

Abstract
Phospholipase C (PLC) enzymes produce second messengers that increase the intracellular Ca2+ concentration and activate protein kinase C (PKC). These enzymes also share a highly conserved arrangement of core domains. However, the contributions of the individual domains to regulation are poorly understood, particularly in isoforms lacking high-resolution information, such as PLCϵ. Here, we used small-angle X-ray scattering (SAXS), EM, and functional assays to gain insights into the molecular architecture of PLCϵ, revealing that its PH domain is conformationally dynamic and essential for activity. We further demonstrate that the PH domain of PLCβ exhibits similar dynamics in solution that are substantially different from its conformation observed in multiple previously reported crystal structures. We propose that this conformational heterogeneity contributes to subfamily-specific differences in activity and regulation by extracellular signals.
Keywords: Phospholipase C, signal transduction, conformational change, calcium, structural biology, phosphatidylinositol lipid, signaling protein, second messenger, PKC
Introduction
Phospholipase C (PLC)2 enzymes hydrolyze phosphatidylinositol lipids at cell membranes, producing inositol phosphates and diacylglycerol, which in turn promote the release of Ca2+ from intracellular stores and activate protein kinase C (PKC) (1). Of the six PLC subfamilies, PLCβ and PLCϵ are required for normal cardiovascular function, and dysregulation of their expression and/or activity can result in cardiac hypertrophy and heart failure (2–6). The activity of these lipases is autoinhibited by various domains and structural elements but can be stimulated up to ∼60-fold following activation of cell surface receptors (1). PLCβ is activated downstream of G protein–coupled receptors (GPCRs) primarily through direct interactions with the heterotrimeric G protein subunits Gαq and Gβγ (1, 7). PLCϵ is also activated downstream of GPCRs by Gβγ (8, 9), by the small GTPases RhoA and Rap1A, and by Ras GTPases following receptor tyrosine kinase activation (10).
Although the structural and molecular mechanisms by which PLCβ is regulated have been characterized at high resolution (1, 7, 11–14), structural insights into PLCϵ regulation are lacking. PLCβ and PLCϵ both contain a pleckstrin homology (PH) domain, followed by four tandem EF-hand repeats (EF1–4), a TIM barrel that houses the active site, and a C2 domain (Fig. 1A) (1, 11). Additional domains that flank this core contribute to subfamily-specific regulation. PLCβ has a ∼400-amino acid C-terminal extension involved in autoinhibition (14), Gαq-dependent activation (12), and membrane association (13, 15–18). PLCϵ contains an N-terminal CDC25 domain with guanine nucleotide exchange activity for Rap1A (19) and two C-terminal Ras association (RA) domains that interact with scaffolding proteins (4) and small GTPases (20, 21).
Figure 1.

Contributions of the PLCϵ PH and RA domains to activity, stability, and liposome binding. A, domain structure of R. norvegicus PLCϵ and Homo sapiens PLCβ3. The residue numbers above the diagrams correspond to predicted domain boundaries. PLCϵ variants used in this study are depicted in brackets. B, deletion of RA domains (PH-C2) or PH domain (EF-COOH) decreases basal specific activity 6- and 15-fold relative to PH-COOH, respectively. Data represent at least four experiments performed in duplicate ± S.D. (error bars) (****, p ≤ 0.0001 based on one-way ANOVA followed by Tukey's multiple-comparison test). C, representative thermal melt curves of PH-COOH (purple hexagons), EF-COOH (green circles), and PH-C2 (blue triangles). Deletion of the RA domains decreases thermal stability by 2.9 ± 0.6 °C, relative to PH-COOH. Data represent at least four experiments performed in duplicate ± S.D. D, representative SDS-polyacrylamide gel of PLCϵ variants following ultracentrifugation in the presence (+) or absence (−) of PE/PIP2 liposomes. Total protein samples (T) contained protein incubated in the presence (+) or absence (−) of liposomes but were not subject to centrifugation. E, band density of PLCϵ variants in the supernatant (S), pellet (P), and total protein (T) in the presence or absence of liposomes were quantified via ImageJ. The ratio of protein present in the supernatant samples versus the respective total protein control samples is shown. No significant differences in lipid binding for PLCϵ variants were observed. Data represent at least three independent experiments.
Crystal structures of PLCβ alone (22), in complex with G protein activators (12, 13, 23, 24) and/or physiologically relevant small molecules (24) show that the PH, EF-hands, TIM barrel, and C2 domains consistently assemble into a compact, globular structure. Deletion of any of these individual domains eliminates basal activity, whereas the C-terminal extension is dispensable (7, 25). Thus, interdomain contacts between the core domains appear essential for function. However, this view is being challenged by studies suggesting that the PLCβ PH domain is flexibly connected to the rest of the core and that this flexibility may be essential for its regulation (26–28). In support of this hypothesis, the related PLCδ enzyme contains a PH domain that is flexibly connected to the core and dispensable for activity but increases processivity by binding PIP2 at the membrane (29–31).
In contrast, relatively little is known about the structure and molecular regulation of PLCϵ. The pathways leading to G protein–mediated activation of PLCϵ have been characterized (5, 10, 20, 32–34), but the activator-binding sites on PLCϵ are largely unknown. It is also not known how the accessory domains contribute to regulation, in part because soluble, catalytically active variants have not been available for study. The only structural information for this enzyme comes from NMR structures of its two RA domains and a crystal structure of the RA2 domain in complex with Ras GTPase (35). Biochemical studies of PLCϵ have shown that its CDC25 and PH domains, its first two EF-hands, and its RA domains are dispensable for lipase activity (35–37). Thus, PLCϵ appears more similar to PLCδ than PLCβ with respect to the requirement for its PH domain in lipase activity.
In this work, we used biochemical assays, small-angle X-ray scattering (SAXS), and negative stain EM to obtain structural insights into the solution architecture of PLCϵ and probe the roles of its PH and RA domains in regulating basal activity. We show that the PH domain, likely together with the first two EF-hands (EF1/2), is conformationally heterogeneous with respect to the rest of the PLC core, which retains a compact structure. In addition, the PH and RA domains have opposing effects on the thermal stability of the enzyme that indicate that one or both RA domains are intimately associated with the core, but the PH domain is not. To compare these results with that of a structurally well-characterized PLC, we also examined the solution structure of the PLCβ3 core domains. In contrast to the structures observed across multiple different crystal forms, we show that the PLCβ PH domain also adopts multiple conformations in solution. Using site-directed mutagenesis and disulfide bond engineering, we found that both the extended and closed conformational states of the PLCβ PH domain retain basal activity and adsorb to liposomes, suggesting that these conformational states are functionally relevant. Thus, it seems likely that all PLC enzymes containing PH domains may be inherently dynamic in solution and that these conformational states may underlie subfamily-specific differences in their autoregulation, their association with cell membranes, and their allosteric regulation by other signaling proteins.
Results
The PLCϵ PH and RA domains have differential impacts on basal activity, thermal stability, and liposome binding
Given the lack of prior knowledge about the tertiary organization of PLCϵ, we investigated the respective contributions of the PH domain and the two RA domains to basal activity, the structural integrity of the enzyme, and binding to model membranes. Because the full-length enzyme presents significant challenges in expression and purification, we expressed and purified three PLCϵ domain deletion variants: PH-COOH, which retains the core domains and both RA domains (residues 837–2282); PH-C2, which lacks both RA domains (residues 837–1972); and EF-COOH, which lacks the PH domain (residues 1035–2282) (Fig. 1A) (1, 8). Using a [3H]PIP2 liposome hydrolysis assay (24, 38, 39), we found that the PH-COOH variant has the highest basal activity (Fig. 1B; 390 ± 123 nmol of IP3/min/nmol of PLCϵ variant). Loss of the RA domains in PH-C2 decreased basal activity 6-fold (65 ± 23 nmol of IP3/min/nmol of PLCϵ variant, p ≤ 0.0001), whereas loss of the PH domain in EF-COOH decreases activity 15-fold (25 ± 12 nmol of IP3/min/nmol of PLCϵ variant, p ≤ 0.0001) (Fig. 1B).
To test whether the loss in specific activity is due to protein destabilization, the melting temperature (Tm) of each variant was determined using differential scanning fluorimetry (DSF) (40). The PH-COOH and EF-COOH variants have similar Tm values of 51.3 ± 0.7 and 50.6 ± 1.6 °C, respectively (Fig. 1C), but PH-C2 has a lower Tm (48.3 ± 1.0 °C, p ≤ 0.005). This suggests that one or both RA domains directly contribute to the thermal stability of the enzyme. The resulting destabilization of the PLC core may be sufficient to account for the decreased basal activity of this variant. We next tested whether differences in basal activity were due to changes in the ability of the variants to bind PIP2-containing liposomes using a pelleting assay (41). We hypothesized that PH-COOH and PH-C2 would show the greatest binding to liposomes, as they retain the PH domain. However, in the presence of liposomes, all three variants were present to similar extents in the pellet, suggesting that differences in activity are not due to differences in binding (Fig. 1, D and E).
