

Abstract
Synovial sarcoma typically presents as periarticular soft tissue mass in adolescent and young adult patients. Very rarely, soft tissue sarcomas may arise primarily within bone posing a significant diagnostic challenge as primary osseous malignancies such as osteosarcoma and metastatic disease are much more common. While tissue sampling with immunohistochemical and genetic testing are required for definitive diagnosis, radiologists and orthopedic oncologists should consider alternate etiologies when typical imaging features of more common bone tumors are not identified. As an example, we present a 33-year-old male referred with a pathologic hip fracture proven to represent primary synovial sarcoma of bone.
Keywords: Primary bone tumor, Synovial sarcoma
Introduction
Primary bone and soft tissue sarcomas affecting adolescent and young adult (20-39 years) patients include numerous mesenchymal neoplasms of varying clinical presentations and biological behaviors [1]. Among these, common sarcomas of bone include osteosarcoma and Ewing sarcoma, while synovial sarcoma, rhabdomyosarcoma, and liposarcoma represent common soft tissue sarcomas in this patient demographic [2]. Differential diagnostic considerations for a musculoskeletal tumor largely depend on patient age, location within the body, and location within the bone. Very rarely, sarcomas which typically arise within soft parts occur primarily within bone [3]. These cases are exceedingly rare with limited reports in the literature often contributing to diagnostic uncertainty. In these complex and unusual cases, definite diagnosis requires biopsy including identification of known immunohistochemical markers or genetic modifications. In this background, we report a 33-year-old male presenting with a pathological hip fracture proven to represent primary synovial sarcoma of bone.
Case report
A 33-year-old male was referred to our tertiary cancer center for evaluation and treatment of a proximal femoral lesion. Past medical history was pertinent only for remote trauma resulting in right femur fracture requiring retrograde intramedullary nail fixation. Two years prior to the current presentation, he was seen by a community orthopedic surgeon for hip pain. Hip arthroscopy at that time identified a labral tear which was repaired (details of the visit and procedure are otherwise unavailable). The patient stated that hip pain worsened over the following years at which point his surgeon recommended magnetic resonance imaging (MRI) of the hip.
Outside unenhanced MRI demonstrated a heterogeneous femoral neck mass with medial cortical destruction and extra-osseous extension of disease concerning for an aggressive bone tumor (Fig. 1). Radiographs demonstrated an ill-defined osteolytic lesion of the femoral neck, intertrochanteric region, and lesser trochanter with medial cortical disruption and a radio-dense extra-osseous mass without appreciable mineralization to suggest osteoid or chondroid matrix (Fig. 2). Given concern for malignancy, staging studies including chest computed tomography (CT) were performed demonstrating evidence of pulmonary metastatic disease (Fig. 3). Two days later, he sustained a pathologic fracture while rolling in bed and was subsequently transferred to our institution for further diagnosis including biopsy and treatment planning.
Fig. 1.

Coronal T1-weighted SE and fat-suppressed T2-weighted TSE MR images demonstrate a T1-hypointense, T2-hyperintense heterogeneous proximal femoral mass with medial cortical destruction, and extra-osseous extension of disease consistent with an aggressive bone tumor.
MR, magnetic resonance; SE, spin echo; TSE, turbo spine echo.
Fig. 2.
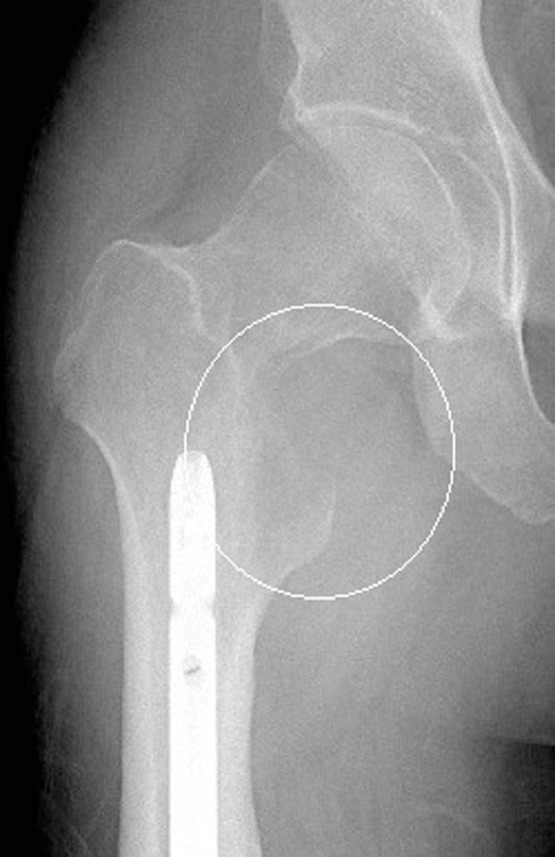
AP radiograph of the right hip demonstrates a geographic, but ill-defined osteolytic lesion without mineralization involving the medial femoral neck and lesser trochanter with a radio-dense soft tissue mass.
