutic approach against acetaminophen-induced hepatocellular necrosis. J. Hepatol. 2013;58(2):297–305. doi: 10.1016/j.jhep.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 58.Vanden Berghe T., Declercq W., Vandenabeele P. NADPH oxidases: new players in TNF-induced necrotic cell death. Mol. Cell. 2007;26(6):769–771. doi: 10.1016/j.molcel.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 59.Lin Y., Choksi S., Shen H.M., Yang Q.F., Hur G.M., Kim Y.S. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J. Biol. Chem. 2004;279(11):10822–10828. doi: 10.1074/jbc.M313141200. [DOI] [PubMed] [Google Scholar]
- 60.Sano M., Minamino T., Toko H., Miyauchi H., Orimo M., Qin Y. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446(7134):444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 61.Wang M., Qanungo S., Crow M.T., Watanabe M., Nieminen A.L. Apoptosis repressor with caspase recruitment domain (ARC) is expressed in cancer cells and localizes to nuclei. FEBS Lett. 2005;579(11):2411–2415. doi: 10.1016/j.febslet.2005.03.040. [DOI] [PubMed] [Google Scholar]