

Summary
Formation of supramolecular assemblies appears to be a general mechanism in immune signalling pathways. These supramolecular assemblies appear to form through a nucleated polymerization mechanism. This review examines selected immune signalling pathways that involve supramolecular assemblies, describes the concepts of protein polymerization, and discusses how those concepts of protein polymerization implicate new elegant ways for signal amplification, setting threshold and noise reduction in these pathways.
Keywords: absent in melanoma 2‐like receptors, inflammasome, killer‐cell immunoglobulin‐like receptor, nucleotide‐binding oligomerization domain‐like receptors, retinoic acid‐inducible gene‐I‐like receptors, signalosome
Introduction
Organisms have evolved complex immune systems that sense and protect them from pathogens. Innate immune systems defend against infection by sensing pathogen patterns, and thus innate immune sensing is considered relatively non‐specific. In addition to the innate defence mechanisms, vertebrates have evolved adaptive immune systems, which sense and respond against specific antigens. A distinctive feature of adaptive immunity is its ability to register a memory, wherein the responses against the second encounter with the same antigen are much more robust and rapid, and often prevent a second invasion by the pathogen.
The cells of immune systems use a large number of receptors to sense the health of host cells. The receptors of the innate immune system are cell surface, endosomal and cytosolic pattern recognition receptors (PRRs).1, 2 They recognize conserved pathogen‐associated molecular patterns present on microbes and intrinsic danger‐associated molecular patterns generated by internal injury. The Toll‐like receptors (TLRs), nucleotide‐binding and oligomerization domain (NOD) ‐like receptors (NLRs), absent in melanoma 2 (AIM2) ‐like receptors (ALRs), retinoic acid‐inducible gene‐I (RIG‐I) ‐like receptors (RLRs), and cGAS/STING are the major PRR families.1, 2 Signalling through PRRs induces nuclear factor‐κB (NF‐κB) activation, interferon expression and caspase‐1 activation. The inflammatory responses induced by NF‐κB activation, antiviral response by interferons, and cytokine maturation and cell death induction by caspase‐1 constitute the crucial innate defence mechanisms.1, 2
Traditionally, B and T lymphocytes have been considered to be the mediators of the adaptive immune system. Recently, natural killer (NK) lymphocytes have been shown to possess immunological memory.3, 4, 5 Therefore, NK cells possess features of both innate and adaptive immunity. The B‐cell receptors (BCR) and T‐cell receptors (TCR) mediate antigen recognition by B cells and T cells, and along with other receptors/co‐receptors trigger cell activation. Somatic DNA rearrangements in individual B and T cells generate highly diverse BCR and TCR repertoires, enabling these cells to recognize and respond to a huge number of distinct antigens. In contrast, germ‐line‐encoded receptors control immune sensing and functions of NK cells.6, 7, 8 Natural killer cells express an array of activating receptors, including the natural cytotoxicity receptors (NKp30, NKp44 and NKp46), SLAM receptors (such as 2B4), DNAM‐1, CD2 and CD44. The NK cells express many major histocompatibility complex class I‐specific inhibitory receptors.6, 7, 8 Human NK cells express two major classes of inhibitory receptors, inhibitory members of killer‐cell immunoglobulin‐like receptors (KIRs) and the NKG2A‐CD94 heterodimer.7, 8 KIR recognizes HLA‐C expression, and NKG2A recognizes HLA‐E expression on other cells. The activation of NK cells is under the tight control of dominant inhibition exerted by these inhibitory receptors.7, 8
A principal means of controlling functions and coordinating cell–cell communications in the immune system is through a set of small molecules referred to as cytokines.9, 10, 11, 12, 13 There are several families of cytokines, including the type I cytokines [interleukin‐2 (IL‐2), IL‐3, IL‐4, IL‐5, IL‐6, IL‐7, IL‐9, IL‐11, IL‐12, IL‐13, IL‐15, IL‐21, IL‐23, IL‐27 and several haematopoietic growth factors], the type II cytokines [type I interferons (IFNs), IFN‐γ, IL‐10 family cytokines], tumour necrosis factor (TNF) ‐related cytokines (TNFs, lymphotoxin and Fas ligands), immunoglobulin superfamily cytokines (IL‐1, IL‐18, IL‐33, IL‐36 and IL‐37) and chemokines.12, 13 These cytokines are produced in response to various stimuli and act by binding to their cognate receptors expressed on target cells. The type I cytokine receptors consist typically of two or more type I transmembrane chains and elicit a variety of immunological events.9, 10, 13 The receptors of type I IFNs are composed of at least two transmembrane polypeptide chains, and generally induce anti‐proliferative and anti‐viral responses and stimulate cytolytic functions of lymphocytes.14 The receptors for IFN‐γ are expressed on all cell types except erythrocytes. The functional human IFN‐γ receptor is composed of two chains, IFNGR‐1 and IFNGR‐2.14, 15 The signals from IFN‐γ receptors promote antiviral activities and exert a range of actions to control the pro‐inflammatory and anti‐inflammatory host responses.14, 15 The signals from IFN‐γ receptors are important activators of macrophages.15 Interleukin‐10 receptor is composed of IL‐10R1 and IL‐10R2 chains and is a principal mediator of anti‐inflammatory responses.16, 17 Most of the members of the TNF receptor family are type I transmembrane glycoproteins, and signals from them control cellular growth, differentiation and survival.18, 19 The receptors of chemokines are specific G‐protein‐coupled receptors (GPCRs) on leucocytes that signal to control multiple immunological events, including immune cell development, transendothelial migration of leucocytes, and activation of phagocytes and lymphocytes.11, 20 The members of the IL‐1 receptor family are receptors for pro‐inflammatory cytokines such as IL‐1β.21 These receptors consist of a primary chain that, upon cytokine binding, usually associates with an accessory chain. The cytosolic domain of IL‐1 receptor family, the Toll‐IL‐1 receptor resistance (TIR) domain, is the same as that of the TLRs, hence IL‐1 receptor family signalling pathways are similar to those of the TLRs.21, 22, 23 The IL‐1 family of cytokines and receptors are the major mediators of the innate response against infection that triggers specific adaptive responses.21, 23
Traditionally, signalling is thought to occur through freely diffusible molecules, which is indeed the case for secondary messengers such as cAMP in GPCR signalling.24 However, recent studies have revealed that the protein components of many pathways signal by assembling into higher‐order complexes that could resemble semi‐solid states.25, 26, 27 The formation of such supramolecular assemblies is often mediated by protein–protein interaction domains of constituent molecules.25, 26 Formation of supramolecular assemblies appears to be a general mechanism employed by many signalling pathways. This has led to the proposal of new sets of rules for describing the basic events, such as signal amplification, setting threshold and spatiotemporal regulations, of signal transduction.25, 26 This review examines selected immune signalling pathways that are seen to involve supramolecular assemblies, discusses briefly the principles of protein assembly reactions, and describes the potential implications of protein polymerization principles in signalling through supramolecular assemblies.
