

Abstract
Ras proteins participate in multiple signal cascades, regulating crucial cellular processes, including cell survival, proliferation, and differentiation. We have previously reported that Ras proteins are modified by sumoylation and that Lys-42 plays an important role in mediating the modification. In the current study, we further investigated the role of Lys-42 in regulating cellular activities of K-Ras. Inducible expression of K-RasV12 led to the activation of downstream components, including c-RAF, MEK1, and extracellular signal–regulated kinases (ERKs), whereas expression of K-RasV12/R42 mutant compromised the activation of the RAF/MEK/ERK signaling axis. Expression of K-RasV12/R42 also led to reduced phosphorylation of several other protein kinases, including c-Jun N-terminal kinase (JNK), Chk2, and focal adhesion kinase (FAK). Significantly, K-RasV12/R42 expression inhibited cellular migration and invasion in vitro in multiple cell lines, including transformed pancreatic cells. Given that K-Ras plays a crucial role in mediating oncogenesis in the pancreas, we treated transformed pancreatic cells of both BxPC-3 and MiaPaCa-2 with 2-D08, a small ubiquitin-like modifier (SUMO) E2 inhibitor. Treatment with the compound inhibited cell migration in a concentration-dependent manner, which was correlated with a reduced level of K-Ras sumoylation. Moreover, 2-D08 suppressed expression of ZEB1 (a mesenchymal cell marker) with concomitant induction of ZO-1 (an epithelial cell marker). Combined, our studies strongly suggest that posttranslational modification(s), including sumoylation mediated by Lys-42, plays a crucial role in K-Ras activities in vivo.
Keywords: Ras protein, posttranslational modification (PTM), signaling, cell invasion, cell migration, transformation, domain structure, lysine residue, K-Ras, sumoylation
Introduction
Ras proteins are the best-studied proto-oncogene products owing to their crucial roles in cell survival, proliferation, and transformation. K-Ras, H-Ras, and N-Ras are three related gene products that are commonly expressed in mammalian cells and have overlapping but distinctive functions (1–4). Ras proteins are membrane-anchored GTPases that participate in multiple signal cascades, mediating numerous biological processes (2, 5, 6). Their activities, subcellular localizations, and stabilities are therefore tightly controlled in normal cells; deregulation of any of these features often causes malignant transformation (1). Ras proteins are regulated by a wide range of posttranslational modifications, including farnesylation, carboxylmethylation, palmitoylation, phosphorylation, ubiquitination, and acetylation (1, 7–15).
The RAS/RAF/MEK/ERK2 signaling pathway plays a central role in mediating cell survival and proliferation, and its dysregulation was detected in more than 50% of human tumors (16). K-Ras mutations found in over 90% of pancreatic ductal adenocarcinoma cancers and 20–30% of nonsmall-cell lung cancers are believed to play a key role in driving these malignancies (17). Great efforts have been made to develop inhibitors that target K-Ras and its regulatory pathways (e.g. farnesyltransferase and geranylgeranyltransferase inhibitors). However, little progress has been made in this regard, partly due to rather unusual alternative K-Ras geranylgeranylation, underscoring the need to explore new posttranslational modification targets for this protein.
K-Ras oncogenic mutations (e.g. V12) occur early in carcinogenesis of major human malignancies, which promotes cell migration and invasion of cancer cells (18, 19). Given that our recent study reveals that Ras proteins are posttranslationally modified by sumoylation and that Lys-42 plays a crucial role in mediating the sumoylation (20), we further examined whether the RasR42 mutant affected cells' ability to promote cell migration and invasion. We found that K-RasR42 displayed a weakened ability to promote cell migration and invasion, which was coupled with reduced activation of FAK as well as protein kinases of the MAPK superfamily.
To date, no compounds that target Ras have been approved for clinic applications, even though Ras proteins were the first, and remain the best-studied, oncoproteins. Given our recent observation that sumoylation plays an important role in regulating Ras activities, we also tested whether a SUMO inhibitor (2-D08) was capable of blocking migration of cells harboring the K-RasV12 mutation. We observed that 2-D08 blocked migration of transformed pancreatic MiaPaCa-2 cells (containing K-RasV12), but not BxPC-3 cells (containing WT K-Ras), in a concentration-dependent manner. This line of study is likely to accelerate the development of therapies that target ubiquitin-like modifications.