PLCϵ variants have molecular envelopes distinct from those predicted from PLCβ crystal structures
The PLCϵ PH domain increases basal activity, but has no effect on thermal stability. We therefore hypothesized that the PH domain may be flexibly attached to the rest of the core. If so, then its molecular envelope should be characteristically different from that predicted by the crystal structures of PLCβ (46% sequence identity for the region spanning EF3 through the C2 domain). We used small-angle X-ray scattering (SAXS) to determine low-resolution ab initio bead models for the PLCϵ PH-COOH, EF-COOH, and PH-C2 variants (Fig. 2, Tables 1 and 2, and Figs. S1–S4) (42). Size-exclusion chromatography (SEC)-SAXS was used to ensure sample homogeneity (Figs. S1 and S2). The minimal variability of radius of gyration (Rg) as determined by Guinier approximation using the SAXS data (Fig. S3) or molecular weights as determined by multi-angle light scattering (MALS) (Fig. S4) across the elution peaks indicates that the samples were monodisperse. Guinier plots for each variant were also consistent with the absence of aggregates. PH-COOH and PH-C2 have Rg values of 43 ± 0.54 and 40 ± 0.48 Å, respectively, whereas EF-COOH had an Rg of 46 ± 2.5 Å (Fig. 2 (D–F) and Table 2). All three PLCϵ variants also had pair distance distribution functions consistent with a predominantly compact shape, but with some extended conformational aspects. The maximal intramolecular dimension (Dmax) for each variant was determined, with values of ∼162, ∼148, and ∼161 Å, for PH-COOH, PH-C2, and EF-COOH, respectively (Fig. 2, G–I). The SAXS data were also used to estimate the molecular masses of the variants using the volume of correlation (Vc) and the Porod volume (Vp) (43). PH-COOH had molecular masses of 118 kDa (Vc) and 126 kDa (Vp), lower than the calculated molecular mass of 165.5 kDa. As an additional control, MALS was used to independently validate the molecular mass of this variant at 168 kDa (Fig. S4). PH-C2 had estimated molecular masses of 123.6 kDa (Vc) and 137 kDa (Vp), comparable with its calculated molecular mass of 130.8 kDa. Finally, EF-COOH had estimated molecular masses of 143.9 kDa (Vc) and 138.3 kDa (Vp), again comparable with the calculated molecular mass of 143.4 kDa (44).
Figure 2.

SAXS envelopes reveal the solution architectures of PLCϵ variants. A–C, raw scattering curves for PLCϵ PH-COOH (A), PH-C2 (B), and EF-COOH (C). D–F, Guinier analyses of low q values, ln(I) (beam intensity) versus q2 (scattering angle) with Rg of PH-COOH, PH-C2, and EF-COOH, respectively. Fitting of the linear regressions to the data is represented by residuals, shown at the bottom of the plots, demonstrating that the proteins are monomeric in solution. G-I, pair distance distribution functions (P(r)) indicating elongated envelopes for PH-COOH, PH-C2, and EF-COOH variants, respectively. Estimated maximum intramolecular distances (Dmax) are provided. Ab initio envelope models (left) and equivalent envelopes rendered as volumes (right) show protrusions in PH-COOH (J) and PH-C2 (K) and lack of density, likely corresponding to the missing RA domains in PH-C2. As a reference, the crystal structure of the PLCβ3 core (colored as in Fig. 1A; PDB entry 3OHM (12)) is fit within the SAXS-derived envelopes such that the PH domain is oriented toward the extended protrusion and the C2 domain toward the additional density on the opposite side of the envelope. In L (EF-COOH), the domain is extended similarly to PH-COOH, but electron density is lost from the protrusion and the center of the molecule due to loss of the PH domain. The PLCβ3 core lacking the PH domain is fit within the density as described for J and K.
Table 1.
SAXS data collection and analysis parameters
| SAS data collection parameters | |
| Instrument | BioCAT facility at the Advanced Photon Source beamline 18ID with Pilatus3 1M (Dectris) detector |
| Wavelength (Å) | 1.033 |
| Beam size (μm2) | 160 (h) × 75 (v) |
| Camera length (m) | 3.5 |
| q measurement range (Å−1) | 0.00365–0.383 |
| Absolute scaling method | NAa |
| Basis for normalization to constant counts | To incident intensity, by ion chamber counter |
| Method for monitoring radiation damage | Automated frame-by-frame comparison of relevant regions |
| Exposure time, number of exposures | 1-s exposure time with a 2-s total exposure period of entire SEC elution |
| Sample configuration | SEC-SAXS. Size separation by an ÄKTA Pure with a Superdex 200 Increase 10/300 GL column. SAXS data measured in a 1.5-mm inner diameter quartz capillary. |
| Sample temperature (°C) | 20 |
| Software employed for SAS data reduction, analysis, and interpretation | |
| SAXS data reduction | Radial averaging; frame comparison, averaging, and subtraction done using BioXTAS RAW 1.4.0 (55) and ATSAS (56) |
| Basic analysis: Guinier, molecular weight, P(r) | Guinier fit and molecular weight using BioXTAS RAW 1.4.0 (55), P(r) function using GNOM (65) |
a Not applicable.
Table 2.
SAXS structural parameters of PLCϵ variants
| PLCϵ PH-COOH | PLCϵ PH-C2 | PLCϵ EF-COOH | |
|---|---|---|---|
| Guinier analysis | |||
| I(0) (cm−1) | 65.49 ± 0.27 | 51.1 ± 0.14 | 53.39 ± 0.48 |
| Rg (Å) | 43.37 ± 0.54 | 40.46 ± 0.48 | 46.44 ± 2.51 |
| q min (Å−1) | 0.0183 | 0.0114 | 0.0083 |
| q range (Å−1) | 0.0183–0.0296 | 0.0114–0.0320 | 0.0083–0.0278 |
| P(r) analysis | |||
| I(0) (cm−1) | 65.77 | 51.20 | 54.47 |
| Rg (Å) | 44.64 | 41.29 | 49.31 |
| Dmax (Å) | 162 | 148 | 161 |
| Porod volume (Å−3) | 213,000 | 213,000 | 277,000 |
| q range (Å−1) | 0.0183–0.184 | 0.0114–0.197 | 0.0083–0.172 |
Comparison of the ab initio SAXS models of PH-COOH and PH-C2 reveals that they are very similar in overall shape (Fig. 2, J and K). PLCϵ PH-COOH has a larger molecular volume (4.57 × 105 Å3) compared with PH-C2 (4.06 × 105 Å3), consistent with the presence of the RA domains in the PH-COOH variant (45). As a comparison, we fit the atomic structure of the PLCβ3 core within the density (including the PH-C2 domains from PDB entry 3OHM (12)), confirming that the PLCϵ envelopes are substantially larger than would be predicted for PLCβ, which is consistent with their larger size (PLCβ3 PH-C2 is ∼60 and ∼77% of the total mass of PLCϵ PH-COOH and PH-C2, respectively). Most interestingly, the SAXS envelopes of PH-COOH and PH-C2 both feature a well-defined, extended protrusion consistent with the extended tail observed in their pair distance distribution functions (Fig. 2, G, H, J, and K). This feature is consistently observed in ab initio models of these variants, as evidenced by the normalized spatial discrepancy (NSD) values of 0.968 ± 0.032 for PH-COOH and 0.924 ± 0.044 for PH-C2 for 15 independently determined models (46). As this feature is observed in both PH-COOH and PH-C2, it cannot correspond to the RA domains (Figs. 1A and 2) and thus most likely represents the PH domain. The ab initio model of the EF-COOH variant shares approximately the same long dimension as the PH-COOH variant. EF-COOH also contains a similar extended protrusion from the main body of the envelope, with an NSD of 0.627 ± 0.039 for 10 independent models (Fig. 2 (F, I, and L) and Table 2). However, electron density appears to be lost both from the protrusion and from the center of the molecule, relative to the models of PH-COOH and PH-C2 (Fig. 2, J–L). This is supported by the decreased molecular volume of the EF-COOH variant (3.48 × 105 Å3) (45). These differences are most likely a direct consequence of deleting the PH domain in EF-COOH.
Negative stain EM reconstruction of PLCϵ variants
To obtain higher resolution and orthogonal structures of the PLCϵ variants, we used negative stain EM (Fig. 3 and Fig. S5) to calculate single-particle reconstructions. Preliminary analysis of the data sets revealed challenges in obtaining dominant 3D class averages for each set of particles. Together with the SAXS data, this is consistent with conformational heterogeneity in the PLCϵ variants and necessitated a conservative approach to data processing and calculating 3D reconstructions (Fig. S5).
Figure 3.

Single-particle negative stain EM reconstructions confirm elongated conformations in PLCϵ variants. A–C, raw, unadjusted micrograph field views with examples of picked particles (circles) for PLCϵ PH-COOH (A), PH-C2 (B), and EF-COOH (C). D–F, the top five reference-free 2D class averages for PH-COOH (D), PH-C2 (E), and EF-COOH (F) variants. G, two representative, refined 3D models of PH-COOH, with the top purple model generated from 9,879 particles and bottom orchid model generated from 13,628 particles. H, representative models of PH-C2, with the top navy model generated from 24,129 particles and the bottom sky blue model generated from 22,343 particles. I, representative models of EF-COOH, with the top sea foam green model generated from 13,416 particles and the bottom lime green model generated from 14,498 particles. J–L, FSC curves for refined 3D models of PH-COOH, PH-C2, and EF-COOH, respectively. The gold-standard FSC (0.143) for all models indicates a final resolution of 25, 20, and 20 Å, respectively. FSC curves reflect the cross-correlation coefficient from aligning two independent half-maps. A representative workflow for the PLCϵ PH-COOH variant, containing A, D, and G, is shown in Fig. S5.