Supramolecular assemblies in immune signalling by cytosolic receptors: intracellular inflammasomes and signalosomes
Although a few TLR members localize to endosomes, other members function at the cell surface.28 The ALRs, NLRs and RLRs are the major intracellular PRRs.1, 29
The ALR family members are the major PRRs for foreign dsDNA. AIM2 is the prototypic member of this family.30, 31, 32 The number of ALR genes in mammals varies; four in humans (AIM2, IFI16 , PHYHIN1 and MNDA) and 14 in mouse.33, 34 ALRs are composed of an N‐terminal pyrin domain and one or two C‐terminal HIN domain(s).22 AIM2 contains a single HIN domain, but IFI16 possesses two tandem HIN domains.22 AIM2 recognizes cytosolic DNA,32, 35 and IFI16 recognizes both cytosolic and nuclear DNA.35, 36, 37
The NLR family members respond to many stimuli.38, 39 NLRs possess more complex domain architectures than ALRs. The NLR family members contain a C‐terminal leucine‐rich repeat domain, a middle NOD, and a member‐specific N‐terminal domain.22 The N‐terminal domains in NLRB, NLRC and NLRP are the BIR domain, caspase recruitment domain (CARD) and pyrin domain (PYD), respectively.22 NLRP is the largest subfamily of NLRs, and NLRP3 is a well‐studied member.40
Members of the RLR family sense and respond to cytosolic viral RNA.41, 42, 43 RIG‐1 and MDA5 are the two well‐studied RLRs.41, 42, 43 They share a high sequence homology and consist of the same domain architecture. They consist of the N‐terminal tandem CARD domain (2CARD), central DExD/H‐box helicase domain, and Zn2+‐binding C‐terminal domain.35, 43 Like many other proteins that possess similar helicase domains, RIG‐1 and MDA5 lack duplex‐unwinding activity.35, 44 Despite similarities in sequences and domain architectures, RIG‐1 and MDA5 have distinct specificities for viral RNAs; they recognize different sets of viruses and viral RNAs.35, 42, 45, 46, 47, 48
The ALRs, NLRs and RLRs appear to share the common feature of self‐associating, and thereby promoting assembly of other molecules, including the effectors, into supramolecular complexes. These supramolecular complexes are called signalosomes, except for those of ALR and NLR members, which are called inflammasomes. It appears that intracellular PRRs reside in auto‐inhibited conformations that are activated upon ligand binding.
ALR‐ and NLR‐mediated inflammasomes
There is no structural study on HIN–PYD interactions in AIM2. However, isolated HIN and PYD domains of AIM2 interact with one another, and the interaction is diminished in the presence of dsDNA.49, 50 Charged residues of PYD's α helices 1 and 2 mediate the binding of isolated HIN with isolated PYD. Therefore, conceivably, full‐length AIM2 is maintained in an auto‐inhibited conformation, wherein the PYD domain docks onto HIN's charged DNA‐binding region.49, 50 The auto‐inhibition could be relieved when AIM2 binds to dsDNA through its HIN domain.49, 50 Conceivably, the long (~50 residues) linker between the HIN and PYD promotes this structural transition in AIM2. Structure of a mouse NLRC4, which lacked CARD (mNLRC4∆CARD), showed that its leucine‐rich repeat domain docks onto the NOD domain to maintain an auto‐inhibited conformation.51 The CARD domain of monomeric NLRC4 fails to recruit caspase‐1, suggesting that the auto‐inhibited NLRC4 sequesters its CARD domain to avoid uncontrolled interactions.51 Conceivably, other NLRs and ALRs are also maintained in auto‐inhibited conformations to avoid uncontrolled signalling.
Upon recognizing dsDNA, AIM2 undergoes self‐association. Recombinant AIM2 forms large aggregates in the presence of dsDNA.32 The full‐length AIM2 could polymerize in vitro into filaments even in the absence of dsDNA, albeit at high protein concentration.52 This observation challenges the auto‐inhibition model of AIM2 regulation and suggests that DNA binding promotes AIM2 polymerization by enhancing AIM2's local concentration.52 Recombinant PYD of AIM2 (AIM2PYD) polymerizes into filaments. Like most of the other components of inflammasomes, purification of monomeric AIM2PYD needs it to be tagged – for example, with MBP – and, immediately upon the removal of the tag, AIM2PYD forms extensive bundles and precipitates.53 Interestingly, bacterial expression of green fluorescent protein (GFP) ‐tagged AIM2PYD generates relatively homogeneous filaments that do not precipitate.53 At high concentration, GFP‐AIM2PYD polymerizes spontaneously.53 Whether or not ectopic expression of AIM2PYD in cells leads to its polymerization and bypasses the requirement of dsDNA for inflammasome assembly is not investigated. Nevertheless, in macrophage cells that express GFP‐tagged AIM2, transfection of DNA induces clustering of AIM2 around the DNA and pyroptotic death of those macrophages.32 These observations indicate that upon dsDNA binding, AIM2 undergoes self‐association through its PYD. AIM2HIN could also contribute to AIM2 self‐association.52
AIM2‐mediated inflammasome assembly and activation require the bipartite signalling adaptor ASC that contains an N‐terminal PYD and a C‐terminal CARD. Interaction of AIM2PYD with ASCPYD is required for inflammasome assembly and activation.31, 32 An inflammasome is a speck‐like structure, as seen by light microscope in cells, which, apart from other components, contains pro‐caspase‐1. These specks are the sites where the recruited caspase‐1 molecules are activated through proximity. Ectopic expression of fluorophore‐tagged individual domains of ASC in ASC‐deficient cells has demonstrated that speck formation occurs in separable steps.54 Upon induction, cells expressing ASCPYD, instead of the full‐length ASC, could form ASCPYD‐containing filaments that do not progress to the ASC specks.54 Both domains of ASC (ASCPYD and ASCCARD) are required for the speck formation in cells. Induction of cells expressing fluorophore‐tagged ASCCARD, instead of the full‐length ASC, could not lead to ASC specks, underscoring the requirement of ASCCARD for post‐filament assembly reactions in cells.54 Isolated AIM2PYD or isolated ASCPYD could polymerize into filaments in vitro.53, 55 When co‐expressed with ASCPYD in bacteria, AIM2PYD forms smaller filaments that are located at one end of the much longer ASCPYD filaments.55 Although ASCPYD alone polymerizes in vitro, its polymerization rate is much faster in the presence of AIM2PYD or AIM2–dsDNA complex.55 Therefore, AIM2 polymers could act as nuclei for recruitment and polymerization of ASC through PYD–PYD interactions. In ASCPYD filaments, the C‐termini of ASCPYD, which link ASCPYD to ASCCARD, are pointed outwards away from the filament core.55 Mixing of purified AIM2PYD, full‐length ASC (ASCFL) and caspase‐1CARD leads to star‐shaped AIM2 inflammasomes.55 The ASC resides at the centre of the stars.55 Unlike the case of AIM2PYD–ASCPYD binary complexes, wherein ASCPYD polymerized into long filaments, ASC does not polymerize into long filaments in these star‐shaped ternary complexes.55 Caspase‐1CARD constitutes the long arms of the stars.55 The stoichiometries of the components in inflammasomes purified from cells are consistent with the properties of in vitro reconstituted inflammasomes.55
These data suggest the following model for the ternary AIM2 inflammasome assembly. Upon recognizing dsDNA, AIM2 assembles on the dsDNA and nucleates ASC polymerization through PYD–PYD interactions. ASC polymerizes into short filaments through PYD–PYD interactions, and ASCCARD initiates polymerization of caspase‐1, through CARD–CARD interactions, into long filaments. Molecular proximity among caspase‐1 molecules in filaments leads to caspase‐1 activation.35, 56, 57 Despite having more complex domain architecture, assembly of NLRP3 inflammasomes, which is ASC‐dependent, occurs by similar mechanisms.35, 55, 56, 57 In this unified assembly mechanism of ASC‐dependent inflammasomes,55 a sensor (AIM2 or NLRP3), upon ligand binding, undergoes aggregation of the sensor nucleate ASC polymerization, and ASC polymers nucleate caspase‐1 (effector) polymerization and thereby activate caspase‐1 through autoproteolysis. In this unified mechanism,55 PYD–PYD interactions engage the sensor with the adaptor ASC, and CARD–CARD interactions engage the adaptor ASC with the effector caspase‐1. Although placed in the NLR family, NLRC4 is seen to act as an adaptor to the NAIP sensors.58, 59, 60, 61, 62 Upon ligand binding, NAIPs recruit NLRC4 to assemble NAIP−NLRC4−caspase‐1 inflammasomes. NAIP−NLRC4−caspase‐1 inflammasomes could assemble in the absence of ASC. However, NLRC4 interacts with ASC, and NAIP inflammasomes and NLRP3 inflammasomes co‐localize in a single speck in macrophage cells infected with Salmonella Typhimurium flagellin.63 These observations indicate that different sensors could use different adaptors for inflammasome assembly and that different inflammasomes could cooperate to elicit cellular responses.
