

Summary
CD4+ Foxp3+ regulatory T (Treg) cells can control both cellular and humoral immune responses; however, when and how Treg cells play a predominant role in regulating autoimmune disease remains elusive. To deplete Treg cells in vivo at given time‐points, we used a mouse strain, susceptible to glucose‐6‐phosphate isomerase peptide‐induced arthritis (GIA), in which the deletion of Treg cells can be controlled by diphtheria toxin treatment. By depleting Treg cells in the GIA mouse model, we found that a temporary lack of Treg cells at both priming and onset exaggerated disease development. Ablation of Treg cells led to the expansion of antigen‐specific CD4+ T cells including granulocyte–macrophage colony‐stimulating factor, interferon‐γ and interleukin‐17‐producing T cells, and promoted both T‐cell and B‐cell epitope spreading, which perpetuated arthritis. Interestingly, specific depletion of cytotoxic T‐lymphocyte antigen‐4 (CTLA‐4) on Treg cells only, was sufficient to protect mice from GIA, due to the expansion of CTLA‐4− Treg cells expressing alternative suppressive molecules. Collectively, our findings suggest that Treg cells, independently of CTLA‐4, act as the key driving force in controlling autoimmune arthritis development.
Keywords: autoimmune arthritis, cytotoxic T‐lymphocyte antigen‐4, epitope spreading, regulatory T cells
Introduction
Autoimmune diseases are complex and normally constitute three stages: an autoimmune priming phase, the clinical inflammatory onset and a chronic destructive development.1 The tipping point leading to the inflammatory attack and the self‐perpetuated chronic development could be influenced by an imbalance between effector and regulatory T cells, leading to epitope spreading regardless of the specificity of the initially insulted antigen and with a subsequent uncontrolled cytokine and pathogenic antibody production. It is possible that this uncontrolled balance also occurs during the chronic phase, explaining the relapsing pattern and sometimes resolution of the disease.2 Foxp3+ regulatory T (Treg) cells are crucial to maintain self‐tolerance and homeostasis. Loss of functional Treg cells causes widespread autoimmunity in both mice and humans.3 However, it remains unclear whether a lack of Treg cells is the driving force to promote the propagation of autoimmune diseases.
Mice lacking the Foxp3 transcription factor spontaneously develop fatal autoimmune disorders due to the expansion of self‐reactive T cells, which are continuously suppressed by Treg cells.4 The Treg cells use a wide range of suppressive mechanisms,5 among which, cytotoxic T‐lymphocyte antigen‐4 (CTLA‐4) has been demonstrated to play a crucial role in maintaining self‐tolerance and immune homeostasis.6 CTLA‐4 is up‐regulated on activated conventional T cells but constitutively expressed on Treg cells. Depletion of Treg cells, blocking of total CTLA‐4 or depletion of Treg‐cell‐specific CTLA‐4, all result in an increase of antigen‐specific follicular T helper cells, germinal centre B cells, memory cells and plasma cells after immunization, illustrating that Treg cells control humoral immune responses through CTLA‐4.7, 8 However, it remains to be determined whether Treg cells are also involved in epitope spreading, which is of major importance in the chronic phase of autoimmune diseases such as rheumatoid arthritis (RA).
Rheumatoid arthritis is a perpetuating autoimmune disease that takes months or even years to progress from the pre‐RA stage to full‐blown RA, which involves autoimmunity to post‐translational modifications of proteins, increases of pro‐inflammatory cytokines and an altered balance between effector and regulatory T cells.9 An antibody response to glucose‐6‐phosphate isomerase (GPI), is detectable in RA, and in a similar way to the mouse model it is possible that these antibodies specifically bind to cartilage and contribute to the development of arthritis.10 Hence, as in RA, the GPI‐triggered disease starts with a systemic autoimmunity that later develops into an inflammatory attack on joints, in the case of GPI in the form of precipitated protein on the joint cartilage surface.10 As GPI‐mediated autoimmunity resembles the initial steps of RA, we used a variant of this model, the GPI peptide‐induced arthritis (GIA) model,11 to investigate whether and how lack of functional Treg cells drives the development of arthritis.
Here, by in vivo depletion of Treg cells at different time‐points during GIA, we have demonstrated that even a transient loss of Treg cells is sufficient to enhance and prolong arthritis progression. The propagation of disease was accompanied by the expansion of antigen‐specific conventional T cells, myeloid cells, plasma cells and most importantly by both T‐cell and B‐cell epitope spreading to a joint‐specific protein, collagen type II. Notably, CTLA‐4 seems to be dispensable in the GIA model as specific depletion of CTLA‐4 on Treg cells was insufficient to reproduce the enhanced arthritis susceptibility seen in Treg cell‐depleted mice.
Materials and methods
Mice
Bacterial artificial chromosome (BAC) transgenic Foxp3DTR (DEREG) and Foxp3tm9(EGFP/Cre/ERT2)Ayr/J mice (Jackson Laboratory, Bar Harbor, ME) and CTLA‐4fl/fl, on the C57BL/6 background6 were additionally backcrossed to C57BL/10Q for at least six generations. To deplete Foxp3+ cells, DEREG mice were intraperitoneally injected with diphtheria toxin (DT) for 2 consecutive days and complete depletion was achieved by the following day.12 To generate inducible Treg‐specific loss of CTLA‐4, CTLA‐4fl/fl mice were intercrossed with Foxp3tm9(EGFP/Cre/ERT2)Ayr/J mice to obtain Foxp3wt/wt CTLA‐4fl/fl or fl/wt (wild‐type; WT) and Foxp3Cre/Cre CTLA‐4fl/fl (iCKO) genotypes. To induce CTLA‐4 depletion on Foxp3+ cells, mice were intraperitoneally injected with 4 mg tamoxifen in corn oil (+ 5% ethanol) on days 0, 1 and 5. Complete depletion was achieved by day 6.13 All mice were maintained under specific pathogen‐free conditions. All experimental animal procedures were approved by the local ethics committee (License No: N35/16).
Arthritis model and evaluation
Arthritis was induced by the hGPIc‐c peptide (NH2‐IWYINCFGCETHAML‐OH) (10 μg/mouse in 50 μl phosphate‐buffered saline) emulsified with an equal volume of complete Freund's adjuvant. Each mouse was intradermally injected with 100 μl emulsion at the base of the tail. Arthritis development was monitored using a macroscopic scoring system.11 Serum antibody was detected by Luminex assay. At the end‐point of animal models, mice were killed and hind paws were collected for section and followed by haematoxylin & eosin and Safranin O staining.