For the PH-COOH variant, 2D classes containing 34,155 particles were used to generate two initial 3D models, each of which was used to independently calculate two refined 25 Å 3D reconstructions (9,879 and 13,628 particles each) (Fig. 3 (A, D, G, and J) and Fig. S6). Similarly, for the PH-C2 variant, 92,982 particles from the 2D classes were used to generate two refined 20 Å 3D reconstructions (24,129 and 22,343 particles each) (Fig. 3 (B, E, H, and K) Fig. S6). These reconstructions confirm that PH-COOH and PH-C2 are monomeric. The two PH-COOH reconstructions have molecular volumes of 2.92 × 105 and 3.40 × 105 Å3, respectively, and maximum diameters of ∼116 Å. The two reconstructions of PH-C2 have molecular volumes of 3.07 × 105 and 2.78 × 105 Å3 and diameters of ∼116 and ∼124 Å (45). Overall, these reconstructions have smaller molecular volumes compared with the SAXS envelopes. This is most likely due to the dehydration and sample flattening that is common in negative stain (47). Although the PH-COOH variant retains the RA domains, there are no obvious differences compared with PH-C2, further supporting our hypothesis that one or both RA domains stably associate with the rest of the core under these experimental conditions (Figs. 1 and 3). Thus, distinctive features protruding from the main body of the reconstructions of both PH-COOH and PH-C2 likely reflect the conformational heterogeneity of the PH domain with respect to the PLCϵ core (Fig. 3 and Fig. S6).
Two refined 20 Å reconstructions of the EF-COOH variant (13,416 and 14,498 particles each) were likewise generated from 2D classes containing 53,028 particles (Fig. 3 (C, F, I, and L) and Fig. S6). Like the other PLCϵ variants, the EM analysis independently confirms that EF-COOH is monomeric. Similar to the trends observed for the PH-COOH and PH-C2 variants, the reconstructions of EF-COOH are smaller overall than its SAXS envelopes. The EF-COOH reconstructions have molecular volumes of 3.13 × 105 and 3.67 × 105 Å3 and maximum diameters of ∼126 and 130 Å (Fig. 3, G–I) (45). Importantly, the EF-COOH reconstructions further confirm that this variant has a different molecular architecture compared with the PH-COOH and PH-C2 variants, due to deletion of the PH domain. This is consistent with the SAXS analysis of these variants (Fig. 2) and supports a model in which the PH domain adopts multiple conformations with respect to the rest of the PLC core.
The PLCβ3 PH domain is also conformationally dynamic
Because our data are consistent with the PH domain adopting multiple conformations in PLCϵ, we assessed the solution structure of the PLCβ3 core by performing SAXS on a variant that includes its PH, EF, TIM barrel, and C2 domains along with an autoinhibitory portion of its C-terminal domain (CTD), PLCβ3-Δ892 (Figs. 1A and 4).
Figure 4.

Characterization of conformationally stabilized PLCβ3-Δ892 variants. A, representative crystal structure of the PLCβ3 core (PH-C2 domains) with the proximal CTD (PDB entry 3OHM (12)), with the domains labeled and colored as in Fig. 1A. The predicted SAXS envelope is shown as a gray 20-Å low-pass filter surrounding the crystal structure. Residues Glu-60 and Val-164 (shown in ball-and-stick representations) were mutated to cysteines for disulfide bond formation to restrict the motion of the PH domain. The X-Y linker and the loop connecting the C2 domain to the proximal CTD are disordered and omitted for clarity. The ends of the X-Y linker are denoted by hot pink asterisks, and the ends of the C2-proximal CTD are denoted by cyan asterisks (26). B, cysteine mutations in PLCβ3-Δ892 (E60C/V164C) alter basal specific activity only under oxidized conditions (***, p ≤ 0.001; **, p ≤ 0.01 based on one-way ANOVA followed by Tukey's multiple-comparison test). Basal specific activity data represents at least two experiments performed in duplicate ± S.D. (error bars). C, cysteine mutations in PLCβ3-Δ892 (E60C/V164C) do not alter thermal stability under reducing or oxidizing conditions. Representative curves are shown for PLCβ3-Δ892 (gray diamonds), reduced E60C/V164C (red inverted triangles), and oxidized E60C/V164C (orange squares). Thermal stability data represent at least six experiments performed in triplicate ± S.D. D, representative SDS-PAGE of PLCβ3-Δ892 variants following ultracentrifugation in the presence (+) or absence (−) of PE/PIP2 liposomes. Total protein samples (T) contained protein incubated in the presence (+) or absence (−) of liposomes but were not subject to centrifugation. E, PLCβ3-Δ892 variants show minimal binding to liposomes. Gels were quantified as described in Fig. 1E, and no significant differences in liposome binding were observed for the PLCβ3-Δ892 variants. All data are mean ± S.D. of at least four independent experiments and analyzed by one-way ANOVA followed by Tukey's multiple-comparison test.
We first compared the experimentally determined SAXS data for PLCβ3-Δ892 with the calculated SAXS data derived from the PLCβ3 crystal structure (containing the PH-proximal CTD domains from PDB entry 3OHM (12); Figs. 4 and 5, Tables 1 and 3, and Figs. S1–S3). The experimental and calculated Guinier plots for PLCβ3-Δ892 generated similar Rg values of 34 and 31 Å, with experimentally determined molecular masses of 87.9 kDa (Vc) and 96.2 kDa (Vp), comparable with the calculated molecular mass of 100.89 kDa. However, the Guinier plots for the two data sets have a χ2 value of 7.8, indicating substantial divergence between the experimental and calculated data (Fig. 5B). The calculated pair distance distribution function for PLCβ3-Δ892 (Fig. 5C) is bell-shaped, as expected for the globular structure observed in crystal structures (Fig. 4A). However, the experimentally determined pair distance distribution function indicates the presence of extended features, as evidenced by the tail at high values of r (Fig. 5C). In addition, the calculated Dmax is 105 Å, compared with the experimentally determined Dmax of 122 Å. This is consistent with PLCβ3-Δ892 having a more extended structure in solution that is inconsistent with published crystal structures.
Figure 5.

PLCβ3-Δ892 SAXS analysis confirms conformational dynamics of its PH domain. A and B, raw scattering curve (A) and Guinier analyses (B) of low q values, ln(I) (beam intensity) versus q2 (scattering angle) with Rg for PLCβ3-Δ892 (gray line) and calculated from the PLCβ3 crystal structure (dashed black line, PDB code 3OHM (12)). Fitting of the experimental linear regression to the experimental data is represented by residuals, as shown in the bottom tenth of the plot, and confirms that the proteins are monomeric in solution. C, pair distance distribution function P(r), with maximum interparticle distance (Dmax) for the experimentally determined and calculated PLCβ3-Δ892 data shown as in B. D, ab initio “dummy atom” envelope models (left) and equivalent envelope rendered as volumes (right) for PLCβ3-Δ892. The PLCβ3 crystal structure is fitted as a reference, with the PH domain oriented toward the extended protrusion and colored as in Fig. 1A. E–G, raw scattering curve (E), Guinier analysis (F), and pair distance distribution function (G) of oxidized PLCβ3-Δ892 E60C/V164C. H, ab initio “dummy atom” envelope models (left) and equivalent envelope rendered as volumes (right) of oxidized PLCβ3-Δ892 E60C/V164C, with the crystal structure fit as in D. I–K, raw scattering curve (I), Guinier analysis (J), and pair distance distribution function (K) of reduced PLCβ3-Δ892 E60C/V164C. L, ab initio “dummy atom” envelope models (left) and equivalent envelope rendered as volume (right) for reduced PLCβ3-Δ892 E60C/V164C, with the crystal structure fit as in D. PLCβ3-Δ892 and reduced PLCβ3-Δ892 E60C/V164C envelopes show similar protrusions, whereas oxidized PLCβ3-Δ892 E60C/V164C appears more compact.
Table 3.