RLR‐mediated signalosomes
Upon dsRNA binding, MDA5 and RIG‐I become activated, interact and activate the mitochondrial adaptor protein MAVS, which then recruits TRAF proteins to activate NF‐κB and IFN responses.22, 56, 64, 65, 66, 67, 68 This signalling involves molecular assembly into higher‐order assemblies termed as signalosomes.22, 35 LGP2 lacks 2CARD domains and was thought to inhibit MDA5‐ and RIG‐I‐induced signalling. However, it now appears that LGP2 associates with dsRNA much faster than MDA5, assists MDA5−dsRNA interactions and promotes antiviral signalling by MDA5.69
MDA5 recognizes long dsRNA46, 47, 70 and assembles into filaments on the dsRNA.71, 72, 73 Structural studies have shown that MDA5 recognizes the interior of dsRNA and stacks head to tail towards the RNA termini to form filaments.73, 74 The isolated helicase C‐terminal domain could assemble into filaments on dsRNA.74 Therefore, the signalling domain 2CARD is dispensable for filament formation by MDA5 on dsRNA. Head‐to‐tail stacked helicase C‐terminal domain forms the core, and 2CARD decorates the outside of the core regions of MDA5 filaments.74 Mutations in MDA5 that impair filament formation, without impairing dsRNA binding, also impair the ability of MDA5 to induce IFN.74 This suggests that MDA5 filament formation on dsRNA is required for its function in cells.
Unlike MDA5, RIG‐I recognizes dsRNA end with 5ʹ triphosphate (5ʹ ppp) or 5ʹ diphosphate (5ʹ pp).44, 48, 73, 74, 75, 76, 77 Like MDA5, upon RNA binding, RIG‐I assembles into filaments.78, 79 RIG‐I filament formation is more efficient on shorter dsRNAs (< 500 bp) than on long RNAs (> 1 kb).77, 78 RIG‐I binds to RNA end in an ATP‐independent manner and, in the presence of ATP, assembles into filaments and translocates into the RNA interior.44, 78 RIG‐I filaments are, on average, shorter than MDA5 filaments. The 2CARD deletion mutant of RIG‐I could form filaments.78 Therefore, like the MDA5 example, the signalling domain 2CARD is dispensable for RIG‐I filament formation on RNA. Impairment of filament formation impacts negatively the signalling activity of RIG‐I, suggesting that RIG‐I filaments contribute to its signalling.78
MAVS becomes more resistant towards solubilization and extraction from mitochondria of virus‐infected cells.66 In virus‐infected cells, MAVS redistributes on mitochondria into speck‐like assemblies.80 MAVS isolated from virus‐infected cells possesses larger MAVS particles. The crude mitochondrial lysate of virus‐infected cells that possess the MAVS particles activates interferon regulatory factor 3 (IRF3).81 Disaggregation of MAVS particles abolishes the signalling ability of the crude mitochondrial lysate of virus‐infected cells.81 Recombinant MAVS polymerizes into filaments that are functional in the sense that they activate IRF3 in cytosolic extracts.81 Super‐resolution microscopy has shown that MAVS redistributes on mitochondria into rod‐shaped assemblies in virus‐infected cells.82 The isolated CARD domain of MAVS (MAVSCARD) polymerizes into filaments that can activate IRF3 in cytosolic extracts.74, 81, 82 Point mutations that disrupt MAVS self‐association also impair its signalling in cells.82 These observations demonstrate that MAVS signals upon undergoing polymerization on mitochondria in virus‐infected cells.
In‐vitro‐generated proteinase K‐resistant MAVS filaments, which are composed largely of MAVSCARD, induce functional MAVS assembly on mitochondria in mitochondrial lysates.81 This suggests that MAVS filament formation is largely driven by its CARD and requires nucleation by pre‐assembled CARD. It appears that pre‐assembled CARD of MDA5, RIG‐I or MAVS itself could nucleate MAVS polymerization on mitochondria.
Isolated 2CARD of MDA5 and RIG‐I is monomeric unless at high concentration.74 Conceivably, MDA5 polymerization on dsRNA brings its 2CARD into close proximity and thereby promotes association among them to nucleate MAVS polymerization on mitochondria.35 Recombinant SNAP‐fused MAVSCARD (MAVSCARD‐S) exists as short filaments and could be refolded, following unfolding, in monomers.74 The monomeric MAVSCARD‐S polymerizes with a very slow kinetics and forms filaments over the course of days.74 A fraction of recombinant MDA52CARD is high‐molecular‐weight aggregates. These high‐molecular‐weight aggregates, not the monomers, of MDA5 nucleate MAVSCARD‐S polymerization.74
Full‐length RIG‐I could stimulate filament formation by MAVS in the presence of dsRNA and ATP, indicating that RIG‐I filaments nucleate MAVS polymerization.78 RIG‐1 deletion mutant lacking 2CARD is incapable of nucleating MAVS polymerization, indicating that RIG‐I filament nucleates MAVS polymerization through its 2CARD.78 It appears that assembly of a minimum of four RIG molecules on dsRNA is required to nucleate MAVS polymerization.78 The MAVS filaments formed upon nucleation by RIG‐I filament, often possess RIG‐I at the end.78 This ability of filaments of full‐length RIG‐I to nucleate MAVS polymerization is independent of K63‐ubiquitination (K63‐Ubn) of RIG‐I's 2CARD78 (see below).
Unlike MDA5, filament formation is dispensable for RIG‐I‐mediated nucleation of MAVS polymerization and signalling.78, 83, 84 RIG‐I2CARD is known to be conjugated with K63‐Ubn and can also bind to unanchored K63‐UBn.84 Isolated RIG‐I2CARD or full‐length RIG‐I assembled at shorter dsRNA (< 40 bp, which is insufficient to accommodate four RIG‐I molecules) could seed MAVS only in the presence of K63‐Ubn.35, 78, 85 K63‐Ubn seems to promote MAVS2CARD tetramerization to seed MAVS polymerization and signalling. The two mechanisms of RIG‐I‐mediated seeding (filament‐mediated and K63‐Ubn‐mediated) of MAVS polymerization and signalling are not mutually exclusive but cooperative.35 It appears that the same surface of 2CARD of RIG‐I mediates filament‐dependent and K63‐Ubn‐dependent seeding of MAVS polymerization and activation.78 Unlike RIG‐I2CARD, there appears to be no stimulatory effect of K63‐Ub4 on MDA52CARD‐mediated seeding of MAVS polymerization.74 The common theme emerging from these studies is that MDA5's or RIG‐I's 2CARD needs to be brought into close proximity and self‐associated to promote MAVS polymerization and signalling. Why RIG‐I has two cooperating mechanisms to achieve this is unknown.