Anti‐CTLA‐4 treatment
To block CTLA‐4, hGPIc‐c peptide‐immunized mice received anti‐CTLA‐4 antibody (9H10) treatment on day 3, 6 and 9, 100 μg/mouse/day, intraperitoneal injection. Control mice received the same amount of anti‐hamster IgG treatment.
Luminex immunoassay
Serum antibodies were detected by Luminex based immunoassay. Briefly, biotinylated peptides or protein were immobilized to beads either through NeutrAvidin (ThermoFisher Scientific, Waltham, MA) or directly, respectively. Serum samples were diluted 1 : 100 (v/v) in phosphate‐buffered saline/ 0·05% Tween‐20 and incubated in 96‐well black plates pre‐coated with bead array for 75 min on a shaker (850 rpm). After washing on a magnetic plate washer (EL406; Biotek, Winooski, VT), samples were incubated with secondary anti‐mouse IgG Fcy‐PE (Jackson Immuno Research, West Grove, PA) for 60 min with shaking, followed by measurement in Bio‐plex Pro 200. The mean fluorescence intensity was used to quantify the interaction of serum antibody with given peptides or proteins.
Antibodies and flow cytometry analysis
The following antibodies were purchased either from Biolegend (San Diego, CA) or BD Biosciences (San Jose, CA): anti‐CD45 (30‐F11), anti‐CD3 (145‐2C11), anti‐CD4 (H129.19), anti‐B220 (RA3‐6B2), anti‐CD11b (M1/70), anti‐CD62L (MEL‐14), anti‐CD44 (Ly24), anti‐programmed cell death protein 1 (PD‐1) (RMPI‐30), anti‐CD40L (MR1), anti‐CTLA‐4 (UC10‐4B9), anti‐Ki‐67 (B56), anti‐interferon‐γ (IFN‐γ) (R46A2), anti‐interleukin‐17 (IL‐17) (TC1118H10.1), anti‐granulocyte–macrophage colony‐stimulating factor (GM‐CSF) (MP1‐22E9), anti‐tumour necrosis factor‐α (TNF‐α) (MP6‐XT22), anti‐IL‐10 (JES5‐16E3). Foxp3 (FJK‐16s), anti‐CD73 (eBioTY/11.8) and anti‐FR4 (eBIo12A5) were purchased from eBioscience (San Diego, CA). Live and dead cells were distinguished by near‐infrared dead cell staining (Molecular Probes, Life Technologies, Carlsbad, CA). After surface staining, cells were fixed and permeabilized (Cytofix/Cytoperm; BD Biosciences); CD40L, IFN‐γ, IL‐17, IL‐10, GM‐CSF and TNF‐α were stained intracellularly. For characterization of Treg cells, the cells were fixed and permeabilized with a transcription factor Fix/Perm kit, then Foxp3, CTLA4 and Ki‐67 were stained. To check antigen‐specific T cells, home‐made biotinylated‐Aq‐hGPIc‐c were labelled with phycoerythrin‐Streptavidin (Molecular Probes) at a molar ratio of 1 : 4 to make tetramer complex. At least 100 000 cells were collected by LSRII flow cytometry and data were analysed using flowjo software (TreeStar, Ashland, OR).
Recall assay
Splenocytes or inguinal lymph node cells were isolated 12–14 days after immunization, plated in 96‐well ELISPOT plates (#MSIPS4W10, MultiScreen Filter Plates; Merck Millipore, Burlington, MA) at 1 × 106 or 5 × 105 cells/well, respectively and stimulated with hGPIc‐c (10 μm), mGPIc‐c (10 μm), galCII259–273 (10 μg/ml), CII259–273 (10 μg/ml) or concanavalin A (ConA, 1 μg/ml) for 24 hr. Both IFN‐γ and IL‐17 secretion were determined. Spots were developed with BCIP/NBT (Sigma‐Aldrich, St Louis, MO). Wells were scanned by an ImmunoScan Elispot reader and analysed with immunospot software (Cellular Technology Ltd, Shaker Heights, OH).
Statistical analysis
Data analysis was performed using prism software (GraphPad, version 7.0; GraphPad, San Diego, CA). Significance was determined by Mann–Whitey's U‐test or Student's t‐test for two‐group comparisons; one‐way analysis of variance (anova) or two‐way anova with Tukey's multiple comparisons post‐test for three‐group comparisons; and Fisher's exact test. P‐values of < 0·05 were considered as significant.
Results
Transient ablation of Treg cells leads to a pro‐inflammatory milieu
DEREG mice express a DT receptor‐enhanced green fluorescent protein (GFP) under the control of the foxp3 gene promoter, as part of a BAC transgene, which enables the depletion of Treg cells at any given time by injection of DT.12 Naive DEREG mice and WT littermates were treated with DT (1 μg/mouse, intraperitoneally) for two consecutive days (i.e. days – 2 and –1) and the complete depletion was achieved the following day (day 0); however, Treg cells repopulated rapidly in the next 2–3 days (Fig. 1a). Notably, GFP− Foxp3+ T cells expanded markedly before the rebound of GFP+ Foxp3+ Treg cells and hence the total frequency of Foxp3+ Treg cells was significantly increased for the following 3–4 weeks after depletion (Fig. 1b, c). Except for the decrease of both CD4+ and CD8+ T cells, other immune cells including B cells, dendritic cells (CD11b− CD11c+), macrophages (CD11b+ F4‐80+), neutrophils (CD11b+ Gr.1hi) and plasma cells (B220− CD138+) were significantly increased in the spleen on day 1 after the first DT treatment in DEREG mice (Fig. 1d). Furthermore, although the frequency of Foxp3+ T cells was still low in DEREG mice on day 1, cytokine‐producing CD4+ T cells were expanded, including GM‐CSF, TNF‐α, and IFN‐γ‐producing T cells (Fig. 1e). However, serum cytokine production was not changed either before or after DT treatment, indicating that the systemic immune homeostasis was not affected (Fig. 1f). Notably, rebounded Treg cells show similar suppression capacity towards CD4+ CD25− effector T cells as original existing ones (see Supplementary material, Fig. S1). Therefore, our data suggest that DT treatment results in a temporal loss of Treg cells, which leads to a local pro‐inflammatory milieu in DEREG mice.