SAXS structural parameters of PLCβ3-Δ892 variants
| PLCβ3-Δ892 | PLCβ3-Δ892 E60C/V164C (oxidized) | PLCβ3-Δ892 E60C/V164C (reduced) | |
|---|---|---|---|
| Guinier analysis | |||
| I(0) (cm−1) | 59.44 ± 0.07 | 11.47 ± 0.34 | 25.33 ± 0.33 |
| Rg (Å) | 33.78 ± 0.37 | 35.13 ± 2.54 | 34.79 ± 0.93 |
| q min (Å−1) | 0.0089 | 0.0159 | 0.0112 |
| q range (Å−1) | 0.0089–0.0384 | 0.0159–0.0363 | 0.0112–0.0372 |
| P(r) analysis | |||
| I(0) (cm−1) | 59.76 | 11.36 | 25.28 |
| Rg (Å) | 34.43 | 35.81 | 35.12 |
| Dmax (Å) | 122 | 120 | 120 |
| Porod volume (Å−3) | 165,000 | 166,000 | 164,000 |
| q range (Å−1) | 0.0089–0.237 | 0.0159–0.227 | 0.0112–0.230 |
The volume of the ab initio envelope of PLCβ3-Δ892 encapsulates the crystal structure of PLCβ3 but is more elongated and features a well-defined, extended protrusion (Fig. 5D). Thus, the overall shape of the PLCβ3-Δ892 SAXS envelope is more consistent with the SAXS envelopes and 3D reconstructions of PLCϵ PH-COOH and PH-C2 than with the low-pass–filtered 20-Å envelope of the PLCβ 3OHM crystal structure (Figs. 2, 3, and 4A). The compact portion of the PLCβ3-Δ892 SAXS envelope also shows additional density beyond the low-pass–filtered crystal structure (Fig. 5D). This most likely corresponds to disordered regions that are not observed in the crystal structure, such as the X-Y linker within the TIM barrel domain. In PLCβ3, this linker is ∼80 residues (∼9 kDa) and is disordered in all published structures (Fig. 4A).
To confirm that the extended protrusion observed in the experimentally determined PLCβ3-Δ892 envelope corresponds to the PH domain, we generated the E60C/V164C double mutant of PLCβ3-Δ892 (Fig. 4A). These two mutations, in combination with an exogenous bismaleimidoethane cross-linking reagent, were previously shown to restrict the motion of the PH domain without impacting basal activity (26). Because the distance between the Cβ atoms of these residues varies between crystal structures, we anticipated that they would be able to form a redox-dependent disulfide bond. Thus, we purified PLCβ3-Δ892 E60C/V164C under either oxidizing or reducing conditions. Using the [3H]PIP2 hydrolysis activity assay, we found that reduced PLCβ3-Δ892 E60C/V164C had a specific activity of 0.33 ± 0.20 nmol of IP3/min/nmol of PLCβ3-Δ892 variant, comparable with that of PLCβ3-Δ892 (0.41 ± 0.20 nmol of IP3/min/nmol of PLCβ3-Δ892 variant) (Fig. 4B). Oxidized PLCβ3-Δ892 E60C/V164C had a specific activity of 2.3 ± 0.37 nmol of IP3/min/nmol of PLCβ3-Δ892 variant, significantly greater than that of PLCβ3-Δ892 (p ≤ 0.001) and reduced PLCβ3-Δ892 E60C/V164C (p ≤ 0.01, Fig. 4B). However, all three PLCβ3-Δ892 variants had essentially identical Tm values as measured by DSF (Fig. 4C), and all variants bound similarly to PE/PIP2 liposomes, as measured using the liposome pelleting assay (Fig. 4, D and E). The increased basal activity of oxidized PLCβ3-Δ892 E60C/V164C is in contrast to a recent report from Kadamur and Ross (26), who found no significant difference in basal activity when the PH domain was conformationally restricted by cross-linking. This difference is most likely due to the use of PLCβ3-Δ892 in this study as opposed to full-length PLCβ3. Full-length PLCβ3 retains the C-terminal extension and thus has increased membrane association and much higher basal activity (1, 7).
The oxidized and reduced PLCβ3-Δ892 E60C/V164C mutants were also analyzed by SAXS and were confirmed to be monomeric and monodisperse in solution (Fig. 5, Table 3, and Figs. S1–S4). Guinier plots of these variants generated Rg values of ∼35 Å (Fig. 5, F and J). The experimentally determined molecular mass of oxidized PLCβ3-Δ892 E60C/V164C was found to be 71 kDa (Vc) and 65.3 kDa (Vp), and reduced PLCβ3-Δ892 E60C/V164C had molecular masses of 69 kDa (Vc) and 53.3 kDa (Vp), all of which are lower than the calculated molecular mass (100.6 kDa). The discrepancy in the SAXS-derived molecular masses versus the calculated molecular masses is likely due to the lower signal/noise ratio of these data sets. Thus, MALS was used to confirm the molecular masses of these variants in solution (Fig. S4). Finally, the pair distance distribution functions of these variants are similar, with Dmax values of ∼120 Å (Fig. 5, G and K). The ab initio envelopes of oxidized and reduced PLCβ3-Δ892 E60C/V164C were then compared with one another and PLCβ3-Δ892 (Fig. 5, D, H, and L). The oxidized PLCβ3-Δ892 E60C/V164C envelope has a more compact, globular structure as compared with the envelope of PLCβ3-Δ892 (Fig. 5, D and H). The molecular volume of the oxidized PLCβ3-Δ892 E60C/V164C envelope is 3.42 × 105 Å3, comparable with that of PLCβ3-Δ892 (3.44 × 105 Å3) (45). A similar result was obtained upon the addition of 15 mm excess bismaleimidoethane to PLCβ3-Δ892 E60C/V164C purified under reducing conditions. The resulting envelope again has a more compact shape than that of PLCβ3-Δ892, with a molecular volume of 3.43 × 105 Å3 (Fig. S7 and Table S1). Thus, the disulfide bond between E60C and V164C limits the motion of PH domain similarly to the previously reported cross-linker (26). In contrast, the envelope for reduced PLCβ3-Δ892 E60C/V164C is most similar to that of PLCβ3-Δ892, as it clearly features the extended protrusion (Fig. 5, D and L). It also shares a similar molecular volume as the other PLCβ3-Δ892 variants (3.49 × 105 Å3). Overall, these results are most consistent with the PLCβ3 PH domain, likely together with EF1/2, adopting multiple conformations in solution that contribute to the final volume of the reconstruction.
Discussion
PLCϵ and PLCβ regulate the intracellular Ca2+ concentration and PKC activation downstream of GPCRs and, in the case of PLCϵ, downstream of receptor tyrosine kinases (1, 10, 11). Numerous structures of PLCβ have been determined over the last 10 years (12–14, 18, 23, 24, 48), but the observed compactness of PLCβ in these structures appears to be misleading, given the growing evidence that its PH domain is conformationally dynamic (26–28), including in this study. Although PLCϵ is anticipated to share a similar core architecture with PLCβ (46% sequence identity for the region including EF3-C2), it was not known whether its PH domain would be situated more similarly to crystal structures of PLCβ or PLCδ with respect to the core or how its C-terminal RA domains would influence the overall conformation of the protein.
Using biochemical assays, SAXS, and single-particle EM, we show that the PLCϵ variants PH-COOH, PH-C2, and EF-COOH retain activity and have molecular architectures consisting of a compact core that features a well-defined protrusion (Figs. 1–3). As both PH-COOH and PH-C2 share this feature, this protrusion is highly unlikely to correspond to the RA domains and instead is most likely a core domain (Figs. 1–3) (9, 36). The loss of density on the opposite end of the SAXS envelope in PH-C2 relative to PH-COOH is consistent with deletion of the RA domains. However, fitting the structure of the PLCβ3 core (PDB entry 3OHM (12)) in the PH-COOH and PH-C2 envelopes such that the C terminus of the C2 domain extends into the additional density also fails to account for the volume of the observed envelopes. Whereas PLCϵ PH-C2 is ∼35 kDa larger than PLCβ3 PH-C2, this is largely due to insertions and a longer X-Y linker and is still insufficient to account for the total volume of the envelope, especially the extended protrusion. Thus, the large envelopes are most consistent with dynamic behavior at the N terminus of the PLCϵ variants. This observation is consistent with the fact that deletion of the PH domain has no effect on thermal stability (Fig. 1C), indicating that interactions between this domain and the rest of the core are transient in nature. We propose that the PH domain in both PLCβ3 and PLCϵ is connected to the rest of the core via the weakly conserved EF1/2 hands and that a hinge between EF2 and EF3 allows the PH domain to adopt multiple conformations with respect to the core. In support of this hypothesis, we used ensemble rigid-body modeling to generate a model based on PLCβ3-Δ892 SAXS data (Figs. 5 and 6). In the resulting computational model, which fits the experimental data with a χ2 of 1.67, the PH domain and EF1/2 are rotated away from the rest of the PLC core and from one another. The PH domain is rotated ∼140° from its position in the crystal structures and translated by ∼70 Å. EF1/2 is also rotated from its position in the crystal structure by ∼155° and translated by ∼20 Å (Fig. 6). This flexibility would therefore allow the PH domain and EF1/2 to adopt multiple conformational changes with respect to one another and to the rest of the PLC core. Although the current model is limited by the resolution of the SAXS envelopes and the EM reconstructions, it represents the simplest model of the data.
Figure 6.