Supramolecular assemblies in transmembrane immune signalling
Signalling through many transmembrane receptors has also been seen to involve supramolecular assemblies. Signalling through TNF receptor involves assembly of the kinases RIP1 and RIP3 into amyloid‐like fibrils.86 The membrane‐bound adaptor protein LAT is phosphorylated during TCR signalling, and the phosphorylated LAT recruits many downstream molecules and appears to nucleate formation of supramolecular signalling complexes.27, 87 Members of the TLR family appear to signal as dimers, and signalling through them involves signalosome assembly around their cytosolic tails.22 KIR represents a unique case, wherein a transmembrane receptor itself polymerizes.88, 89 KIR undergoes Zn2+‐dependent polymerization into filaments at the plasma membrane at NK cell–target cell synapses.88, 89
TLR‐mediated signalosomes
All TLRs possess the TIR domain in their cytosolic portions and signal by recruiting various TIR domain‐containing adaptor proteins.22, 90, 91 All TLRs except TLR3 signal by recruiting the TIR domain‐containing adaptor protein MyD88.90, 91 MyD88 interacts directly with the TLR dimers, except TLR4. TLR4 interacts with MyD88 indirectly with the bridging adaptor MAL. TLR4 signalling involves the adaptor TRAM, which is needed to recruit another TIR domain‐containing adaptor, TRIF. In addition to a TIR domain, MyD88 also possesses a death domain. MyD88 recruits, through death domain–death domain homotypic interactions, IL‐1 receptor‐associated kinases (IRAKs), leading to activation of NF‐κB.92, 93, 94, 95 TLR3 signals exclusively through the TIR domain‐containing adaptor TRIF.90, 91, 96, 97
Upon ligand binding, TLRs form M‐shaped dimers that promote TIR domain dimerization in the cytosol.22, 98, 99, 100 The dimerized TIR domains could provide scaffolds for the assembly of further signalling complexes. The TIR domain of MAL (MALTIR) polymerizes into filaments.101 TLR4TIR alone does not polymerize.101 However, TLR4TIR co‐polymerizes with MALTIR. Although whether or not such filaments are formed in cells is unclear, it is conceivable that the interactions forming TIR filaments could be important for promoting downstream signalling.101 MyD88's death domains alone form heterogeneous oligomers. However, in the presence of IRAK's death domains, MyD88's death domains assemble into stable complexes called myddosomes.102, 103 In these myddosomes, the death domains are arranged in a left‐handed helical tower, wherein six MyD88 death domains are situated at the top, four IRAK‐4 death domains are in the middle, and four IRAK‐2 death domains are at the bottom.22, 103 The S34Y mutant of MyD88 cannot be assembled in myddosomes in vitro and cannot elicit signalling from most TLRs.97, 104 Therefore, conceivably, myddosome assembly is required for TLR signalling.97
Polymerization of transmembrane receptor
Ligand binding‐induced dimerization of a receptor tyrosine kinase that triggers the intrinsic kinase activity present in the cytosolic domain of the receptor was a seminal discovery.24 It now appears that dimerization or oligomerization is a general principle in transmembrane signalling.105, 106, 107, 108 Inhibitory KIR represents a unique case wherein a transmembrane receptor undergoes polymerization. KIR polymerization is dependent on the metal ion Zn2+ and leads to linear filaments of ~100–200 nm lengths.88, 89 Zn2+‐induced KIR polymerization could represent a new transmembrane signalling mode, wherein a transmembrane receptor polymerizes in higher‐order assemblies that are much larger than the known dimers or oligomers of transmembrane receptors.88, 89 Conceivably, KIR polymers represent a novel mode of signalosome assembly, wherein polymers of a transmembrane receptor provide scaffolds to promote cytosolic signalosome assembly. KIR functions at NK cell‐target cell synapses to block NK cell activation towards HLA‐C expressing target cells.7, 8 KIR clusters rapidly in an actin‐independent manner and blocks the proximal steps of NK cell activation at those synapses.7, 8, 109 Zn2+‐dependent polymerization may contribute to actin‐independent clustering and signalling of KIR.88, 89 KIR prevents proximal activation signals.7, 8 Consistently, they signal rapidly.110 By holding the bound molecules in close proximity, KIR polymerization may provide the necessary nucleus (see below), so may bypass the unfavourable nucleation events, for rapid assembly of signalosomes in the cytosol. KIR polymerization itself may not be limited by an unfavourable nucleation event, as Zn2+ drives it. Furthermore, cooperative interactions will make recruitment events robust, and promote enzymatic reactions by promoting proximity between enzymes and substrates and by stabilizing active enzyme conformations. Such mechanisms may be necessary, as the key enzymes of KIR signalling,7, 8 SHP‐1 and c‐Abl, are maintained in the cytosol in inactive conformation and become active upon recruitment to inhibitory synapses. Lastly, signalosomes may promote sustained signalling without continuous KIR–HLA‐C binding and so may contribute to signal amplification.88, 89
Mechanisms of protein assembly reactions
Protein assembly reactions are often described in terms of two basic models, nucleation‐dependent polymerization (NDP) and isodesmic polymerization.111, 112, 113, 114, 115, 116
Nucleation‐dependent polymerization
An NDP reaction consists of kinetically distinct steps (Fig. 1a). The initial steps (nucleation) are slower than the later steps (polymerization or assembly). A mathematical description seeks the forward and reverse rate constants of each step.117, 118 Considering the initial steps as being near equilibrium reduces the kinetic problem to an equilibrium one and so greatly simplifies the analysis. In a thermodynamic description, the initial steps of an NDP reaction consist of a number of unfavourable equilibria (Fig. 1a). This makes the initiation (nucleation) of polymerization difficult. The corresponding free energy barrier has to be climbed and crossed for the polymerization to proceed (Fig. 1b). The peak of this free energy barrier corresponds to a species whose formation makes further polymerization thermodynamically favourable. This high energy, and hence scarce, species represents the nucleus, whose formation is the bottleneck for the overall polymerization (Fig. 1b).
Figure 1.
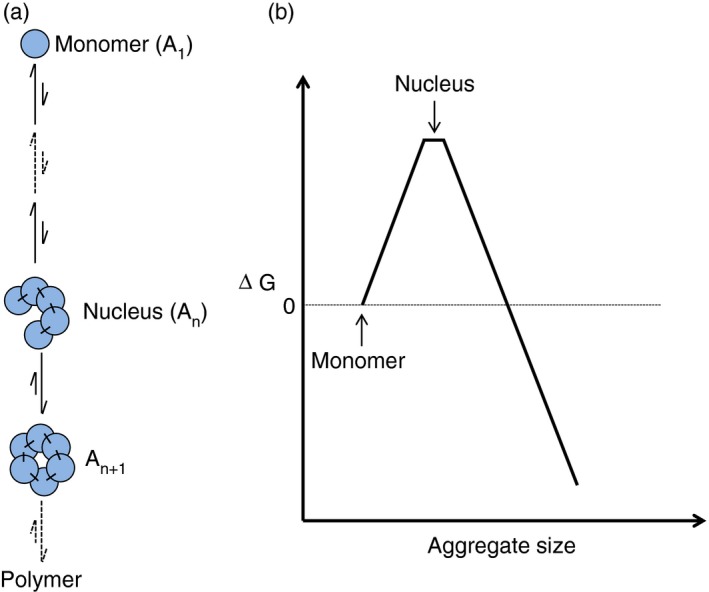
Nucleation‐dependent polymerization. (a) Schematic of a nucleation‐dependent polymerization reaction showing the nucleation and the growth phases. (b) Free energy barrier associated with a nucleation‐dependent polymerization reaction.
The slope of the free energy curve at any aggregate size is given by the concentration multiplied by ratio of the association rate constant (k on) to the dissociation rate constant (k off).118 In the nucleation phase, the dissociation rate constants are greater than the association rate constants (Fig. 1a). After nucleation, the slope of the free energy curve reverses its direction, and the association rate constants become greater than the dissociation rate constants for all the post‐nucleation steps (Fig. 1a). Therefore, in terms of reaction kinetics, the nucleus is the smallest aggregate for which k on is greater than k off.118
An NDP reaction has three distinct features.115, 118, 119 (i) The polymerization kinetics has an initial lag phase that could be described by a t 2 function. The lag phase arises because k off > k on during the initial (nucleation) phase. The length of the lag phase is proportional to the steepness of the initial portion of the energy curve and is dependent on protein concentration and size of nucleus.120 (ii) There is a critical concentration for polymerization. The lag time strongly depends on protein concentration. The lag time increases with decreasing protein concentration, implying that no polymerization would occur at a sufficiently low protein concentration. This protein concentration is referred to as the critical concentration. The critical concentration can vary from one protein to another. (iii) The lag phase is eliminated if a small amount of polymer is provided at the beginning of the reaction. This process is called seeding.