Figure 1.
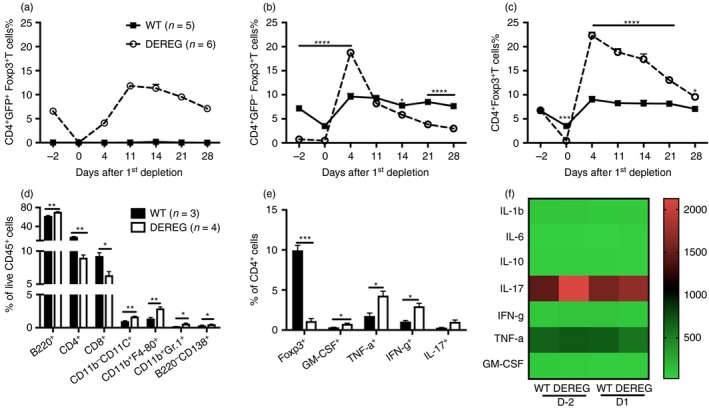
Transient ablation of regulatory T (Treg) cells leads to a pro‐inflammatory milieu. Diphtheria toxin (DT) was administered in DEREG and wild‐type (WT) littermate control mice for two consecutive days, that is day 2 and 1. The frequencies of CD4+ GFP+ Foxp3+ T cells (a), CD4+ GFP− Foxp3+ T cells (b), and CD4+ Foxp3+ T cells (c) in blood were followed for 28 days. The frequencies of different immune cell populations (d) and different cytokine‐producing CD4+ T cells (e) in the spleen 1 day after the first administration of DT were determined by flow cytometry. Sera samples on day 2 and day 1 were subjected to cytokine‐plex analysis by luminex immunoassay (f). The scale indicates the concentration of cytokines (pg/ml). Error bars represent mean ± SEM and significant differences between DEREG and WT mice were determined by unpaired t‐test.
Treg cells control arthritis development in GIA
To investigate the role of Treg cells more specifically during the priming of autoreactive T cells, we used the DEREG mice and tested the GIA model. GIA is an acute autoimmune disease model with typical arthritis symptoms appearing on days 10–12 and remission within 30 days after immunization.14 In contrast to the rebound expansion of Treg cells in naive DEREG mice after DT treatment, there was no obvious increase in Treg cells during the disease development in the GIA model between DEREG and control mice upon DT treatment (see Supplementary material, Fig. S2). The obvious question then was whether the short‐term loss of Treg cells on GIA before priming or upon disease onset could have an impact on arthritis. DT was administered at either days –2 and –1 to achieve complete depletion at day 0, or at days 8 and 9 to achieve complete depletion at day 10, the onset of disease. In both cases, DEREG mice developed more severe acute arthritis symptoms and also developed a chronic phase of the disease, which was not observed in littermate controls (Fig. 2a, b). Moreover, arthritic joints from DEREG mice were marked by synovial infiltration, hyperplasia, cartilage destruction and bone erosion, whereas joints from WT mice were largely intact with few infiltrates but the cartilage was almost gone on day 50 after immunization (Fig. 2c).
Figure 2.
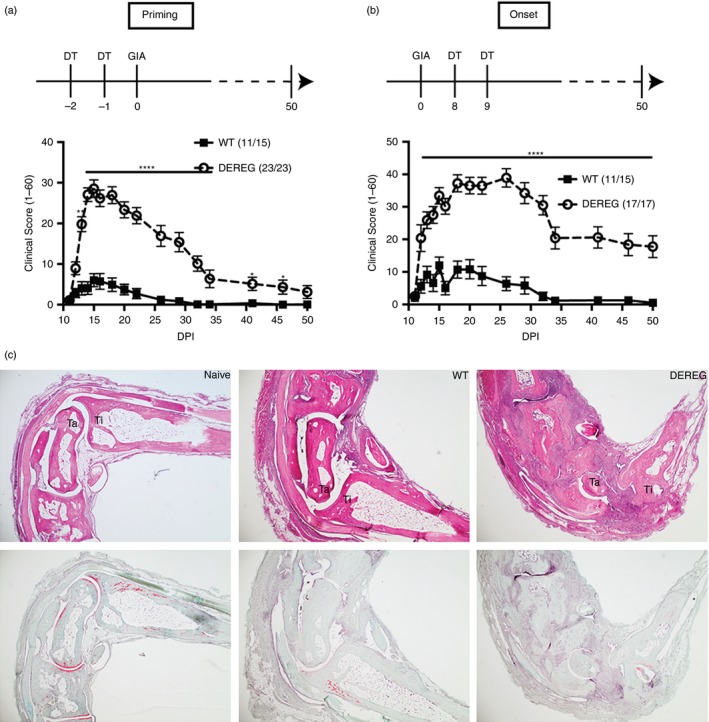
Regulatory T (Treg) cells control arthritis development in glucose‐6‐phosphate isomerase peptide‐induced arthritis (GIA). Diphtheria toxin (DT) was injected in DEREG and littermate control mice on days 2 and 1 (priming) or days 8 and 9 (onset) after immunization with hGPIc‐c to deplete Treg cells in the GIA model. The clinical score (1–60) is shown as mean ± SEM with Treg cell depletion occurring either at the priming (a) or onset phase (b). (b) Data from two pooled independent experiments. Numbers in the brackets indicate the incidence of the disease. Representative images of haematoxylin & eosin staining (top) and Safranin O staining (bottom) of decalcified joint tissues of wild‐type (WT) and DEREG mice on day 50 after immunization. Naive mice were used as control (c).
To investigate whether Treg cells played a critical role in the chronic phase as well, we treated DEREG and littermate control mice in the GIA model with DT at the remission stage. As shown in the Supplementary material (Fig. S3), there was a relapse but no difference in severity comparing between the DEREG and WT mice. Taken together, our data show that a transient lack of Treg cells in the priming or onset phase have a long‐term impact on autoimmune arthritis, increasing severity as well as chronicity.