Fig. 3.

Unenhanced axial chest CT demonstrates multiple bilateral noncalcified pulmonary nodules consistent with metastatic disease.
CT, computed tomography.
Upon arrival, he was admitted to the hospital for pain control, CT-guided core needle biopsy of the proximal femur, and percutaneous fixation of femoral neck fracture. Histopathology including hematoxylin and eosin staining demonstrated a malignant spindle cell neoplasm with hyperchromatic nuclei and a high nuclear:cytoplasmic ratio, but rare mitotic figures and absence of necrosis (Fig. 4). Immunohistochemical staining was positive for CKAE1/3CAM, CK7, CK8/18, CD99, and Bcl-2 and weakly positive for EMA. Tumor morphology and IHC suggested synovial sarcoma and therefore the specimen was sent for additional molecular testing which confirmed the presence of SS18/SSX1 fusion transcript as detected by RT-DNA amplification consistent with synovial sarcoma. He completed 4 cycles of neoadjuvant chemotherapy (doxorubicin and olaratumab) followed by uncomplicated proximal femoral replacement with endoprosthesis reconstruction. Unfortunately, during perioperative chemotherapy holiday, re-staging chest CT demonstrated progression of pulmonary metastases warranting a change in adjuvant chemotherapy to high-dose ifosfamide × 6 cycles.
Fig. 4.

Histopathology and immunohistochemical staining of synovial sarcoma.
(A) Spindled cells of similar appearance with hyperchromatic nuclei and sparse cytoplasm exhibiting a “blue” appearance with scant stromal collagen (20x H&E).
(B) Strong positive staining for CKAE1/3CAM.
(C) Weakly positive staining for CK7.
(D) Weakly positive staining for CD99.
(E) Positive staining for Bcl-2.
H&E, hematoxylin and eosin. (Color version of figure is available online.)
Discussion
Synovial sarcoma most commonly presents as a periarticular soft tissue mass in adolescent and young adult patients [4]. A large population study from the Armed Forces Institute of Pathology reported a mean age of 32 years with equivalent incidence among males and females [5]. At diagnostic imaging, synovial sarcoma commonly appears as a heterogeneous hemorrhagic and enhancing soft tissue mass on MRI which may demonstrates internal calcifications (approximately one-third of cases) at diagnostic radiography or CT [4]. Not a tumor of synovium, rather a monophasic or biphasic spindle cell tumor with variable epithelial differentiation, synovial sarcoma typically arises in close proximity to a joint (within 5 cm) and most often affects the lower extremity, commonly around the hip or knee [4]. Other primary sites of disease may include the hands/feet, pleura, retroperitoneum, and head/neck. Synovial sarcomas are aggressive tumors with guarded prognosis due to increased risk of local tumor recurrence and metastatic disease to regional lymph nodes or lungs, present at diagnosis in up to 25% of patients [2].
Few sarcomas which typically arise within soft tissues are known to arise primarily within bone [3]. Most affect long bones such as the femur or tibia. Leiomyosarcoma and fibrosarcoma are most common with limited reports of synovial sarcoma of bone currently available in the literature [6], [7], [8], [9], [10], [11], [12]. In this case, the center of the mass was felt to be within the femur favoring a primary bone tumor rather than a soft tissue mass invading bone. In adolescent and young adult patients, osteosarcoma and Ewing sarcoma are most common and would comprise the primary differential considerations for an aggressive bone tumor in this patient demographic. Lack of mineralization argued against osteosarcoma, while Ewing sarcoma most often demonstrates permeative or moth-eaten osteolysis. When coupled with the presence of pulmonary metastases, the lesion was felt to be most suggestive of a bone metastasis from an unknown primary neoplasm.
While rarity contributes to difficulty in preoperative diagnosis of synovial sarcoma of bone, the imaging characteristics commonly seen in soft tissue synovial sarcoma are also infrequently present when arising in bone furthering diagnostic uncertainty. Internal hemorrhage and calcifications are less commonly present with most appearing as a nonspecific ill-defined osteolytic lesion without mineralization. The proximal femoral tumor presented herein demonstrates small foci of cystic degeneration or necrosis, while appearing purely osteolytic on radiographs without evidence of internal calcifications. Cortical disruption and extra-osseous extension of disease are typical features of synovial sarcoma of bone [3]. However, these findings are nonspecific and therefore diagnosis relies on histopathology, immunohistochemical staining, and identification of diagnostic genetic modifications.