The polymerization of many proteins is described well by the theory of NDP reactions. However, for many proteins, the polymerization kinetics is much more abrupt and could not be described by the predicted t 2 dependence.115 The kinetics is described well by exponential time dependence. To explain this exponential time dependence, the theory of NDP reactions has been extended to include secondary nucleation events such as fragmentation and branching.117, 118, 121, 122, 123
Isodesmic (linear) polymerization
In isodesmic polymerization,111, 114, 115, 124 there are no distinct nucleation and elongation phases. Each association event involves the same bond, and hence, the rate of polymerization is independent of aggregate size. Therefore, an isodesmic polymerization could be considered to be similar to the elongation phase of an NDP reaction. Kinetics of an isodesmic polymerization does not show a lag phase. There is no critical concentration barrier for an isodesmic polymerization reaction.
An isodesmic polymerization reaction generally does not display the features of an NDP reaction. However, it is not often easy to differentiate between them.124 The distinction between the two reaction types is subtle and rests entirely upon nucleus size and k on and k off. Under certain situations, an isodesmic polymerization reaction can display the features of an NDP reaction. Since an isodesmic polymerization could display at least two of the three NDP features, a polymerization reaction needs to possess all three features to be considered as an NDP reaction. The NDP features are more prominent at lower protein concentrations. At very high protein concentrations, the nucleation could become favourable and lag phase and dependence of kinetics on protein concentrations may disappear. Finally, it is worth noting that the NDP and isodesmic mechanisms represent two extremes, and it is conceivable for a polymerization reaction to possess features of both.
Implications of protein assembly reactions in signalling
The traditional view of signalling, wherein signal transduction is thought to occur through freely diffusible molecules, does not fully describe signalling through supramolecular assemblies such as signalosomes and inflammasomes.26 How these supramolecular assemblies form and promote signalling is largely unclear. As noted above, in the cases of pattern recognition receptors, domains with intrinsic propensity of self/cross‐association, present in cytosolic receptors and/or adaptors, control signalosome/inflammasome assembly. During TCR and BCR signalling, signalosome assembly is initiated by phosphorylation of specific adaptor proteins, which creates docking sites for downstream molecules. KIR represents a unique example, wherein a transmembrane receptor itself polymerizes.88, 89 Conceivably, KIR polymers provide a novel mode of signalosome assembly, wherein polymers of a transmembrane receptor provide scaffolds to promote cytosolic signalosome assembly. By viewing the formation of supramolecular assemblies in terms of the NDP mechanism,89, 115 one can imagine new mechanisms for signal amplification, setting threshold and spatiotemporal regulation in signal transduction.
A nucleated assembly could ensure that engagement of a small number of sensors would suffice to activate an appropriately high number of effector molecules to reach the response threshold. For example, a given AIM2 oligomer, formed on dsDNA, could nucleate polymerization of many ASC molecules into filament, and the surface of each ASC filament could provide many secondary nucleation sites for polymerization, and thereby activation, of many more caspase‐1. The domains that mediate formation of supramolecular assemblies are often parts of or bound to an enzyme.26 Recruitment to supramolecular assemblies would greatly increase the local concentration of these enzymes and so would promote proximity‐driven auto‐activation or catalysis towards the substrate. Filament formation by caspase‐1 in inflammasomes could promote proximity‐driven auto‐activation of caspase‐1. In myddosomes, IRAK2 is a substrate for IRAK4, and proximity between the two in myddosomes could enable more efficient IRAK4 catalysis. Under basal state, many enzymes are maintained in auto‐inhibited conformations. Recruitment of such enzymes into supramolecular assemblies could stabilize them in active conformation. Conceivably, once assembled, the signalosome could signal without the sustained presence of the trigger. For example, KIR signals upon polymerization, and polymerized KIR cannot bind to its HLA‐C ligand.88, 89 Therefore, conceivably, once polymerized, KIR dissociates from its ligand and signals without sustained ligand binding. This points towards the possibility of signal amplification by serial engagement, wherein a given ligand molecule could initiate signalling by many receptors.
Formation of supramolecular assemblies through the NDP mechanism, wherein unfavourable initiation constitutes a kinetic and thermodynamic bottleneck for the overall reaction, could set an appropriately high threshold to avoid stochastic receptor triggering. This would eliminate the chances of signalling by transient fluctuations in concentration, diffusion and conformation, and would ensure that signalling is triggered only upon persistent and strong stimulation. Although upon over‐expression AIM2 polymerizes spontaneously in cells, it polymerizes only in the presence of dsDNA at physiological expression levels.52 It therefore appears that dsDNA binding increases the local AIM2 concentration to overcome the threshold set by unfavourable nucleation. A comparison of signalling rates has revealed that TNF signalling, which involves supramolecular assemblies, occurs at a much slower rate than signalling by GPCRs or Receptor tyrosine kinases (RTK)s,26, 125 suggesting that the requirement of supramolecular assemblies has a high signalling threshold. Further, the cooperativity in the formation of higher‐order assemblies could explain all‐or‐none responses after the threshold is met. For example, conceivably, formation of supramolecular assemblies at the cytosolic side of TLR4 could explain the all‐or‐none responses of macrophages against lipopolysaccharide.126
The strong concentration dependence of k on, and hence lag time, could effectively control the rapidity of a response. The kinetics of TNF‐induced NF‐κB activation is slower at low ligand dose than at higher ligand dose.26, 125 Therefore, it appears that the same threshold could be achieved with different kinetics based on strength of stimuli. This is important because this would allow the cells to sense the urgency and respond with appropriate rapidity. KIR is organized in nanoclusters in resting NK cells.127, 128 Conceivably, the molecular proximity in KIR nanoclusters and Zn2+ involvement make the nucleation favourable for rapid KIR polymerization. Alternatively, Zn2+‐dependent KIR polymerization could be a rapid isodesmic polymerization reaction.
How signalling through these supramolecular assemblies is terminated is unclear. Inflammasomes may not be degraded efficiently in mammalian cells and are released into extracellular space upon pyroptosis. The released inflammasomes are functional and perform IL‐1β processing, and could be engulfed by macrophages to induce activation of inflammasomes in recipient cells.129, 130 Therefore, supramolecular assemblies implicate unique signalling mechanisms not only within a cell but also between cells.
Summary
Traditionally, signalling pathways have been thought to involve freely diffusible components. However, it is now evident that signalling can occur through semi‐solid‐like supramolecular protein assemblies with minimal free diffusion of the constituents. The ALRs, NLRs and RLRs recognize intracellular ligands and signal by triggering a series of association reactions that lead to functional supramolecular assemblies. The supramolecular assemblies mediated by members of ALRs and NLRs are called inflammasomes, wherein molecular proximity promotes caspase‐1 auto‐activation, the supramolecular assemblies mediated by RLRs are called signalosomes that trigger IFN response and NF‐κB activation. Further, signalling through many transmembrane receptors is seen to involve signalosome assembly around the cytosolic side of the plasma membrane. Although TLR4 is proposed to signal as a dimer, its signalling involves signalosome (called myddosome) assembly around its cytosolic tail. During TCR and BCR signalling, signalosome assembly is initiated by phosphorylation of specific adaptor proteins that creates docking sites for downstream molecules. KIR represents a unique example. KIR itself polymerizes into Zn2+‐dependent filaments at the plasma membrane in the context of NK cell‐target cell synapses. Conceivably, KIR filaments mediate signalling by acting as scaffolds for signalosome assembly at the cytosolic side of NK cell‐target cell synapses.
Protein assembly reactions are often described in terms of two basic models, NDP and isodesmic polymerization. In an NDP reaction, the initial steps (nucleation) are slower than the later ones (growth). From the thermodynamic viewpoint, the nucleation phase consists of a number of unfavourable equilibria. After nucleation, high enthalpy gain from association makes further polymerization (growth) thermodynamically favourable. In terms of kinetics, dissociation rate constants are greater than association rate constants during nucleation, and after nucleation, association rate constants become greater than dissociation rate constants. The kinetics of an NDP reaction shows an initial lag phase, and there exists a critical protein concentration below which no polymers form. In isodesmic polymerization, there are no kinetically distinct phases and all association steps involve the same bond.