Transient Treg cell depletion enhances antigen‐specific T‐cell responses
To understand why and how short‐term lack of Treg cells causes a long‐term impact on autoimmune arthritis, we investigated whether the loss of Treg cells affects T‐cell homeostasis. To do this we depleted Treg cells at the onset of GIA, at days 8 and 9, and the mice were killed at the peak of the disease, at days 13–14. Using tetramer staining, we detected an almost 10‐fold increase of hGPIc‐c specific T cells in the draining lymph nodes of DEREG mice compared with WT mice (Fig. 3a). Moreover, among these hGPIc‐c specific T cells in draining lymph nodes, 90% were CD44+ CD62L− effector T cells in DEREG mice but only 30% in WT mice (Fig. 3b). In addition, the frequencies of various cytokine (GM‐CSF, IFN‐γ and IL‐17) ‐producing antigen‐specific T cells (CD4+ CD40L+) were significantly elevated in both spleen and draining lymph nodes in DEREG mice (Fig. 3c, d). Furthermore, we wanted to investigate whether loss of Treg cells led to the recognition of additional joint antigens, besides the immunizing hGPIc‐c one. Therefore, we analysed the presence of antigen‐specific T cells against hGPIc‐c, mGPIc‐c, native CII259–273 and glycosylated CII259–273 (gal‐CII259–273). As expected, there was a massive expansion of hGPIc‐c‐specific and mGPIc‐c‐specific T cells producing both IFN‐γ and IL‐17 on day 12. However, more importantly, CII259–273‐specific as well as gal‐CII259–273‐specific T cells were also significantly increased in DEREG mice (Fig. 3e). Upon disease remission at day 30, antigen‐specific T cells against hGPIc‐c and mGPIc‐c dropped around three‐ to four‐fold in both DEREG and WT mice, but there was no obvious decrease of antigen‐specific T cells against CII259–273 and gal‐CII259–273 in draining lymph nodes of DEREG mice (Fig. 3f). Hence, our results suggest that depletion of Treg cells not only triggers the expansion of antigen‐specific T cells but also induces pathogenic effector T cells recognizing additional joint epitopes such as collagen type II, which might contribute to the chronicity of the disease in this setting.
Figure 3.
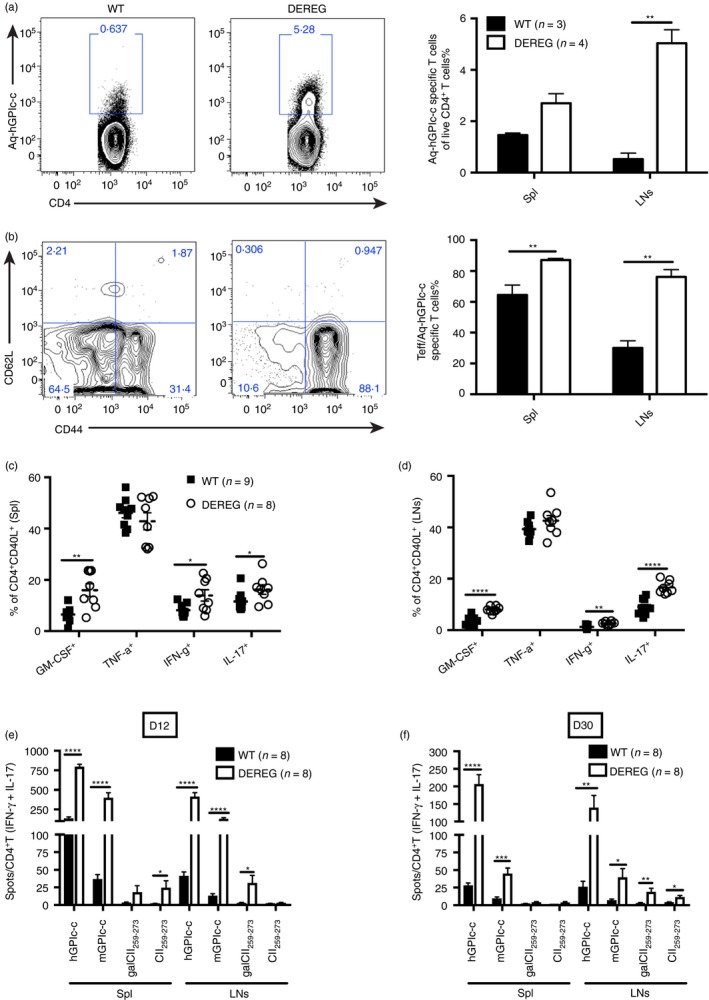
Transient regulatory T (Treg) cell depletion enhances antigen‐specific T‐cell responses. Diphtheria toxin (DT) was administered on the onset phase of glucose‐6‐phosphate isomerase peptide‐induced arthritis (GIA) (i.e. days 8 and 9) and DEREG and littermate control mice were killed on days 13–14. The representative FACS plots show the hGPIc‐c‐specific CD4+ T cells in DEREG and littermate controls in draining lymph nodes (LNs) and the frequencies of hGPIc‐c‐specific CD4+ T cells in spleen (Spl) and LNs are shown (a). Frequencies of CD44+ CD62L− effector T (Teff) cells among hGPIc‐c‐specific T cells (b). Cytokine‐producing T cells out of CD4+ CD40L+ antigen‐specific T cells in spleen (c) and LNs (d) were determined by flow cytometry. Antigen‐specific T cells responding to hGPIc‐c, mGPIc‐c, gal‐CII259–273 and CII259–273 with IFN‐γ or IL‐17 in both spleen and LNs were determined by ELISPOT on day 12 (e) and day 30 after immunization (f). Numbers of spots were normalized to per 1 × 105 CD4+ T cells. (a) and (b) show represented experiments out of three independent experiments; (c–f) show data from two pooled independent experiments. Error bars represent mean ± SEM and significant differences between DEREG and wild‐type (WT) mice were determined by unpaired t‐test.
Treg cell depletion promotes B‐cell epitope spreading
As we observed a dramatic enhancement of antigen‐specific T‐cell responses, both in strength and width, we sought to investigate whether Treg cell depletion also influences the humoral response. Indeed, plasma cells (B220− CD138+) increased around fourfold in the draining lymph nodes of DEREG mice, whereas the increase in the spleen was not as pronounced (Fig. 4a). However, surprisingly, no difference in serum IgG antibodies against hGPIc‐c was detected between DEREG and WT littermates at any time‐point during disease development (Fig. 4b). Yet serum antibodies against the full hGPI and mGPI proteins were dramatically increased in DEREG mice, especially in the chronic phase of the diseases, that is after day 30. We also detected additional antibody responses against other epitopes of hGPI such as hGPI301–315 and hGPI340–354, which locate close to the hGPIc‐c (hGPI325–339) peptide, though these responses were stronger in WT littermates. Furthermore, we analysed serum antibodies against the major epitopes of collagen type II, namely triple helical C1 and J115 and found that the mean fluorescence intensity (MFI) of serum antibodies against the C1 epitope increased profoundly in DEREG mice over the course of the disease (Fig. 4c). Therefore, our data show that Treg cell depletion also facilitates B‐cell epitope spreading, especially in the chronic phase of the disease.