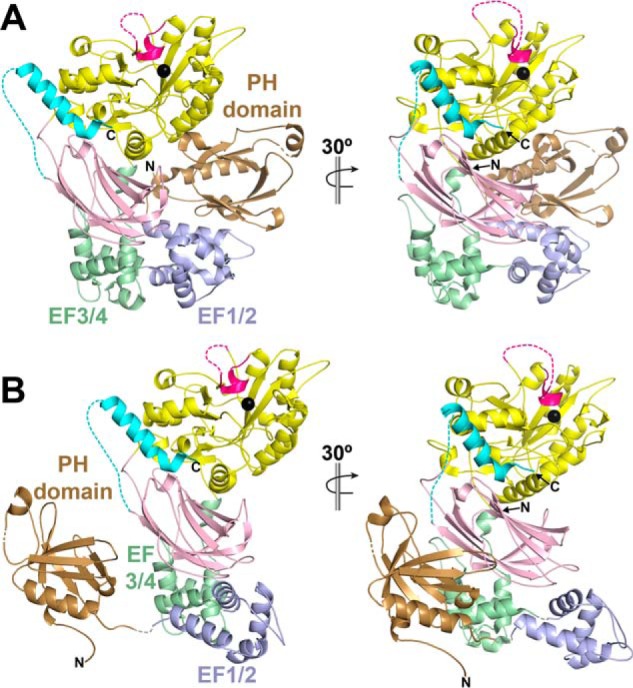
A rigid-body ensemble model of PLCβ3-Δ892 is consistent with conformational heterogeneity of the PH domain and EF1/2. A, crystal structures of the PLCβ subfamily reveal a compact structure, wherein the PH domain is in close proximity to the TIM barrel domain and EF-hands 1 and 2 (EF1/2). Domains are colored as in Fig. 1A, with the exception of EF1/2, which are shown in periwinkle blue. Dashed lines, regions disordered in the crystal structure, with the catalytic Ca2+ ion shown as a black sphere. B, a rigid-body ensemble model that better fits the experimentally determined SAXS data was determined using SASREF (χ2 of 1.67 (61)). In this model, the PH domain (residues 11–146), EF1/2 (residues 147–221), and the remaining domains of PLCβ3-Δ892 (residues 222–892) were treated as independent, rigid bodies. Additional restraints included restricting the PH domain C terminus to within 5 Å of the N terminus of EF1/2 and the C terminus of EF1/2 within 5 Å of the N terminus of EF3 and the rest of the enzyme. In the resulting model, the PH domain is translated by ∼140° and translated ∼70 Å from its position in the crystal structure. This large motion is facilitated by EF1/2, which is rotated by ∼155° and translated ∼20 Å from its position in the crystal structure. EF1/2 hands are weakly conserved compared with the rest of core domains across PLC enzymes and variably ordered in crystal structures of PLCβ. Finally, the residues linking EF2 and EF3 may serve as a hinge region, facilitating the rotation of EF1/2 and allowing the PH domain to sample multiple conformations.
We also showed that the PLCϵ PH domain is important for lipase activity (Fig. 1B). The C-terminal RA domains of PLCϵ also make a substantial contribution to basal activity (Fig. 1B), but because these domains contribute to stability as measured by DSF (Fig. 1C), the loss of activity is most simply explained by decreased structural integrity of the core. As the RA2 domain is the primary binding site for activators such as Rap1A and Ras (20, 21), this may provide a potential mechanism by which small GTPases could allosterically activate phosphatidylinositol hydrolysis. In future studies, the role of the N-terminal CDC25 domain will need to be similarly assessed.
Although the PLCβ3 core is compact in crystal structures (Fig. 4) (12, 24), its SAXS envelope features a similar extended protrusion observed in the PLCϵ PH-COOH and PH-C2 envelopes (Figs. 2, 3, and 5). To determine whether this extended protrusion is in fact the PH domain, we used SAXS to study the PLCβ3-Δ892 E60C/V164C variant under reducing and oxidizing conditions, which would restrain the motion of the PH domain (Fig. 5) (26). The molecular envelope of the reduced PLCβ3-Δ892 E60C/V164C mutant is most similar to that of PLCβ3-Δ892, whereas oxidation or cross-linking results in a more compact molecular envelope lacking a defined extended protrusion (Fig. 5 and Fig. S7). The more compact structures observed following oxidation or cross-linking are more consistent with the conformation of PLCβ3 in crystal structures. Thus, we believe that these studies are the first to provide direct structural evidence for multiple conformations of the PLCβ PH domain and possibly EF1/2. However, the respective contributions of these conformational states to basal activity and membrane association require further study (26).
Extension of the PH domain from the catalytic core may be important for PLCβ and PLCϵ regulation by Gβγ subunits (8, 9, 49, 50). Despite multiple studies over several decades, the Gβγ-binding site on PLCβ has remained elusive (27, 28, 51, 52). Previous studies have suggested that Gβγ-dependent regulation of PLCβ is modulated by interactions between these two proteins and the membrane (28, 53). It is therefore possible that the PH domain is extended when PLCβ is in contact with membranes. Interdomain contacts between the PH domain and the other core domains, including their respective orientation at the membrane, may also contribute to regulation (28). More recently, the Gβγ-binding site in PLCβ was proposed to be blocked in the closed conformation of the PH domain, suggesting that conformational flexibility of the domain is required for Gβγ binding and activation (26). Our data are fully consistent with this model. The mechanism of Gβγ-dependent activation of PLCϵ is only beginning to be investigated (9), but because our data are consistent with PLCϵ also containing a conformationally dynamic PH domain, we propose that it may likewise contribute to Gβγ-dependent regulation.
Experimental procedures
PLCϵ and PLCβ expression and purification
cDNA encoding Rattus norvegicus PLCϵ PH-COOH (residues 837–2282), PH-C2 (residues 832–1972), and EF-COOH (residues 1035–2282) were subcloned into the pFastBac-HTA vector (Invitrogen) and confirmed by sequencing over the entire coding region. Proteins were expressed in baculovirus-infected Sf9 cells, harvested 48–72 h postinfection, and flash-frozen. PLCβ3-Δ892 (residues 10–892) was expressed as described previously (14). PLCβ3-Δ892 E60C/V164C was generated using site-directed mutagenesis, confirmed by sequencing over the entire coding region, and expressed as described (14).
PLCϵ variants were purified as described (9), with some modifications for proteins used in single-particle EM and liposome-pelleting assays. After lysis and ultracentrifugation, the supernatant was applied to a 5-ml HisTrap FF column (GE Healthcare) equilibrated with Buffer A (20 mm HEPES, pH 8, 300 or 500 mm NaCl, 20 mm imidazole, 10 mm β-mercaptoethanol, 0.1 mm EDTA, and 0.1 mm EGTA) and eluted with a gradient of 0–100% Buffer A supplemented with 500 mm imidazole pH 8. Fractions containing protein were pooled, buffer-exchanged with an equal volume of Buffer E (20 mm HEPES, pH 8, 50 mm NaCl, 2 mm DTT, 0.1 mm EDTA, and 0.1 mm EGTA), and loaded onto a 1-ml MonoQ 5/50 GL column (GE Healthcare) pre-equilibrated with Buffer E. Protein was eluted with a gradient of 0–100% MonoQ-S Buffer E containing 500 mm NaCl. Fractions containing protein were again pooled, buffer-exchanged, and concentrated as described above before loading on a 1-ml MonoS 5/50 GL column (GE Healthcare) pre-equilibrated with Buffer E. The protein was eluted, pooled, and concentrated as described above before final purification over tandem Superdex 200 Increase columns (GE Healthcare).
PLCβ3-Δ892 and reduced PLCβ3-Δ892 E60C/V164C were purified as described previously (54). Oxidized PLCβ3-Δ892 E60C/V164C was purified similarly, but reducing agents were omitted in each step.
Differential scanning fluorimetry
DSF assays were carried out as described previously (40, 54), with minor modifications. Purified PLCϵ (0.2–0.6 mg/ml) and PLCβ variants (0.5 mg/ml) were incubated with 5× SYPRO orange dye in the presence of 5 mm CaCl2. All assays were performed in duplicate or triplicate with protein from at least two independent purifications.
Basal activity assays
PLCϵ and PLCβ activity was measured as described previously (24, 38, 54). The PLCϵ PH-COOH variant was assayed at a final concentration of 0.075 ng/μl, PH-C2 at 0.1–1 ng/μl, and EF-COOH at 0.5 ng/μl. PLCβ3-Δ892 was assayed at a final concentration of 10 ng/μl, and PLCβ3-Δ892 E60C/V164C (oxidized or reduced) was assayed at 12 ng/μl. Oxidized PLCβ3-Δ892 E60C/V164C was assayed in the absence of reducing agents. All assays were performed at least in duplicate with protein from at least two independent purifications.
SEC-SAXS/MALS-DLS data collection and analysis
SEC-SAXS was performed at BioCAT (Sector 18) Advanced Photon Source equipped with an ÄKTA Pure FPLC and a Pilatus3 1M detector (Dectris) with a beam size of 160 μm × 75 μm (Table 1). Data were collected at room temperature, with a 12-KeV X-ray (1.033-Å wavelength) and ∼3.5-m sample-to-detector distance (q range = 0.00365 to 0.383 Å−1). All purified PLCϵ and PLCβ3-Δ892 variants were diluted to a final concentration of 2–3 mg/ml in 20 mm HEPES, pH 8, 200 mm NaCl, 2 mm DTT, 0.1 mm EDTA, and 0.1 mm EGTA. Samples were centrifuged at 16,000 × g for 5 min at 4 °C before data collection. Protein samples were eluted on a Superdex 200 Increase column (GE Healthcare; Figs. S1 and S2). The eluate was passed through the UV monitor and then the SAXS flow cell, which consists of a 1.5-mm quartz capillary with 10-μm walls. Data were collected every 2 s with 1-s exposure times. For PLCϵ PH-COOH, frames corresponding to the leading edge of the elution peak as opposed to the highest point of the peak were selected for SAXS analysis. The usual practice of averaging frames corresponding to the highest point in the peak manifested symptoms of aggregation in the Guinier analysis due to possible capillary fouling (Fig. S1A). For all other PLCϵ and all PLCβ3-Δ892 variants, frames corresponding to the protein peak were used for SAXS analysis (Fig. S1, B–F). A buffer file was generated for all samples by averaging exposures flanking the elution peak and was used for background subtraction (Fig. S1).