By viewing formation of supramolecular assemblies in terms of the NDP mechanism, one can imagine new elegant mechanisms for signal amplification, setting threshold and spatiotemporal regulation in signal transduction. Formation of supramolecular assemblies through an NDP mechanism, wherein initiation is unfavourable, could set an appropriately high threshold to avoid stochastic receptor triggering. Nucleated polymerization could ensure that a small number of ligand‐bound receptors could nucleate assembly of many more downstream molecules to amplify signal. The cooperativity of an NDP reaction could explain the all‐or‐none response seen in signalling pathways that involve supramolecular assemblies. An NDP reaction shows a strong dependence on concentration. The lag phase and reaction rate of an NDP reaction show a strong dependence on concentration. At very high concentration, an NDP reaction loses its characteristic features and behaves like an isodesmic reaction. This concentration dependence could be a way of modulating the signalling responses as per the requirement. The kinetics of TNF signalling is faster at higher ligand dose than the lower dose. KIR is organized in nanoclusters in resting NK cells. Conceivably, the molecular proximity in KIR nanoclusters and Zn2+ involvement makes the nucleation favourable for rapid polymerization. Experimental evidence for these new mechanisms of threshold, all‐or‐none host defence programmes, have just begun to emerge, and the future should witness more developments.
Disclosures
The authors have no conflicts of interest to declare.
References
- 1. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol 2015; 33:257–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chow J, Franz KM, Kagan JC. PRRs are watching you: Localization of innate sensing and signaling regulators. Virology 2015; 479–480:104–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol 2016; 16:112–23. [DOI] [PubMed] [Google Scholar]
- 4. Lau CM, Sun JC. The widening spectrum of immunological memory. Curr Opin Immunol 2018; 54:42–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. O'Leary JG, Goodarzi M, Drayton DL, von Andrian UH. T cell‐ and B cell‐independent adaptive immunity mediated by natural killer cells. Nat Immunol 2006; 7:507–16. [DOI] [PubMed] [Google Scholar]
- 6. Bryceson YT, Chiang SC, Darmanin S, Fauriat C, Schlums H, Theorell J et al Molecular mechanisms of natural killer cell activation. J Innate Immun 2011; 3:216–26. [DOI] [PubMed] [Google Scholar]
- 7. Long EO. Negative signaling by inhibitory receptors: the NK cell paradigm. Immunol Rev 2008; 224:70–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol 2013; 31:227–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Atanasova M, Whitty A. Understanding cytokine and growth factor receptor activation mechanisms. Crit Rev Biochem Mol Biol 2012; 47:502–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Whitty A, Raskin N, Olson DL, Borysenko CW, Ambrose CM, Benjamin CD et al Interaction affinity between cytokine receptor components on the cell surface. Proc Natl Acad Sci U S A 1998; 95:13165–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stephens B, Handel TM. Chemokine receptor oligomerization and allostery. Prog Mol Biol Transl Sci 2013; 115:375–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bazan JF. Emerging families of cytokines and receptors. Curr Biol 1993; 3:603–6. [DOI] [PubMed] [Google Scholar]
- 13. Dinarello CA. Historical insights into cytokines. Eur J Immunol 2007; 37(Suppl 1):S34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Weerd NA, Nguyen T. The interferons and their receptors – distribution and regulation. Immunol Cell Biol 2012; 90:483–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Green DS, Young HA, Valencia JC. Current prospects of type II interferon γ signaling and autoimmunity. J Biol Chem 2017; 292:13925–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Walter MR. The molecular basis of IL‐10 function: from receptor structure to the onset of signaling. Curr Top Microbiol Immunol 2014; 380:191–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin‐10 and the interleukin‐10 receptor. Annu Rev Immunol 2001; 19:683–765. [DOI] [PubMed] [Google Scholar]
- 18. Ward‐Kavanagh LK, Lin WW, Sedy JR, Ware CF. The TNF receptor superfamily in co‐stimulating and co‐inhibitory responses. Immunity 2016; 44:1005–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sedger LM, McDermott MF. TNF and TNF‐receptors: from mediators of cell death and inflammation to therapeutic giants – past, present and future. Cytokine Growth Factor Rev 2014; 25:453–72. [DOI] [PubMed] [Google Scholar]
- 20. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol 2014; 32:659–702. [DOI] [PubMed] [Google Scholar]
- 21. Boraschi D, Tagliabue A. The interleukin‐1 receptor family. Semin Immunol 2013; 25:394–407. [DOI] [PubMed] [Google Scholar]
- 22. Yin Q, Fu TM, Li J, Wu H. Structural biology of innate immunity. Annu Rev Immunol 2015; 33:393–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cohen P. The TLR and IL‐1 signalling network at a glance. J Cell Sci 2014; 127:2383–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hunter T. Signaling – 2000 and beyond. Cell 2000; 100:113–27. [DOI] [PubMed] [Google Scholar]
- 25. Kagan JC, Magupalli VG, Wu H. SMOCs: supramolecular organizing centres that control innate immunity. Nat Rev Immunol 2014; 14:821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu H. Higher‐order assemblies in a new paradigm of signal transduction. Cell 2013; 153:287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Su X, Ditlev JA, Hui E, Xing W, Banjade S, Okrut J et al Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 2016; 352:595–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barton GM, Kagan JC. A cell biological view of Toll‐like receptor function: regulation through compartmentalization. Nat Rev Immunol 2009; 9:535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010; 140:805–20. [DOI] [PubMed] [Google Scholar]
- 30. Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H et al An orthogonal proteomic‐genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol 2009; 10:266–72. [DOI] [PubMed] [Google Scholar]
- 31. Hornung V, Ablasser A, Charrel‐Dennis M, Bauernfeind F, Horvath G, Caffrey DR et al AIM2 recognizes cytosolic dsDNA and forms a caspase‐1‐activating inflammasome with ASC. Nature 2009; 458:514–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fernandes‐Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009; 458:509–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brunette RL, Young JM, Whitley DG, Brodsky IE, Malik HS, Stetson DB. Extensive evolutionary and functional diversity among mammalian AIM2‐like receptors. J Exp Med 2012; 209:1969–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cridland JA, Curley EZ, Wykes MN, Schroder K, Sweet MJ, Roberts TL et al The mammalian PYHIN gene family: phylogeny, evolution and expression. BMC Evol Biol 2012; 12:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sohn J, Hur S. Filament assemblies in foreign nucleic acid sensors. Curr Opin Struct Biol 2016; 37:134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li T, Diner BA, Chen J, Cristea IM. Acetylation modulates cellular distribution and DNA sensing ability of interferon‐inducible protein IFI16. Proc Natl Acad Sci U S A 2012; 109:10558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kerur N, Veettil MV, Sharma‐Walia N, Bottero V, Sadagopan S, Otageri P et al IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma‐associated herpesvirus infection. Cell Host Microbe 2011; 9:363–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Franchi L, Warner N, Viani K, Nunez G. Function of Nod‐like receptors in microbial recognition and host defense. Immunol Rev 2009; 227:106–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhong Y, Kinio A, Saleh M. Functions of NOD‐like receptors in human diseases. Front Immunol 2013; 4:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci 2016; 41:1012–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chow KT, Gale M Jr, Loo YM. RIG‐I and other RNA sensors in antiviral immunity. Annu Rev Immunol 2018; 36:667–94. [DOI] [PubMed] [Google Scholar]