Figure 4.
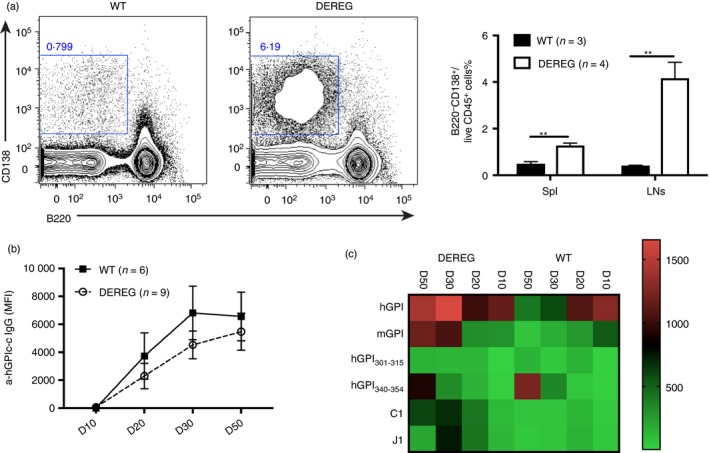
Regulatory T (Treg) cell depletion promotes B‐cell epitope spreading. Diphtheria toxin (DT) was administered on the onset phase of glucose‐6‐phosphate isomerase peptide‐induced arthritis (GIA) (i.e. day 8 and 9) and DEREG and wild‐type (WT) littermates were killed on day 13–14 to check the plasma cells. The representative FACS plots show plasma cells (B220− CD138+) in DEREG and WT mice and the frequencies of plasma cells in spleen and lymph nodes (LNs) are shown (a). Serum was collected on days 10, 20, 30 and 50 during the disease progression and the antibody response against different proteins and peptides was analysed by Luminex immunoassay. The mean fluorescence intensity (MFI) of serum antibodies against hGPIc‐c is shown (b). The MFI of serum antibodies against proteins (hGPI and mGPI) and peptides (hGPI301–315, hGPI340–354, C1 and J1) is shown as mean in the heat map (c). (a) shows one representative experiment out of three independent experiments. Error bars represent mean ± SEM and significant differences between DEREG and WT mice were determined by unpaired t‐test.
Treg cells without CTLA‐4 still possess suppressive capacity
CTLA‐4 has been shown to be of major importance for the suppressive capacity of Treg cells. Hence, we wondered whether CTLA‐4 also played a dominant role in the development of GIA. To answer this question, we used previously described conditional knockout mice (iCKO) where CTLA‐4 can be specifically depleted on Foxp3+ cells using tamoxifen.13 CTLA‐4 was specifically depleted on Treg cells in iCKO mice either before priming or upon disease onset and arthritis development in the GIA model was followed. Surprisingly, Treg cells lacking CTLA‐4 were able to suppress the development of arthritis at both settings, whereas CTLA‐4‐sufficient Treg cells in WT littermates were unable to suppress GIA (Fig. 5a, b). There was no difference in the serum antibody responses against the hGPIc‐c, C1 and J1 epitopes but there was a trend of a lower response towards hGPI and mGPI proteins in iCKO mice compared with littermate controls (Fig. 5c, d). Furthermore, we analysed cytokine‐producing antigen‐specific T cells but there was no difference in GM‐CSF and IFN‐γ‐secreting cells, in contrast, TNF‐α and IL‐17‐producing T cells were even significantly increased in WT littermates, correlating with more severe disease in WT mice (Fig. 5e). As seen previously in another model of arthritis, specific depletion of CTLA‐4 on Treg cells in iCKO mice increased the frequency of Foxp3+ cells and led to the up‐regulation of other suppressive molecules such as PD‐1, together with an anergic phenotype in Treg cells of iCKO mice marked by CD73hi and folate receptor 4 (FR4)hi (Fig. 5f). Therefore, our data suggest that Treg cells specifically lacking CTLA‐4 can nonetheless suppress the development of arthritis due to a quantitative increase in the Treg cells and qualitative compensation by other suppressive molecules such as PD‐1.
Figure 5.