Raw scattering data were reduced using BioXTAS RAW 1.4.0 (55) and ATSAS (56). The low variability in the distribution of Rg corresponding to the individual frames as seen plotted with the “scattering chromatograms” (integrated intensity of individual exposures plotted against frame number) demonstrates the monodisperse nature of the samples (Fig. S3). For oxidized PLCβ3-Δ892 E60C/V164C, automated Guinier approximation for individual frames was not possible due to low signal to noise, and we therefore used averaged frames 188–193 to enable reliable Guinier approximation and calculation of the Rg. We used MALS data and the molecular mass distribution across the elution peak to confirm the monodisperse nature of PLCϵ PH-COOH, oxidized PLCβ3-Δ892 E60C/V164C, and reduced PLCβ3-Δ892 E60C/V164C (Fig. S4). For the SEC-MALS-DLS (dynamic light scattering) experiments, the samples were loaded on the SEC column with an exclusion limit of 1.25 MDa (WTC-030S5, Wyatt Technologies), which was followed by a UV detector, a MALS detector, and a DLS detector (DAWN Helios II, Wyatt Technologies). This experiment enabled us to not only establish that the samples were monodisperse but also to acquire a more reliable estimate for the molecular mass of samples analyzed using the ASTRA software (Wyatt Technologies) that were ambiguous with SAXS. I(q) versus q curves generated after buffer subtraction and averaging selected exposures were used for more detailed analyses of the SAXS data. Rg, I(0), and Dmax for each protein were obtained using PRIMUS (57). 10–15 ab initio models were calculated using DAMMIF, and then aligned and averaged with DAMAVER (58). For all PLCϵ and PLCβ3-Δ892 variants, the mean NSD for all calculated models ranged from 0.560 ± 0.018 to 0.968 ± 0.032. One ab initio model was excluded from both the PLCϵ EF-COOH and oxidized PLCβ3-Δ892 E60C/V164C data sets (discard criteria NSD > mean + 2 × S.D.). Final ab initio envelope structures were generated using DAMMIN (59). Plots were generated from buffer-subtracted averaged data (raw scattering and Guinier plots) or pair distance distribution (P(r) plots) and plotted using GraphPad Prism version 7.0.
Theoretical SAXS data for the PLCβ3 core crystal structure was generated using its coordinate file (PDB entry 3OHM (12)). CRYSOL3.0 (56, 60) was used to compare the calculated SAXS data with the experimentally determined data for PLCβ3-Δ892. SASREF (61) was used to generate a computational model that fit the empirical SAXS data. For this model, the following interdomain distance restraints were placed. The C terminus of the PH domain (residues 10–146) was within 5 Å of the N terminus of the EF1/2 hands (residues 147–221), and the C terminus of the EF1/2 hands was within 5 Å of EF3-proximal CTD (residues 222–892). The theoretical SAXS scattering curve was then compared with the empirically determined data. Plots were generated from buffer-subtracted averaged data (raw scattering and Guinier plots) or averaged envelope data (P(r) plots) and plotted using GraphPad Prism version 7.0.
Negative stain EM sample preparation and imaging
For each PLCϵ variant, 3 μl of freshly purified protein (0.015–0.018 mg/ml) was applied to glow-discharged carbon-coated 400-mesh copper grids (Electron Microscopy Sciences) and stained with 0.75% uranyl formate (47). Samples were imaged with a CM200 field electron gun transmission electron microscope (Phillips) operated at 200 kV. Micrographs were collected with a US4000 4k × 4k CCD camera (Gatan), using a 1-s exposure time, defocus range of 5 μm, and at a nomal magnification of 50,000×, resulting in a sampling of 2.31 Å/pixel at the specimen level.
Single-particle 3D EM reconstructions
Data were processed with a beta release of RELION 2.1 (62). Raw micrographs were CTF-corrected, and particles were autopicked before reference-free 2D class averaging. For PLCϵ PH-COOH, 47,300 particles were extracted, and the 2D classes contained 34,155 particles. For PLCϵ PH-C2, 124,747 particles were extracted, and the 2D classes contained 91,982 particles. For PLCϵ EF-COOH, 58,549 particles were extracted, and the 2D classes contained 53,028 particles. To minimize any potential model bias, de novo initial models were generated for PH-COOH, PH-C2, and EF-COOH from their respective 2D class averages. From the respective selected 2D classes for each variant, we generated two particle subsets containing ∼100 particles per 2D class to build de novo initial models with the RELION-2.1b1 stochastic gradient descent method. One initial model per subset was chosen at random and used as a reference for 3D class averaging of particles from the best 2D classes. Four 3D classes were generated per initial model. When possible, the dominant 3D class from each of the two sets of four 3D classes was chosen for 3D auto-refinement, resulting in two final, refined structures. The representative workflow is shown in Fig. S5. Fourier shell correlation (FSC) curves corresponding to refined models were calculated using standard methods (63).
Liposome pelleting assays
200 μm hen egg white phosphatidylethanolamine and 100 μm porcine brain phosphatidylinositol 4,5-bisphosphate (Avanti Polar Lipids) were mixed and dried under N2. Lipids were resuspended in sonication buffer (50 mm HEPES, pH 7, 80 mm KCl, 3 mm EGTA, and 2 mm DTT) and sonicated in two 30-s bursts (14, 38). For oxidized PLCβ3-Δ892 E60C/V164C, DTT was omitted from the sonication buffer. Liposomes (65 μl) were combined with either 2 μg of total PLCϵ variant (0.15 nmol of PLCϵ PH-COOH, 0.19 nmol of PLCϵ PH-C2, 0.17 nmol of PLCϵ EF-COOH) or 1 μg of total PLCβ3-Δ892 variant (0.125 nmol) and sonication buffer to a final volume of 100 μl. Control binding reactions contained protein and buffer only, and total protein controls were incubated with liposomes or buffer. All samples were incubated for 1 h on ice. All samples, excluding total protein control samples, were then centrifuged at 119,000 × g for 1 h. Total protein control samples were incubated on ice for the duration of the ultracentrifugation. For centrifuged samples, the supernatant was removed, and the pellet was resuspended in 100 μl of sonication buffer. The PLCϵ pellets were also subjected to sonication to resuspend the pellet. For analysis by SDS-PAGE and ImageJ, 16 μl of the supernatant, resuspended pellet, or total protein control was denatured with 4 μl of 5× SDS loading dye, and 5 μl of the total sample was analyzed by SDS-PAGE. All gels were stained with Bio-Safe Coomassie Blue (Bio-Rad), and band density was quantified with ImageJ.
Statistical methods
All plots were generated using GraphPad Prism version 7.0. One-way ANOVA was performed with Prism 7.0 and followed by Tukey post hoc multiple comparisons as noted in the figure legends. All error bars represent S.D.
Figure creation
Single-particle EM 3D envelopes were rendered with UCSF Chimera (45). SAXS molecular envelopes were generated from dummy atom coordinate files with e2pdb2mrc.py (64) using a low-pass filter to 20 Å and rendered in Chimera. All ribbon diagrams and dummy atom models were generated using PyMOL (Schrödinger, LLC, New York). Figures were created using Adobe Illustrator and Photoshop.
Author contributions
A. M. L. designed the overall experimental approach. E. E. G.-K., M. S., M. V. C., and C. C. cloned, expressed, and purified all PLCβ and PLCϵ proteins. S. C., E. E. G.-K., M. V. C., and M. S. carried out all SAXS experiments, and E. E. G.-K., M. V. C., and M. S performed DSF and activity assays. E. E. G.-K., M. V. C., and A. B. carried out liposome pelleting assays. E. E. G.-K., F. S. V., W. J., and A. M. L. designed and carried out all electron microscopy studies. A. M. L., E. E. G.-K., F. S. V., M. S., and M. V. C. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank A. V. Smrcka (University of Michigan) for the R. norvegicus PLCϵ cDNA and technical and editorial advice. We thank J. Tesmer (Purdue University) and M. Tantama (Purdue University) for helpful discussions. This research used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. This project was supported by NIGMS, National Institutes of Health (NIH), Grant 9 P41 GM103622. Use of the Pilatus 3 1M detector was provided by NIGMS, NIH, Grant 1S10OD018090-01.