- 42. Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez‐Sobrido L et al Distinct RIG‐I and MDA5 signaling by RNA viruses in innate immunity. J Virol 2008; 82:335–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Loo YM, Gale M Jr. Immune signaling by RIG‐I‐like receptors. Immunity 2011; 34:680–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Myong S, Cui S, Cornish PV, Kirchhofer A, Gack MU, Jung JU et al Cytosolic viral sensor RIG‐I is a 5’‐triphosphate‐dependent translocase on double‐stranded RNA. Science 2009; 323:1070–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K et al Differential roles of MDA5 and RIG‐I helicases in the recognition of RNA viruses. Nature 2006; 441:101–5. [DOI] [PubMed] [Google Scholar]
- 46. Kato H, Takeuchi O, Mikamo‐Satoh E, Hirai R, Kawai T, Matsushita K et al Length‐dependent recognition of double‐stranded ribonucleic acids by retinoic acid‐inducible gene‐I and melanoma differentiation‐associated gene 5. J Exp Med 2008; 205:1601–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Feng Q, Hato SV, Langereis MA, Zoll J, Virgen‐Slane R, Peisley A et al MDA5 detects the double‐stranded RNA replicative form in picornavirus‐infected cells. Cell Rep 2012; 2:1187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F et al RIG‐I‐mediated antiviral responses to single‐stranded RNA bearing 5’‐phosphates. Science 2006; 314:997–1001. [DOI] [PubMed] [Google Scholar]
- 49. Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L et al Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 2012; 36:561–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jin T, Perry A, Smith P, Jiang J, Xiao TS. Structure of the absent in melanoma 2 (AIM2) pyrin domain provides insights into the mechanisms of AIM2 autoinhibition and inflammasome assembly. J Biol Chem 2013; 288:13225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hu Z, Yan C, Liu P, Huang Z, Ma R, Zhang C et al Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 2013; 341:172–5. [DOI] [PubMed] [Google Scholar]
- 52. Morrone SR, Matyszewski M, Yu X, Delannoy M, Egelman EH, Sohn J. Assembly‐driven activation of the AIM2 foreign‐dsDNA sensor provides a polymerization template for downstream ASC. Nat Commun 2015; 6:7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lu A, Li Y, Yin Q, Ruan J, Yu X, Egelman E et al Plasticity in PYD assembly revealed by cryo‐EM structure of the PYD filament of AIM2. Cell Discov 2015; 1:pii: 15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dick MS, Sborgi L, Ruhl S, Hiller S, Broz P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun 2016; 7:11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR et al Unified polymerization mechanism for the assembly of ASC‐dependent inflammasomes. Cell 2014; 156:1193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lu A, Wu H. Structural mechanisms of inflammasome assembly. FEBS J 2015; 282:435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hauenstein AV, Zhang L, Wu H. The hierarchical structural architecture of inflammasomes, supramolecular inflammatory machines. Curr Opin Struct Biol 2015; 31:75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 2011; 477:592–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP et al Differential activation of the inflammasome by caspase‐1 adaptors ASC and Ipaf. Nature 2004; 430:213–8. [DOI] [PubMed] [Google Scholar]
- 60. Poyet JL, Srinivasula SM, Tnani M, Razmara M, Fernandes‐Alnemri T, Alnemri ES. Identification of Ipaf, a human caspase‐1‐activating protein related to Apaf‐1. J Biol Chem 2001; 276:28309–13. [DOI] [PubMed] [Google Scholar]
- 61. Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci U S A 2013; 110:14408–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H et al The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011; 477:596–600. [DOI] [PubMed] [Google Scholar]
- 63. Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P et al Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A 2014; 111:7403–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H et al IPS‐1, an adaptor triggering RIG‐I‐ and Mda5‐mediated type I interferon induction. Nat Immunol 2005; 6:981–8. [DOI] [PubMed] [Google Scholar]
- 65. Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R et al Cardif is an adaptor protein in the RIG‐I antiviral pathway and is targeted by hepatitis C virus. Nature 2005; 437:1167–72. [DOI] [PubMed] [Google Scholar]
- 66. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF‐κB and IRF 3. Cell 2005; 122:669–82. [DOI] [PubMed] [Google Scholar]
- 67. Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus‐triggered IFN‐β signaling. Mol Cell 2005; 19:727–40. [DOI] [PubMed] [Google Scholar]
- 68. Yoneyama M, Fujita T. Recognition of viral nucleic acids in innate immunity. Rev Med Virol 2010; 20:4–22. [DOI] [PubMed] [Google Scholar]
- 69. Bruns AM, Leser GP, Lamb RA, Horvath CM. The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5‐RNA interaction and filament assembly. Mol Cell 2014; 55:771–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Peisley A, Jo MH, Lin C, Wu B, Orme‐Johnson M, Walz T et al Kinetic mechanism for viral dsRNA length discrimination by MDA5 filaments. Proc Natl Acad Sci U S A 2012; 109:E3340–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Peisley A, Lin C, Wu B, Orme‐Johnson M, Liu M, Walz T et al Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci U S A 2011; 108:21010–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Berke IC, Modis Y. MDA5 cooperatively forms dimers and ATP‐sensitive filaments upon binding double‐stranded RNA. EMBO J 2012; 31:1714–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Berke IC, Yu X, Modis Y, Egelman EH. MDA5 assembles into a polar helical filament on dsRNA. Proc Natl Acad Sci U S A 2012; 109:18437–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wu B, Peisley A, Richards C, Yao H, Zeng X, Lin C et al Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 2013; 152:276–89. [DOI] [PubMed] [Google Scholar]
- 75. Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W et al Recognition of 5’ triphosphate by RIG‐I helicase requires short blunt double‐stranded RNA as contained in panhandle of negative‐strand virus. Immunity 2009; 31:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M et al Antiviral immunity via RIG‐I‐mediated recognition of RNA bearing 5’‐diphosphates. Nature 2014; 514:372–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Baum A, Sachidanandam R, Garcia‐Sastre A. Preference of RIG‐I for short viral RNA molecules in infected cells revealed by next‐generation sequencing. Proc Natl Acad Sci U S A 2010; 107:16303–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Peisley A, Wu B, Yao H, Walz T, Hur S. RIG‐I forms signaling‐competent filaments in an ATP‐dependent, ubiquitin‐independent manner. Mol Cell 2013; 51:573–83. [DOI] [PubMed] [Google Scholar]
- 79. Patel JR, Jain A, Chou YY, Baum A, Ha T, Garcia‐Sastre A. ATPase‐driven oligomerization of RIG‐I on RNA allows optimal activation of type‐I interferon. EMBO Rep 2013; 14:780–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Onoguchi K, Onomoto K, Takamatsu S, Jogi M, Takemura A, Morimoto S et al Virus‐infection or 5'ppp‐RNA activates antiviral signal through redistribution of IPS‐1 mediated by MFN1. PLoS Pathog 2010; 6:e1001012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion‐like aggregates to activate and propagate antiviral innate immune response. Cell 2011; 146:448–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xu H, He X, Zheng H, Huang LJ, Hou F, Yu Z et al Structural basis for the prion‐like MAVS filaments in antiviral innate immunity. Elife 2014; 3:e01489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jiang X, Kinch LN, Brautigam CA, Chen X, Du F, Grishin NV et al Ubiquitin‐induced oligomerization of the RNA sensors RIG‐I and MDA5 activates antiviral innate immune response. Immunity 2012; 36:959–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A et al Reconstitution of the RIG‐I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 2010; 141:315–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Peisley A, Wu B, Xu H, Chen ZJ, Hur S. Structural basis for ubiquitin‐mediated antiviral signal activation by RIG‐I. Nature 2014; 509:110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS et al The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012; 150:339–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Balagopalan L, Kortum RL, Coussens NP, Barr VA, Samelson LE. The linker for activation of T cells (LAT) signaling hub: from signaling complexes to microclusters. J Biol Chem 2015; 290:26422–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kumar S, Rajagopalan S, Sarkar P, Dorward DW, Peterson ME, Liao HS et al Zinc‐induced polymerization of killer‐cell Ig‐like receptor into filaments promotes its inhibitory function at cytotoxic immunological synapses. Mol Cell 2016; 62:21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kumar S. Natural killer cell cytotoxicity and its regulation by inhibitory receptors. Immunology 2018; 154:383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. O'Neill LA, Bowie AG. The family of five: TIR‐domain‐containing adaptors in Toll‐like receptor signalling. Nat Rev Immunol 2007; 7:353–64. [DOI] [PubMed] [Google Scholar]