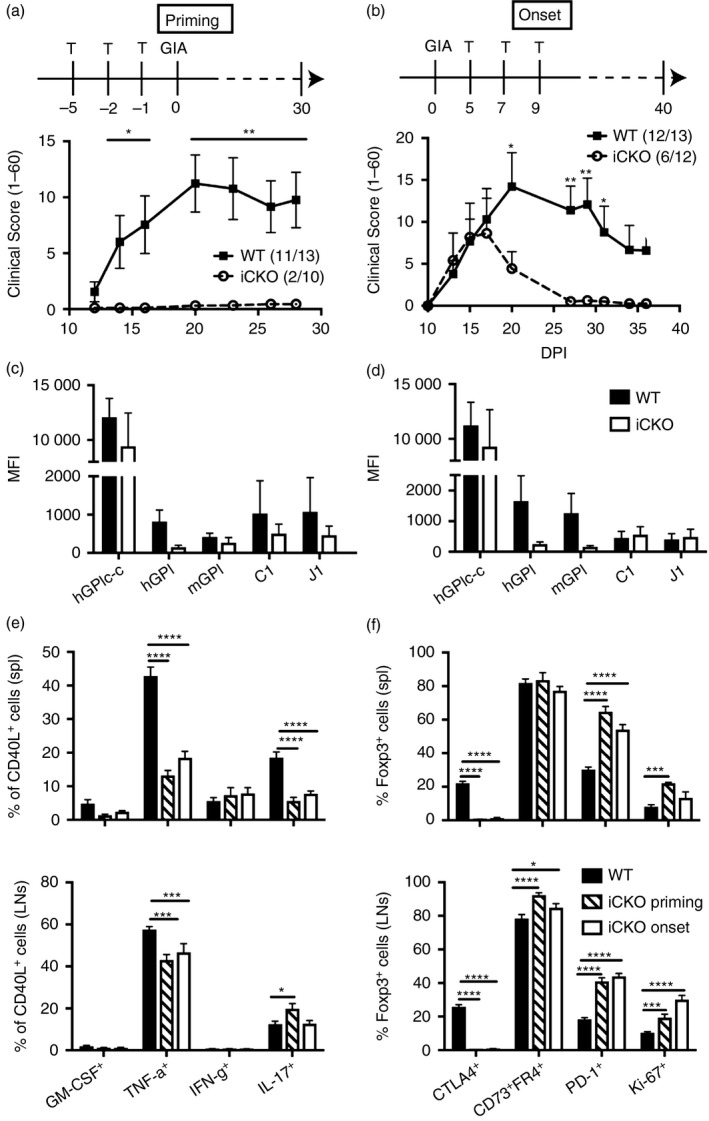
Reglatory T (Treg) cells lacking cytotoxic T‐lymphocyte antigen‐4 (CTLA‐4) can suppress autoimmune arthritis. CTLA‐4 was selectively depleted on Treg cells in conditional knockout (iCKO) mice with tamoxifen before immunization (i.e. days –5, –2 and –1) or upon onset (i.e. days 5, 7 and 9). The clinical score (1–60) is shown upon CTLA‐4 depletion either at the priming phase (a) or onset phase (b). Numbers in the brackets indicate the incidence of the disease. Sera were collected on day 30 and serum IgG against peptides (hGPIc‐c, C1 and J1) and proteins (hGPI and mGPI) were detected by Luminex immunoassay and the values are displayed as MFI at the priming (c) and onset (d) phases, respectively. The frequencies of granulocyte–macrophage colony‐stimulating factor (GM‐CSF), interleukin‐17 (IL‐17) and interfeorn (IFN‐γ) ‐producing T cells amongst CD4+ CD40L+ cells from spleen and draining lymph nodes (e) and the percentages of CTLA‐4+, CD73+ FR4+, PD‐1+ and Ki‐67+ cells among CD4+ Foxp3+ cells from spleen and lymph nodes (f) were checked by flow cytometry on day 15. Error bars represent mean ± SEM and significant differences were determined by unpaired t‐test between two groups and two‐way analysis of variance between three groups.
Anti‐CTLA‐4 antibody treatment does not affect GIA development
Considering that Ctla‐4‐deficient mice suffer from a lymphoproliferative syndrome that is fatal in complete KO mice16 and much milder in iCKO mice,13 we used an anti‐CTLA‐4 antibody to block CTLA‐4 expression instead of depleting it, in order to reduce the risk of violating the fragile immune homeostasis. With anti‐CTLA‐4 antibody treatment of WT mice, there was a tendency to enhanced GIA development, but this was not significant compared with control antibody‐treated mice (Fig. 6a). In line with the similar disease development, there was no difference in titres of serum antibodies against either hGPI or mGPI protein nor against the CII epitopes C1 and J1 (Fig. 6b). There was also no difference in IFN‐γ‐ and IL‐17‐producing antigen‐specific T cells in either spleen or lymph nodes upon hGPIc‐c or mGPIc‐c restimulation (Fig. 6c, d). Therefore, our results suggest that CTLA‐4 does not play an important role in the development of GIA.
Figure 6.
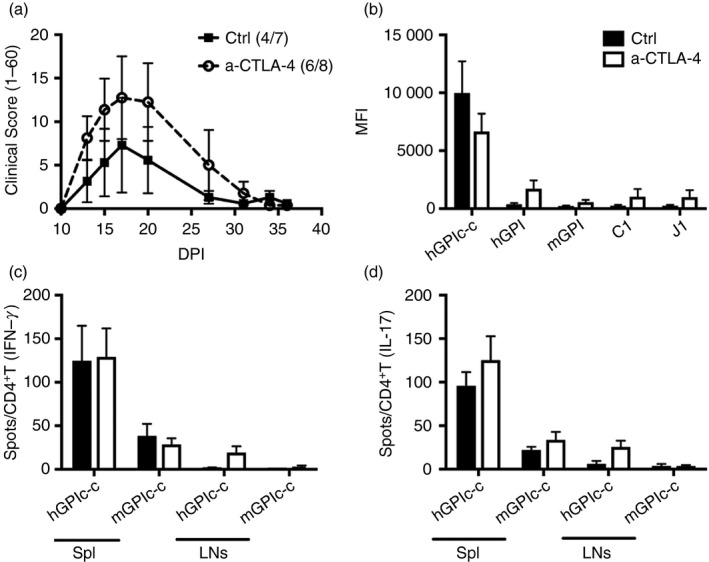
Anti‐cytotoxic T‐lymphocyte antigen‐4 (CTLA‐4) antibody treatment does not affect glucose‐6‐phosphate isomerase peptide‐induced arthritis (GIA) development. Immunized mice were treated with either anti‐CTLA‐4 or control antibody on days 3, 6 and 9. The clinical score (1–60) and mean fluorescence intensity (MFI) of serum antibody responses against proteins (hGPI and mGPI) and peptides (hGPIc‐c, C1 and J1) are shown in (a) and (b), respectively. Antigen‐specific CD4+ T cells against hGPIc‐c and mGPIc‐ were determined by ELISPOT and the number of interferon‐γ (IFN‐γ) (c) and interleukin‐17 (IL‐17) (d) spots in both spleen and lymph nodes were normalized to per 1 × 105 CD4+T cells. Error bars represent mean ± SEM. No significant difference was observed between the two groups by unpaired t‐test.
Discussion
In this study, our findings emphasize that a short‐term imbalance between Treg cells and effector T cells during activation of autoreactive T cells results in a long‐term impact on autoimmune arthritis. The loss of Treg cells is a trigger for a chain reaction including activation of autoreactive T cells, T‐cell and B‐cell epitope spreading towards joint epitopes, effector T‐cell proliferation and cytokine overproduction, all of which eventually contribute to the increase in arthritis severity.