Results
Inducible expression of K-RasR42 suppresses RAF/MEK/ERK signaling
We have previously observed that Lys-42 mediates sumoylation of Ras proteins, which appears to be important for their activity (20). Lys-42 is located between switch I (amino acids 32–38) and II (amino acids 59–67) domains that mediate the interaction with its regulators and effectors (Fig. 1A). Thus, we speculate that Lys-42 mutation would have an impact on Ras functions in vivo. To further determine whether Lys-42 was crucial for promoting RAS activation and its downstream signaling, K-RasV12 and K-RasV12/R42 expression was subjected to induction by doxycycline (Dox). We observed that inducible expression of K-RasV12 was strongly associated with the activation of downstream components, including c-Raf, MEK, and ERK, and that the activation of downstream signaling components was K-Ras concentration–dependent (Fig. 1B). On the other hand, expression of K-RasV12/R42 greatly suppressed the activation of c-Raf, MEK1, and ERKs, albeit its expression was comparable with that of K-RasV12 after the addition of Dox, strongly supporting the notion that Lys-42 is crucial for the activation of Ras as well as its major signaling cascade of RAF/MEK/ERK. Because Ras is known to interact with PI3K (21), we measured the activation kinetics of AKT after induced expression of K-RasV12 and K-RasV12/R42. Intriguingly, AKT activation was not affected in cells expressing K-RasV12/R42 as compared with those expressing oncogenic K-RasV12.
Figure 1.
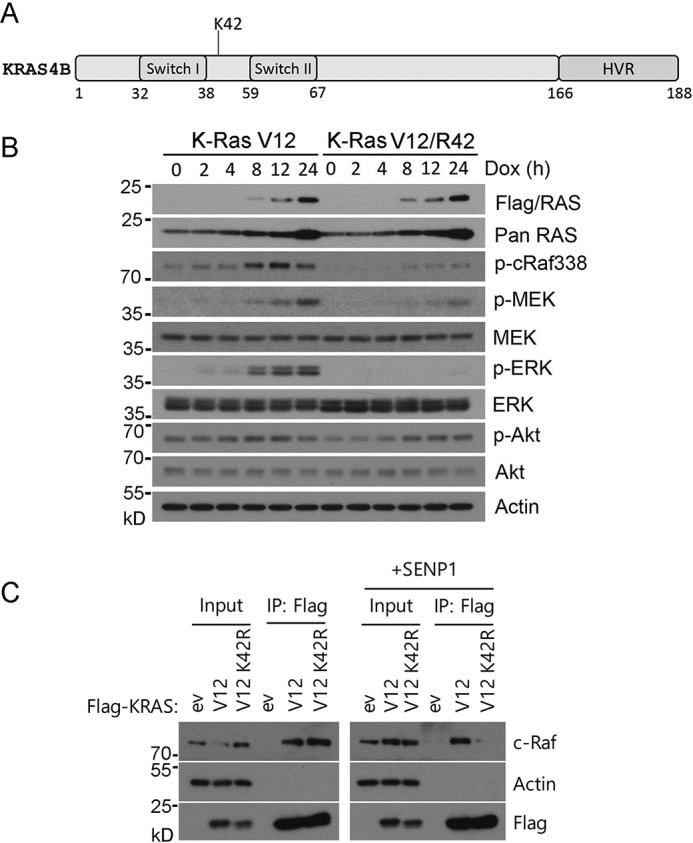
Lys-42 is crucial for K-Ras downstream signaling. A, schematic representation of major domains of K-Ras protein. Lys-42 is located between switch I (amino acids 32–38) and II (amino acids 59–67) domains that mediate the interaction with its regulators and effectors. The hypervariable (HVR) domain between amino acids 165 and 188 in the C terminus specifies the membrane localization through extensive posttranslational modifications. B, Tet293/K-RasV12 or Tet293/K-RasV12/R42 cells were cultured in the presence or absence of Dox. At various times of culture, cells were collected and lysed. Equal amounts of cell lysates were blotted with antibodies to FLAG (K-Ras), pan-Ras, phospho-cRaf338 (p-cRaf388), phospho-MEK (p-MEK), pan-MEK, phospho-ERK (p-ERK), pan-ERK, phospho-AKT (p-AKT), pan-AKT, and β-actin. C, HEK293T cells were transfected with plasmids expressing K-RasV12, K-RasV12/R42, or vector (ev) alone. These cells were also co-transfected with or without SENP1 for 24 h, after which cells were lysed and equal amounts of cell lysates were immunoprecipitated with the anti-FLAG antibody. FLAG immunoprecipitates (IP), along with cell lysate inputs, were blotted for c-Raf, β-actin, and FLAG. In all relevant panels, molecular weight markers are also indicated.