This work was supported by National Institutes of Health Grant 1R01 HL141076-01, American Heart Association Scientist Development Grant 16SDG29920017, and American Cancer Society Institutional Research Grant IRG-14-190-56 to the Purdue University Center for Cancer Research (to A. M. L). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PLC
- phospholipase C
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- IP3
- inositol 1,4,5-triphosphate
- GPCR
- G protein–coupled receptor
- RTK
- receptor tyrosine kinase(s)
- DSF
- differential scanning fluorimetry
- SAXS
- small-angle X-ray scattering
- MALS
- multiangle light scattering
- DLS
- dynamic light scattering
- PKC
- protein kinase C
- PH
- pleckstrin homology
- RA
- Ras association
- Tm
- melting temperature
- SEC
- size-exclusion chromatography
- PDB
- Protein Data Bank
- NSD
- normalized spatial discrepancy
- 2D and 3D
- two- and three-dimensional, respectively
- CTD
- C-terminal domain
- PE
- phosphatidylethanolamine
- PDB
- Protein Data Bank
- FSC
- Fourier shell correlation
- ANOVA
- analysis of variance
- CTF
- contrast transfer function.
References
- 1. Kadamur G., and Ross E. M. (2013) Mammalian phospholipase C. Annu. Rev. Physiol. 75, 127–154 10.1146/annurev-physiol-030212-183750 [DOI] [PubMed] [Google Scholar]
- 2. Filtz T. M., Grubb D. R., McLeod-Dryden T. J., Luo J., and Woodcock E. A. (2009) Gq-initiated cardiomyocyte hypertrophy is mediated by phospholipase Cβ1b. FASEB J. 23, 3564–3570 10.1096/fj.09-133983 [DOI] [PubMed] [Google Scholar]
- 3. Grubb D. R., Crook B., Ma Y., Luo J., Qian H. W., Gao X. M., Kiriazis H., Du X. J., Gregorevic P., and Woodcock E. A. (2015) The atypical 'b' splice variant of phospholipase Cβ1 promotes cardiac contractile dysfunction. J. Mol. Cell Cardiol. 84, 95–103 10.1016/j.yjmcc.2015.04.016 [DOI] [PubMed] [Google Scholar]
- 4. Zhang L., Malik S., Kelley G. G., Kapiloff M. S., and Smrcka A. V. (2011) Phospholipase Cϵ scaffolds to muscle-specific A kinase anchoring protein (mAKAPβ) and integrates multiple hypertrophic stimuli in cardiac myocytes. J. Biol. Chem. 286, 23012–23021 10.1074/jbc.M111.231993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang L., Malik S., Pang J., Wang H., Park K. M., Yule D. I., Blaxall B. C., and Smrcka A. V. (2013) Phospholipase Cϵ hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell 153, 216–227 10.1016/j.cell.2013.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smrcka A. V. (2015) Regulation of phosphatidylinositol-specific phospholipase C at the nuclear envelope in cardiac myocytes. J. Cardiovasc. Pharmacol. 65, 203–210 10.1097/FJC.0000000000000195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lyon A. M., and Tesmer J. J. (2013) Structural insights into phospholipase C-β function. Mol. Pharmacol. 84, 488–500 10.1124/mol.113.087403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wing M. R., Houston D., Kelley G. G., Der C. J., Siderovski D. P., and Harden T. K. (2001) Activation of phospholipase C-ϵ by heterotrimeric G protein βγ-subunits. J. Biol. Chem. 276, 48257–48261 10.1074/jbc.C100574200 [DOI] [PubMed] [Google Scholar]
- 9. Madukwe J. C., Garland-Kuntz E. E., Lyon A. M., and Smrcka A. V. (2018) G protein βγ subunits directly interact with and activate phospholipase Cϵ. J. Biol. Chem. 293, 6387–6397 10.1074/jbc.RA118.002354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smrcka A. V., Brown J. H., and Holz G. G. (2012) Role of phospholipase Cϵ in physiological phosphoinositide signaling networks. Cell. Signal. 24, 1333–1343 10.1016/j.cellsig.2012.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gresset A., Sondek J., and Harden T. K. (2012) The phospholipase C isozymes and their regulation. Subcell. Biochem. 58, 61–94 10.1007/978-94-007-3012-0_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Waldo G. L., Ricks T. K., Hicks S. N., Cheever M. L., Kawano T., Tsuboi K., Wang X., Montell C., Kozasa T., Sondek J., and Harden T. K. (2010) Kinetic scaffolding mediated by a phospholipase C-β and Gq signaling complex. Science 330, 974–980 10.1126/science.1193438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lyon A. M., Dutta S., Boguth C. A., Skiniotis G., and Tesmer J. J. (2013) Full-length Gαq-phospholipase C-β3 structure reveals interfaces of the C-terminal coiled-coil domain. Nat. Struct. Mol. Biol. 20, 355–362 10.1038/nsmb.2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lyon A. M., Tesmer V. M., Dhamsania V. D., Thal D. M., Gutierrez J., Chowdhury S., Suddala K. C., Northup J. K., and Tesmer J. J. (2011) An autoinhibitory helix in the C-terminal region of phospholipase C-β mediates Gαq activation. Nat. Struct. Mol. Biol. 18, 999–1005 10.1038/nsmb.2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park D., Jhon D. Y., Lee C. W., Ryu S. H., and Rhee S. G. (1993) Removal of the carboxyl-terminal region of phospholipase C-β1 by calpain abolishes activation by Gαq. J. Biol. Chem. 268, 3710–3714 [PubMed] [Google Scholar]
- 16. Kim C. G., Park D., and Rhee S. G. (1996) The role of carboxyl-terminal basic amino acids in Gqα-dependent activation, particulate association, and nuclear localization of phospholipase C-β1. J. Biol. Chem. 271, 21187–21192 10.1074/jbc.271.35.21187 [DOI] [PubMed] [Google Scholar]
- 17. Ilkaeva O., Kinch L. N., Paulssen R. H., and Ross E. M. (2002) Mutations in the carboxyl-terminal domain of phospholipase Cβ 1 delineate the dimer interface and a potential Gαq interaction site. J. Biol. Chem. 277, 4294–4300 10.1074/jbc.M109612200 [DOI] [PubMed] [Google Scholar]
- 18. Singer A. U., Waldo G. L., Harden T. K., and Sondek J. (2002) A unique fold of phospholipase C-β mediates dimerization and interaction with Gαq. Nat. Struct. Biol. 9, 32–36 10.1038/nsb731 [DOI] [PubMed] [Google Scholar]
- 19. Jin T. G., Satoh T., Liao Y., Song C., Gao X., Kariya K., Hu C. D., and Kataoka T. (2001) Role of the CDC25 homology domain of phospholipase Cϵ in amplification of Rap1-dependent signaling. J. Biol. Chem. 276, 30301–30307 10.1074/jbc.M103530200 [DOI] [PubMed] [Google Scholar]
- 20. Kelley G. G., Reks S. E., Ondrako J. M., and Smrcka A. V. (2001) Phospholipase Cϵ: a novel Ras effector. EMBO J. 20, 743–754 10.1093/emboj/20.4.743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kelley G. G., Reks S. E., and Smrcka A. V. (2004) Hormonal regulation of phospholipase Cϵ through distinct and overlapping pathways involving G12 and Ras family G-proteins. Biochem. J. 378, 129–139 10.1042/bj20031370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hicks S. N., Jezyk M. R., Gershburg S., Seifert J. P., Harden T. K., and Sondek J. (2008) General and versatile autoinhibition of PLC isozymes. Mol. Cell 31, 383–394 10.1016/j.molcel.2008.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jezyk M. R., Snyder J. T., Gershberg S., Worthylake D. K., Harden T. K., and Sondek J. (2006) Crystal structure of Rac1 bound to its effector phospholipase C-β2. Nat. Struct. Mol. Biol. 13, 1135–1140 10.1038/nsmb1175 [DOI] [PubMed] [Google Scholar]
- 24. Lyon A. M., Begley J. A., Manett T. D., and Tesmer J. J. (2014) Molecular Mechanisms of phospholipase C β3 autoinhibition. Structure 22, 1844–1854 10.1016/j.str.2014.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang W., and Neer E. J. (2001) Reassembly of phospholipase C-β2 from separated domains: analysis of basal and G protein-stimulated activities. J. Biol. Chem. 276, 2503–2508 10.1074/jbc.M003562200 [DOI] [PubMed] [Google Scholar]
- 26. Kadamur G., and Ross E. M. (2016) Intrinsic pleckstrin homology (PH) domain motion in phospholipase C-β exposes a Gβγ protein binding site. J. Biol. Chem. 291, 11394–11406 10.1074/jbc.M116.723940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Drin G., Douguet D., and Scarlata S. (2006) The pleckstrin homology domain of phospholipase Cβ transmits enzymatic activation through modulation of the membrane-domain orientation. Biochemistry 45, 5712–5724 10.1021/bi052317n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Han D. S., Golebiewska U., Stolzenberg S., Scarlata S. F., and Weinstein H. (2011) A dynamic model of membrane-bound phospholipase Cβ2 activation by Gβγ subunits. Mol. Pharmacol. 80, 434–445 10.1124/mol.111.073403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Garcia P., Gupta R., Shah S., Morris A. J., Rudge S. A., Scarlata S., Petrova V., McLaughlin S., and Rebecchi M. J. (1995) The pleckstrin homology domain of phospholipase C-δ 1 binds with high affinity to phosphatidylinositol 4,5-bisphosphate in bilayer membranes. Biochemistry 34, 16228–16234 10.1021/bi00049a039 [DOI] [PubMed] [Google Scholar]