There is no consensus regarding both numerical and functional abnormalities of Treg cells in RA,17 though it has been shown that Treg cells modulate arthritis in mice, as depletion or absence enhances disease whereas transfer ameliorates disease, some of the settings are not physiological.18, 19 In our study, we used DEREG mice to elucidate the role of Treg cells in autoimmune arthritis. The advantage of BAC‐transgenic DEREG mice is that they allow for the specific depletion of Treg cells through the administration of DT at different time‐points; however, the drawback of this system is the rapid rebound of Treg cells following withdrawal of DT treatment, which has been described previously20 and was also observed in our study (Fig. 1c). Nonetheless, this ‘drawback’ provides unique opportunities to mimic human situations. Once there is a deficit of Treg cells, conventional T cells are activated and secrete cytokines such as GM‐CSF and TNF‐α. It has been shown that GM‐CSF secreted by RA synovial CD4+ T cells promotes the differentiation of inflammatory dendritic cells, which possess the potent capacity to induce additional GM‐CSF‐producing T cells in a positive feedback loop.21 TNF‐α has been shown to play a central role in regulating pro‐inflammatory cytokines, chemokines and growth factors expressed in arthritic joints, which makes it an important therapeutic target for RA.22 Interestingly, TNF‐α can disable Treg cell function through dephosphorylation of Foxp3, and inhibition of TNF‐α restores Treg cell suppressive function in RA.23 Therefore, lack of Treg cells could be a trigger in establishing a vicious feedback loop in autoimmune settings.
The isomerase GPI is not a joint‐specific protein but is ubiquitously expressed, yet immunization with a peptide spanning from 325–339 of GPI triggers joint‐specific autoimmune arthritis, which depends on both T and B cells.11 The explanation could be that GPI is uniquely precipitated on the joint surface and the activation of GPI‐reactive T and B cells occurs in joint‐draining lymph nodes.10, 14 The GIA model is an acute (self‐limited in time) arthritis model with remission occurring within 30 days in WT mice. However, ablation of Treg cells at either the priming or onset phase, results in a previously unobserved chronicity of the disease (Fig. 2a, b). Our observations have also confirmed the previous findings that depletion of Treg cells, using anti‐CD25 antibody, control the transition from acute self‐limiting to non‐remitting destructive GPI protein‐induced arthritis.24 Of importance, we have demonstrated that Treg cells not only suppress effector T‐cell responses but also control both T‐cell and B‐cell epitope spreading in GIA. Glycosylated CII259–273 and non‐glycosylated CII259–273 are the dominant T‐cell epitopes in CIA and RA patients.25 Raposo et al.26 recently showed that post‐translational modification of T‐cell epitopes such as CII259–273 can escape central tolerance induction, which explains why T‐cell responses against the glycosylated version of CII259–273 are more prone in RA patients. Our results (Fig. 3e, f) have shown that depletion of Treg cells triggers T‐cell responses against both glycosylated CII259–273 and non‐glycosylated CII259–273. In addition, we observed that serum antibody responses (Fig. 4c) against the major CII epitope in RA patients: C1 and J1, were elevated upon loss of Treg cells, especially in the chronic phase of GIA. Therefore, our study has demonstrated that the original insulted autoantigen (i.e. hGPIc‐c) might not be the same antigen as that to which the chronic autoimmune arthritis development is oriented (i.e. CII). Most likely, the epitope spreading towards autoreactive T and B cells, including both CII and autologous GPI, represents a response to endogenous proteins derived from the joints. This is not a new concept in autoimmune diseases, as it has been elegantly shown that epitope spreading initiates the chronic progression in T‐cell‐mediated demyelinating models of multiple sclerosis.27 Taken together, exploring the cellular and molecular basis of epitope spreading in the chronic phase of autoimmune diseases is crucial to understand the pathogenesis of these diseases and ultimately to design antigen‐specific treatments.
CTLA‐4 is one of the major suppressive mechanisms of Treg cells. Deficiency of CTLA‐4 has been shown to exaggerate arthritis in CIA and the myelin oligodendrocyte glycoprotein (MOG) protein‐induced EAE model28 but not in the MOG‐peptide induced EAE model.29 In the GIA model, CTLA‐4 did not seem to play a crucial role because Treg cells specifically depleted of CTLA‐4 but not WT Treg cells could suppress GIA and blocking of CTLA‐4 with blocking antibody did not affect the disease development. Instead, it appeared that it was the total number of Treg cells that decided disease outcome. As CTLA‐4 blocking antibodies also affect mainly Treg cells, it would be interesting to study the role of CTLA‐4 on conventional T cells in the GIA model. It is known that a functional overlap between CTLA‐4 and Foxp3 can drive regulatory programmes but identification of their divergent pathways is also crucial for understanding different immune pathways.30 As shown in Fig. 5(f), specific depletion of CTLA‐4 on Treg cells induced a quantitative expansion of Treg cells and a qualitative compensation by alternative suppressive and anergic T‐cell phenotypes. Therefore, as autoimmune diseases are such complex diseases, one needs to gather comprehensive information including the local milieu, initiating and propagating autoantigens and which immune cells are involved to fully understand all aspects and phases of disease.
Collectively, our findings emphasize that Treg cells, operating during the priming of autoreactive effector T cells, are the key to control autoimmune arthritis development. To restore the immunological homeostasis focusing on the number of Treg cells in a specifically determined narrow time window is an intriguing approach to induce a state of tolerance and control autoimmune diseases.
Author contributions
MY and RH designed the research; MY, KK, CH and BX performed the research; MY analysed the data; MY, KK, IG and KW discussed the data; MY and RH wrote the manuscript.
Disclosure
No conflict of interest.
Grant support
This work was supported by grants from the Konung Gustaf V:s 80‐Årsfond (MY), the Karolinska Institute Foundation (MY), the STINT (CH2016‐6690) (MY), the Swedish Science Research Council (Vetenskapsrådet) (RH), the Swedish Foundation for Strategic Research (RB13‐0156) (RH), the KA Wallenberg foundation (KAW 2015.0063) (RH), and the European Union Innovative Medicine Initiative project BeTheCure (RH).
Supporting information
Figure S1. Repopulated regulatory T cells show similar suppressive capacity.
Figure S2. Dynamics of regulatory T cells in the glucose‐6‐phosphate isomerase peptide‐induced arthritis model.
Figure S3. Regulatory T cells are dispensable in the chronic phase of glucose‐6‐phosphate isomerase peptide‐induced arthritis.
Acknowledgements
We thank Carlos Palestro and Kristina Palestro for taking good care of animals, Erik Lönnblom for providing opinions for Luminex immunoassay and Changrong Ge for demonstrating the R programme.