To determine the mechanism(s) by which Lys-42 mediates K-Ras signaling, we determined whether it may be involved in regulating protein-protein interaction. HEK293 cells were transfected with plasmids expressing FLAG-tagged K-RasV12 or K-RasV12/R42 for 24 h. We also co-transfected the cells with or without SENP1 expression plasmid (for an isopeptidase specific for removing SUMO moiety from protein substrates). Equal amounts of cell lysates from various transfectants were immunoprecipitated with the anti-FLAG antibody. FLAG immunoprecipitates, along with cell lysate inputs, were blotted with antibodies to c-Raf, actin, and FLAG. We observed that, compared with that of K-RasV12, expression of K-RasV12/R42 compromised its physical interaction with c-Raf in the presence of ectopically expressed SENP1 (Fig. 1C). These results again strongly support the notion that Lys-42 plays a crucial role in mediating K-Ras signaling.
Lys-42 affects several signaling pathways mediated by K-Ras
Given that Ras proteins are crucial for numerous biological functions, including cell proliferation, migration, and stress responses, we next examined whether K-RasR42 expression affected signaling pathways other than the RAF/MEK/ERK signaling axis. Through the analysis of a kinase array, we observed that K-RasR42 expression down-regulated phosphorylation/activity of a few well-studied protein kinases (Fig. 2 (A and B) and Figs. S1 and S2). For example, p38 and JNK phosphorylation was reduced in cells expressing K-RasR42, suggesting that K-Ras sumoylation may also play a role in mediating stress responses. Moreover, K-RasR42 also compromised phosphorylation of Chk2 and FAK, two protein kinases involved in DNA damage responses and cell adhesion, respectively.
Figure 2.

Lys-42 is important for activation of protein kinases mediating various biological responses. A, selected dot blots of protein kinase array using paired cellular lysates as shown. Representative protein kinases (except for AKT and GSK3) exhibiting significant differences between K-RasV12 and K-RasV12/R42 are shown. EV, empty vector; CT, control for the array analysis. See Fig. S1 for additional information and details. B, dot blot signals as shown in A (and Fig. S3) were quantified and were then normalized to signals in cells transfected with empty vector. Relative signal intensity is represented by plus signs.
K-RasR42 suppresses cell migration and invasion
K-Ras oncogenic mutations (e.g. Val-12) occur early in carcinogenesis of major human malignancies, which is known to promote cell migration and invasion of cancer cells (18, 19). Because Lys-42 mutation greatly compromised Ras signaling, we measured whether expression of K-RasR42 would affect cell migration promoted by the oncogenic counterpart in a conventional wound-healing assay. We observed that NIH3T3 cells transfected with FLAG-K-RasV12 displayed rapid closing of the wound gap due to active cell migration compared with that of cells transfected with FLAG-K-RasV12/R42 or vector alone (Fig. 3, A and B), indicating that Lys-42 mutation significantly compromised its ability to promote cell migration. Expression of transfected K-Ras was efficient (Fig. 3C). To confirm that sumoylation plays a role in cell migration, we ectopically expressed K-RasV12 or K-RasV12/R42 in MCF7 cells. We observed that expression of K-RasV12/R42 mutant also suppressed MCF7 cell migration promoted by K-RasV12 (Fig. 3, D and E). This suppression was associated with down-regulation of the MAPK signaling pathway coupled with reduced expression of Snail and Claudin-1 (Fig. 3F), two gene products involved in cell migration and epithelial and mesenchymal transition (EMT) (22). Expression of the K-RasV12/R42 mutant also somewhat affected cell proliferation, which did not seem to be associated with increased cell death or cell cycle arrest (Figs. S3 and S4).
Figure 3.