- 30. Cifuentes M. E., Honkanen L., and Rebecchi M. J. (1993) Proteolytic fragments of phosphoinositide-specific phospholipase C-δ1: catalytic and membrane binding properties. J. Biol. Chem. 268, 11586–11593 [PubMed] [Google Scholar]
- 31. Essen L. O., Perisic O., Cheung R., Katan M., and Williams R. L. (1996) Crystal structure of a mammalian phosphoinositide-specific phospholipase C δ. Nature 380, 595–602 10.1038/380595a0 [DOI] [PubMed] [Google Scholar]
- 32. Oestreich E. A., Malik S., Goonasekera S. A., Blaxall B. C., Kelley G. G., Dirksen R. T., and Smrcka A. V. (2009) Epac and phospholipase Cϵ regulate Ca2+ release in the heart by activation of protein kinase Cϵ and calcium-calmodulin kinase II. J. Biol. Chem. 284, 1514–1522 10.1074/jbc.M806994200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oestreich E. A., Wang H., Malik S., Kaproth-Joslin K. A., Blaxall B. C., Kelley G. G., Dirksen R. T., and Smrcka A. V. (2007) Epac-mediated activation of phospholipase Cϵ plays a critical role in β-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes. J. Biol. Chem. 282, 5488–5495 10.1074/jbc.M608495200 [DOI] [PubMed] [Google Scholar]
- 34. Dusaban S. S., Purcell N. H., Rockenstein E., Masliah E., Cho M. K., Smrcka A. V., and Brown J. H. (2013) Phospholipase Cϵ links G protein-coupled receptor activation to inflammatory astrocytic responses. Proc. Natl. Acad. Sci. U.S.A. 110, 3609–3614 10.1073/pnas.1217355110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bunney T. D., Harris R., Gandarillas N. L., Josephs M. B., Roe S. M., Sorli S. C., Paterson H. F., Rodrigues-Lima F., Esposito D., Ponting C. P., Gierschik P., Pearl L. H., Driscoll P. C., and Katan M. (2006) Structural and mechanistic insights into ras association domains of phospholipase C ϵ. Mol. Cell. 21, 495–507 10.1016/j.molcel.2006.01.008 [DOI] [PubMed] [Google Scholar]
- 36. Seifert J. P., Wing M. R., Snyder J. T., Gershburg S., Sondek J., and Harden T. K. (2004) RhoA activates purified phospholipase C-ϵ by a guanine nucleotide-dependent mechanism. J. Biol. Chem. 279, 47992–47997 10.1074/jbc.M407111200 [DOI] [PubMed] [Google Scholar]
- 37. Seifert J. P., Zhou Y., Hicks S. N., Sondek J., and Harden T. K. (2008) Dual activation of phospholipase C-ϵ by Rho and Ras GTPases. J. Biol. Chem. 283, 29690–29698 10.1074/jbc.M805038200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ghosh M., and Smrcka A. V. (2004) Assay for G protein-dependent activation of phospholipase Cβ using purified protein components. Methods Mol. Biol. 237, 67–75 [DOI] [PubMed] [Google Scholar]
- 39. Seifert J. P., Snyder J. T., Sondek J., and Harden T. K. (2006) Direct activation of purified phospholipase Cϵ by RhoA studied in reconstituted phospholipid vesicles. Methods Enzymol. 406, 260–271 10.1016/S0076-6879(06)06019-8 [DOI] [PubMed] [Google Scholar]
- 40. Mezzasalma T. M., Kranz J. K., Chan W., Struble G. T., Schalk-Hihi C., Deckman I. C., Springer B. A., and Todd M. J. (2007) Enhancing recombinant protein quality and yield by protein stability profiling. J. Biomol. Screen 12, 418–428 10.1177/1087057106297984 [DOI] [PubMed] [Google Scholar]
- 41. Glukhova A., Hinkovska-Galcheva V., Kelly R., Abe A., Shayman J. A., and Tesmer J. J. (2015) Structure and function of lysosomal phospholipase A2 and lecithin:cholesterol acyltransferase. Nat. Commun. 6, 6250 10.1038/ncomms7250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Skou S., Gillilan R. E., and Ando N. (2014) Synchrotron-based small-angle X-ray scattering of proteins in solution. Nat. Protoc. 9, 1727–1739 10.1038/nprot.2014.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Korasick D. A., and Tanner J. J. (2018) Determination of protein oligomeric structure from small-angle X-ray scattering. Protein Sci. 27, 814–824 10.1002/pro.3376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mylonas E., and Svergun D. I. (2007) Accuracy of molecular mass determination of proteins by small-angle X-ray scattering. J. Appl. Crystallogr. 40, s245–s249 10.1107/S002188980700252X [DOI] [Google Scholar]
- 45. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 46. Trewhella J., Duff A. P., Durand D., Gabel F., Guss J. M., Hendrickson W. A., Hura G. L., Jacques D. A., Kirby N. M., Kwan A. H., Perez J., Pollack L., Ryan T. M., Sali A., Schneidman-Duhovny D., et al. (2017) 2017 publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution: an update. Acta Crystallogr. D Struct. Biol. 73, 710–728 10.1107/S2059798317011597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ohi M., Li Y., Cheng Y., and Walz T. (2004) Negative staining and image classification: powerful tools in modern electron microscopy. Biol. Proced. Online 6, 23–34 10.1251/bpo70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Harden T. K., Hicks S. N., and Sondek J. (2009) Phospholipase C isozymes as effectors of Ras superfamily GTPases. J. Lipid Res. 50, S243–S248 10.1194/jlr.R800045-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Camps M., Hou C., Sidiropoulos D., Stock J. B., Jakobs K. H., and Gierschik P. (1992) Stimulation of phospholipase C by guanine-nucleotide-binding protein βγ subunits. Eur. J. Biochem. 206, 821–831 10.1111/j.1432-1033.1992.tb16990.x [DOI] [PubMed] [Google Scholar]
- 50. Smrcka A. V., and Sternweis P. C. (1993) Regulation of purified subtypes of phosphatidylinositol-specific phospholipase Cβ by G protein α and βγ subunits. J. Biol. Chem. 268, 9667–9674 [PubMed] [Google Scholar]
- 51. Feng J., Roberts M. F., Drin G., and Scarlata S. (2005) Dissection of the steps of phospholipase Cβ 2 activity that are enhanced by Gβγ subunits. Biochemistry 44, 2577–2584 10.1021/bi0482607 [DOI] [PubMed] [Google Scholar]
- 52. Guo Y., Philip F., and Scarlata S. (2003) The pleckstrin homology domains of phospholipases C-β and -δ confer activation through a common site. J. Biol. Chem. 278, 29995–30004 10.1074/jbc.M301438200 [DOI] [PubMed] [Google Scholar]
- 53. Runnels L. W., Jenco J., Morris A., and Scarlata S. (1996) Membrane binding of phospholipases C-β1 and C-β2 is independent of phosphatidylinositol 4,5-bisphosphate and the α and βγ subunits of G proteins. Biochemistry 35, 16824–16832 10.1021/bi961606w [DOI] [PubMed] [Google Scholar]
- 54. Hudson B. N., Hyun S. H., Thompson D. H., and Lyon A. M. (2017) Phospholipase Cβ3 membrane adsorption and activation are regulated by its C-terminal domains and phosphatidylinositol 4,5-bisphosphate. Biochemistry 56, 5604–5614 10.1021/acs.biochem.7b00547 [DOI] [PubMed] [Google Scholar]
- 55. Hopkins J. B., Gillilan R. E., and Skou S. (2017) BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Crystallogr. 50, 1545–1553 10.1107/S1600576717011438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Franke D., Petoukhov M. V., Konarev P. V., Panjkovich A., Tuukkanen A., Mertens H. D. T., Kikhney A. G., Hajizadeh N. R., Franklin J. M., Jeffries C. M., and Svergun D. I. (2017) ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 50, 1212–1225 10.1107/S1600576717007786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., and Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 10.1107/S0021889803012779 [DOI] [Google Scholar]
- 58. Volkov V. V., and Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 10.1107/S0021889803000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879. Correction (1999) Biophys. J. 77, 2896 10.1016/S0006-3495(99)77121-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Svergun D., Barberato C., and Koch M. H. J. (1995) CRYSOL: a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 10.1107/S0021889895007047 [DOI] [Google Scholar]
- 61. Petoukhov M. V., and Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 10.1529/biophysj.105.064154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kimanius D., Forsberg B. O., Scheres S. H., and Lindahl E. (2016) Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 5, e18722 10.7554/eLife.18722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Scheres S. H. (2016) Processing of structurally heterogeneous cryo-EM data in RELION. Methods Enzymol. 579, 125–157 10.1016/bs.mie.2016.04.012 [DOI] [PubMed] [Google Scholar]
- 64. Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., and Ludtke S. J. (2007) EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 10.1016/j.jsb.2006.05.009 [DOI] [PubMed] [Google Scholar]
- 65. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 10.1107/S0021889892001663 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.