Evaluation of the gross tumor specimen often demonstrates cystic and hemorrhagic regions scattered among viable neoplastic soft tissue. Histologically, synovial sarcomas are comprised of varying populations of spindle cells and epithelial cells which define the subtype of disease [2]. Monophasic synovial sarcoma consists of uniform sheets of spindle cells with a pericytic distribution and a hemangiopericytoma-like vascular network. Biphasic tumors demonstrate spindle cells alternating with nests of differentiated epithelial cells and a similar vascular network. Finally, poorly differentiated synovial sarcomas are most aggressive demonstrating large, spindle, or round cell morphology and occasionally multinucleated giant cells. Definitive diagnosis is confirmed by positive IHC staining for TLE1, cytokeratins, EMA, BCL, and CD99 and the presence of the chromosomal translocation t(X;18)(p11:q11) involving the SS18 gene and SSX1, SSX2, or SSX4 [13].
Conclusion
Primary osseous and soft tissue sarcomas are rare in comparison with other malignancies afflicting adolescent and young adults. An uncommon presentation of a rare disease poses an even greater challenge to accurate preoperative diagnosis. When encountering a suspicious musculoskeletal tumor, radiologists and orthopedic oncologists should be mindful of atypical etiologies while considering more common lesions in this patient population.
Footnotes
The authors declare that they have no financial disclosures or conflicts of interest.
References
- 1.American Cancer Society Website Cancers that develop in young adults. 2018. https://www.cancer.org/cancer/cancer-in-young-adults/cancers-in-young-adults.html [cited 2018 Sept 18]; available at.
- 2.Kransdorf M.J., Murphey M.D. Imaging of soft tissue tumors. 2nd ed. Lippincott Williams & Wilkins; Philadelphia, PA: 2006. Malignant soft tissue tumors; pp. 7–37. [Google Scholar]
- 3.Sbaraglia M., Righi A., Gambarotti M., Vanel D., Picci P., Dei Tos A.P. Soft tissue tumors rarely presenting primary in bone; diagnostic pitfalls. Surg Pathol. 2017;10:705–730. doi: 10.1016/j.path.2017.04.013. [DOI] [PubMed] [Google Scholar]
- 4.Murphey M.D., Gibson M.S., Jennings B.T., Crespo-Rodríguez A.M., Fanburg-Smith J., Gajewski D.A. Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26:1543–1565. doi: 10.1148/rg.265065084. [DOI] [PubMed] [Google Scholar]
- 5.Kransdorf M.J. Malignant soft-tissue tumors in a large referral population: distribution of diagnoses by age, sex, and location. Am J Roentgenol. 1995;164:129–134. doi: 10.2214/ajr.164.1.7998525. [DOI] [PubMed] [Google Scholar]
- 6.Zulkarnaen M., Pan K.L., Shanmugam P.S., Ibrahim Z.A., Chan W.H. Intraosseous synovial sarcoma of the proximal femur: case report. Malays Orthop J. 2012;6(1):49–52. doi: 10.5704/MOJ.1203.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakajo M., Ohkubo K., Nandate T., Shirahama H., Yanagi M., Anraku M. Primary synovial sarcoma of the sternum: computed tomography and magnetic resonance imaging findings. Radiat Med. 2005;23:208–212. [PubMed] [Google Scholar]
- 8.Cohen I.J., Issakov J., Avigad S., Stark B., Meller I., Zaizov R. Synovial sarcoma of bone delineated by spectral karyotyping. Lancet. 1997;350:1679–1680. doi: 10.1016/S0140-6736(05)64278-X. [DOI] [PubMed] [Google Scholar]
- 9.Beck S.E., Nielsen G.P., Raskin K.A., Schwab J.H. Intraosseous synovial sarcoma of the proximal tibia. Int J Surg Oncol. 2011;2011 doi: 10.1155/2011/184891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hiraga H., Nojima T., Isu K., Yamashiro K., Yamawaki S., Nagashima K. Histological and molecular evidence of synovial sarcoma of bone. A case report. J Bone Joint Surg Am. 1999;81(4):558–563. doi: 10.2106/00004623-199904000-00014. [DOI] [PubMed] [Google Scholar]
- 11.Jung S.C., Choi J.A., Chung J.H., Oh J.H., Lee J.W., Kang H.S. Synovial sarcoma of primary bone origin: a rare case in a rate site with atypical features. Skelet Radiol. 2007;36(1):67–71. doi: 10.1007/s00256-006-0185-2. [DOI] [PubMed] [Google Scholar]
- 12.Wu X., Popovic M., Chow E., Zhang X. Primary synovial sarcoma of the distal femur: a rare case report. J Pain Manage. 2014;1(7):95–98. [Google Scholar]
- 13.Obeidin F., Alexiev B. Soft tissue, other tumors, synovial sarcoma. 2018. http://www.pathologyoutlines.com/topic/softtissuesynovialsarc.html [cited 2018 Oct 1]; available at.