- 91. O'Neill LA. The interleukin‐1 receptor/Toll‐like receptor superfamily: 10 years of progress. Immunol Rev 2008; 226:10–8. [DOI] [PubMed] [Google Scholar]
- 92. Dunne A, Carpenter S, Brikos C, Gray P, Strelow A, Wesche H et al IRAK1 and IRAK4 promote phosphorylation, ubiquitination, and degradation of MyD88 adaptor‐like (Mal). J Biol Chem 2010; 285:18276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Keating SE, Maloney GM, Moran EM, Bowie AG. IRAK‐2 participates in multiple toll‐like receptor signaling pathways to NFκB via activation of TRAF6 ubiquitination. J Biol Chem 2007; 282:33435–43. [DOI] [PubMed] [Google Scholar]
- 94. Gosu V, Basith S, Durai P, Choi S. Molecular evolution and structural features of IRAK family members. PLoS ONE 2012; 7:e49771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Suzuki N, Saito T. IRAK‐4–a shared NF‐κB activator in innate and acquired immunity. Trends Immunol 2006; 27:566–72. [DOI] [PubMed] [Google Scholar]
- 96. Kawasaki T, Kawai T. Toll‐like receptor signaling pathways. Front Immunol 2014; 5:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gay NJ, Gangloff M, O'Neill LA. What the Myddosome structure tells us about the initiation of innate immunity. Trends Immunol 2011; 32:104–9. [DOI] [PubMed] [Google Scholar]
- 98. Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4‐MD‐2 complex. Nature 2009; 458:1191–5. [DOI] [PubMed] [Google Scholar]
- 99. Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG et al Crystal structure of the TLR1‐TLR2 heterodimer induced by binding of a tri‐acylated lipopeptide. Cell 2007; 130:1071–82. [DOI] [PubMed] [Google Scholar]
- 100. Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM et al Structural basis of toll‐like receptor 3 signaling with double‐stranded RNA. Science 2008; 320:379–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ve T, Vajjhala PR, Hedger A, Croll T, DiMaio F, Horsefield S et al Structural basis of TIR‐domain‐assembly formation in MAL‐ and MyD88‐dependent TLR4 signaling. Nat Struct Mol Biol 2017; 24:743–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM et al An oligomeric signaling platform formed by the Toll‐like receptor signal transducers MyD88 and IRAK‐4. J Biol Chem 2009; 284:25404–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lin SC, Lo YC, Wu H. Helical assembly in the MyD88‐IRAK4‐IRAK2 complex in TLR/IL‐1R signalling. Nature 2010; 465:885–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. George J, Motshwene PG, Wang H, Kubarenko AV, Rautanen A, Mills TC et al Two human MYD88 variants, S34Y and R98C, interfere with MyD88‐IRAK4‐myddosome assembly. J Biol Chem 2011; 286:1341–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Heldin CH. Dimerization of cell surface receptors in signal transduction. Cell 1995; 80:213–23. [DOI] [PubMed] [Google Scholar]
- 106. Maruyama IN. Activation of transmembrane cell‐surface receptors via a common mechanism? The “rotation model”. BioEssays 2015; 37:959–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Pediani JD, Ward RJ, Marsango S, Milligan G. Spatial intensity distribution analysis: studies of G protein‐coupled receptor oligomerisation. Trends Pharmacol Sci 2018; 39:175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ng HK, Chow BK. Oligomerization of family B GPCRs: exploration in inter‐family oligomer formation. Front Endocrinol (Lausanne) 2015; 6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Faure M, Barber DF, Takahashi SM, Jin T, Long EO. Spontaneous clustering and tyrosine phosphorylation of NK cell inhibitory receptor induced by ligand binding. J Immunol 2003; 170:6107–14. [DOI] [PubMed] [Google Scholar]
- 110. Abeyweera TP, Merino E, Huse M. Inhibitory signaling blocks activating receptor clustering and induces cytoskeletal retraction in natural killer cells. J Cell Biol 2011; 192:675–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Oosawa F, Kasai M. A theory of linear and helical aggregations of macromolecules. J Mol Biol 1962; 4:10–21. [DOI] [PubMed] [Google Scholar]
- 112. Flyvbjerg H, Jobs E, Leibler S. Kinetics of self‐assembling microtubules: an “inverse problem” in biochemistry. Proc Natl Acad Sci U S A 1996; 93:5975–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Goldstein RF, Stryer L. Cooperative polymerization reactions. Analytical approximations, numerical examples, and experimental strategy. Biophys J 1986; 50:583–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Romberg L, Simon M, Erickson HP. Polymerization of Ftsz, a bacterial homolog of tubulin. is assembly cooperative? J Biol Chem 2001; 276:11743–53. [DOI] [PubMed] [Google Scholar]
- 115. Kumar S, Udgaonkar JB. Mechanisms of amyloid fibril formation by proteins. Curr Sci 2010; 98:639–56. [Google Scholar]
- 116. Jain S, Udgaonkar J. B. Prion protein aggregation. Curr Sci 2011; 101:1311–27. [Google Scholar]
- 117. Bishop MF, Ferrone FA. Kinetics of nucleation‐controlled polymerization. A perturbation treatment for use with a secondary pathway. Biophys J 1984; 46:631–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ferrone F. Analysis of protein aggregation kinetics. Methods Enzymol 1999; 309:256–74. [DOI] [PubMed] [Google Scholar]
- 119. Harper JD, Lansbury PT Jr. Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time‐dependent solubility of amyloid proteins. Annu Rev Biochem 1997; 66:385–407. [DOI] [PubMed] [Google Scholar]
- 120. Kodaka M. Interpretation of concentration‐dependence in aggregation kinetics. Biophys Chem 2004; 109:325–32. [DOI] [PubMed] [Google Scholar]
- 121. Wegner A. Spontaneous fragmentation of actin filaments in physiological conditions. Nature 1982; 296:266–7. [DOI] [PubMed] [Google Scholar]
- 122. Wegner A, Savko P. Fragmentation of actin filaments. Biochemistry 1982; 21:1909–13. [DOI] [PubMed] [Google Scholar]
- 123. Ferrone FA, Hofrichter J, Sunshine HR, Eaton WA. Kinetic studies on photolysis‐induced gelation of sickle cell hemoglobin suggest a new mechanism. Biophys J 1980; 32:361–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Frieden C. Protein aggregation processes: in search of the mechanism. Protein Sci 2007; 16:2334–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Tay S, Hughey JJ, Lee TK, Lipniacki T, Quake SR, Covert MW. Single‐cell NF‐κB dynamics reveal digital activation and analogue information processing. Nature 2010; 466:267–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Gioannini TL, Weiss JP. Regulation of interactions of Gram‐negative bacterial endotoxins with mammalian cells. Immunol Res 2007; 39:249–60. [DOI] [PubMed] [Google Scholar]
- 127. Oszmiana A, Williamson DJ, Cordoba SP, Morgan DJ, Kennedy PR, Stacey K et al The size of activating and inhibitory killer Ig‐like receptor nanoclusters is controlled by the transmembrane sequence and affects signaling. Cell Rep 2016; 15:1957–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Pageon SV, Cordoba SP, Owen DM, Rothery SM, Oszmiana A, Davis DM. Superresolution microscopy reveals nanometer‐scale reorganization of inhibitory natural killer cell receptors upon activation of NKG2D. Sci Signal 2013; 6:ra62. [DOI] [PubMed] [Google Scholar]
- 129. Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G et al The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol 2014; 15:727–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Baroja‐Mazo A, Martin‐Sanchez F, Gomez AI, Martinez CM, Amores‐Iniesta J, Compan V et al The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 2014; 15:738–48. [DOI] [PubMed] [Google Scholar]