References
- 1. Holmdahl R, Malmstrom V, Burkhardt H. Autoimmune priming, tissue attack and chronic inflammation – the three stages of rheumatoid arthritis. Eur J Immunol 2014; 44:1593–9. [DOI] [PubMed] [Google Scholar]
- 2. Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest 2015; 125:2228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol 2010; 11:7–13. [DOI] [PubMed] [Google Scholar]
- 4. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 2007; 8:191–7. [DOI] [PubMed] [Google Scholar]
- 5. Shevach EM. Mechanisms of Foxp3+ T regulatory cell‐mediated suppression. Immunity 2009; 30:636–45. [DOI] [PubMed] [Google Scholar]
- 6. Wing K, Onishi Y, Prieto‐Martin P, Yamaguchi T, Miyara M, Fehervari Z et al CTLA‐4 control over Foxp3+ regulatory T cell function. Science 2008; 322:271–5. [DOI] [PubMed] [Google Scholar]
- 7. Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA‐4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity 2014; 41:1026–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen‐specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA‐4. Immunity 2014; 41:1013–25. [DOI] [PubMed] [Google Scholar]
- 9. Firestein GS, McInnes IB. Immunopathogenesis of rheumatoid arthritis. Immunity 2017; 46:183–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Matsumoto I, Maccioni M, Lee DM, Maurice M, Simmons B, Brenner M et al How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint‐specific autoimmune disease. Nat Immunol 2002; 3:360–5. [DOI] [PubMed] [Google Scholar]
- 11. Pizzolla A, Wing K, Holmdahl R. A glucose‐6‐phosphate isomerase peptide induces T and B cell‐dependent chronic arthritis in C57BL/10 mice: arthritis without reactive oxygen species and complement. Am J Pathol 2013; 183:1144–55. [DOI] [PubMed] [Google Scholar]
- 12. Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G et al Selective depletion of Foxp3+ regulatory T cells induces a scurfy‐like disease. J Exp Med 2007; 204:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klocke K, Holmdahl R, Wing K. CTLA‐4 expressed by FOXP3+ regulatory T cells prevents inflammatory tissue attack and not T‐cell priming in arthritis. Immunology 2017; 152:125–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang M, Haase C, Viljanen J, Xu B, Ge C, Kihlberg J et al Cutting edge: processing of oxidized peptides in macrophages regulates T cell activation and development of autoimmune arthritis. J Immunol 2017; 199:3937–42. [DOI] [PubMed] [Google Scholar]
- 15. Lindh I, Snir O, Lonnblom E, Uysal H, Andersson I, Nandakumar KS et al Type II collagen antibody response is enriched in the synovial fluid of rheumatoid joints and directed to the same major epitopes as in collagen induced arthritis in primates and mice. Arthritis Res Ther 2014; 16:R143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA‐4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA‐4. Immunity 1995; 3:541–7. [DOI] [PubMed] [Google Scholar]
- 17. Morita T, Shima Y, Wing JB, Sakaguchi S, Ogata A, Kumanogoh A. The proportion of regulatory T cells in patients with rheumatoid arthritis: a meta‐analysis. PLoS ONE 2016; 11:e0162306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nguyen LT, Jacobs J, Mathis D, Benoist C. Where FoxP3‐dependent regulatory T cells impinge on the development of inflammatory arthritis. Arthritis Rheum 2007; 56:509–20. [DOI] [PubMed] [Google Scholar]
- 19. Morgan ME, Flierman R, van Duivenvoorde LM, Witteveen HJ, van Ewijk W, van Laar JM et al Effective treatment of collagen‐induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum 2005; 52:2212–21. [DOI] [PubMed] [Google Scholar]
- 20. Mayer CT, Lahl K, Milanez‐Almeida P, Watts D, Dittmer U, Fyhrquist N et al Advantages of Foxp3+ regulatory T cell depletion using DEREG mice. Immun Inflamm Dis 2014; 2:162–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reynolds G, Gibbon JR, Pratt AG, Wood MJ, Coady D, Raftery G et al Synovial CD4+ T‐cell‐derived GM‐CSF supports the differentiation of an inflammatory dendritic cell population in rheumatoid arthritis. Ann Rheum Dis 2016; 75:899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feldmann M, Maini RN. Anti‐TNF α therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol 2001; 19:163–96. [DOI] [PubMed] [Google Scholar]
- 23. Nie H, Zheng Y, Li R, Guo TB, He D, Fang L et al Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF‐α in rheumatoid arthritis. Nat Med 2013; 19:322–8. [DOI] [PubMed] [Google Scholar]
- 24. Frey O, Reichel A, Bonhagen K, Morawietz L, Rauchhaus U, Kamradt T. Regulatory T cells control the transition from acute into chronic inflammation in glucose‐6‐phosphate isomerase‐induced arthritis. Ann Rheum Dis 2010; 69:1511–8. [DOI] [PubMed] [Google Scholar]
- 25. Dzhambazov B, Holmdahl M, Yamada H, Lu S, Vestberg M, Holm B et al The major T cell epitope on type II collagen is glycosylated in normal cartilage but modified by arthritis in both rats and humans. Eur J Immunol 2005; 35:357–66. [DOI] [PubMed] [Google Scholar]
- 26. Raposo B, Merky P, Lundqvist C, Yamada H, Urbonaviciute V, Niaudet C et al T cells specific for post‐translational modifications escape intrathymic tolerance induction. Nat Commun 2018; 9:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med 2005; 11:335–9. [DOI] [PubMed] [Google Scholar]
- 28. Klocke K, Sakaguchi S, Holmdahl R, Wing K. Induction of autoimmune disease by deletion of CTLA‐4 in mice in adulthood. Proc Natl Acad Sci U S A 2016; 113:E2383–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Paterson AM, Lovitch SB, Sage PT, Juneja VR, Lee Y, Trombley JD et al Deletion of CTLA‐4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J Exp Med 2015; 212:1603–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Walker LS. Treg and CTLA‐4: two intertwining pathways to immune tolerance. J Autoimmun 2013; 45:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Repopulated regulatory T cells show similar suppressive capacity.
Figure S2. Dynamics of regulatory T cells in the glucose‐6‐phosphate isomerase peptide‐induced arthritis model.
Figure S3. Regulatory T cells are dispensable in the chronic phase of glucose‐6‐phosphate isomerase peptide‐induced arthritis.