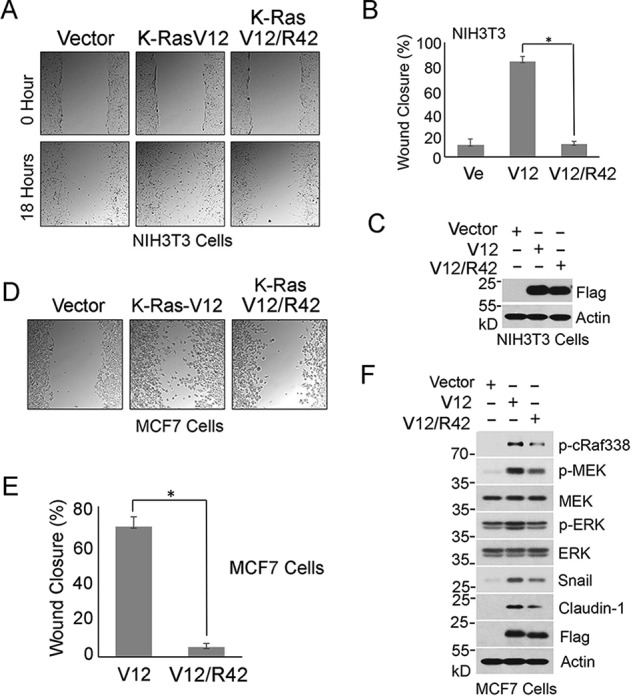
Lys-42 is essential for mediating cell migration. A, NIH3T3 cells transfected with FLAG-K-RasV12, FLAG-K-RasV12/R42 expression plasmid, or vector alone for 24 h were subjected to conventional wound-healing assays. Representative images of cells at 0 and 18 h postscratching are shown. B, percentage of wound closure as described in A was quantified. Data are summarized from three independent experiments. Ve, vector-transfected control. C, equal amounts of lysates from cells transfected with various expression plasmids as described in B were blotted with antibodies to FLAG and β-actin, respectively. D, MCF cells were transfected with a plasmid construct expressing FLAG-K-RasV12 or FLAG-K-RasV12/R42 for 24 h. Empty plasmid vector was also used for transfection as control. Images at 18 h postscratching for various transfections were taken. Representative images are shown. E, percentage of wound closure as described in D was quantified. Data are summarized from three independent experiments. Ve, vector-transfected control. F, MCF7 cells were transfected with a plasmid construct expressing K-RasV12 or K-RasV12/R42 for 24 h. Empty plasmid vector was also used for transfection as control. Equal amounts of cell lysates were blotted for phospho-cRaf388 (p-cRaf388), phospho-MEK (p-MEK), MEK, phospho-ERK (p-ERK), ERK, Snail, claudin-1, FLAG, and β-actin. In all relevant panels, molecular weight markers are also indicated. Error bars, S.D. *, p < 0.05.
Given the crucial role of K-Ras in tumor development in pancreas (23), we then performed wound-healing experiments using a pancreatic cell line (MiaPaCa-2). We transfected MiaPaCa-2 cells with K-Ras and various mutant constructs. We observed that expression of WT K-Ras significantly stimulated cell migration, which was further promoted by Val-12 mutation (Fig. 4, A and B). In contrast, expression of K-RasR42 mutant suppressed cell migration, and the effect was more pronounced in cells expressing K-RasV12/R42, strongly suggestive of a role of Lys-42 in mediating K-Ras biological and pathological activities.
Figure 4.
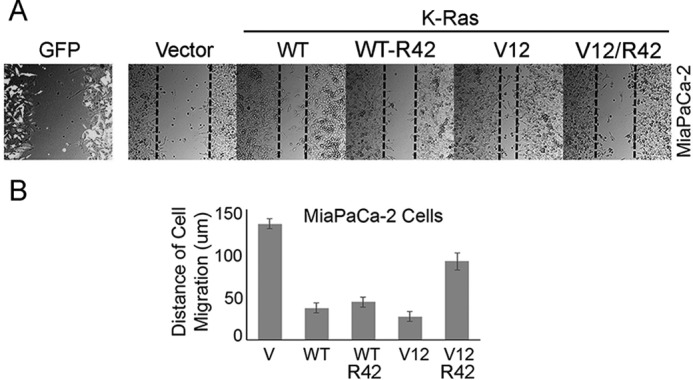
Lys-42 is essential for mediating migration of transformed pancreatic cells. A, MiaPaCa-2 cells were transfected with various K-Ras expression plasmids as shown for 24 h. GFP expression plasmid was used for co-transfection as control. Transfected cells were then subjected to a wound-healing assay as described under “Experimental procedures.” Representative images of cells at 0 and 18 h postscratching were shown. Transfected cells stained with the anti-GFP antibody were also shown. B, percentage of wound closure as described in A was quantified. Data are summarized from three independent experiments. V, vector-transfected control. Error bars, S.D.
We further examined whether Arg-42 affected the ability of K-Ras to promote cell invasion. Tet293 cells stably transfected with K-RasV12 or K-RasV12/R42 expression plasmid, along with parental cells, were subjected to a transwell migration assay. We observed that compared with the parental control cells, significantly increased invasiveness was observed in Tet293-K-RasV12 cells and that Tet293-K-RasV12/R42 cells exhibited greatly reduced transwell migration similar to that of parental cells (Fig. 5, A and B). Moreover, the addition of Dox to Tet293-K-RasV12 cells, but not Tet293-K-RasV12/R42 cells, induced expression of Snail (Fig. 5C), a gene product involved in epithelial–mesenchymal transition (24). Consistent with these observation, transfection of K-RasV12 expression plasmid, but not K-RasV12/R42 or vector, greatly promoted transwell migration of NIH3T3 cells (Fig. 6, A and B). Combined, these observations strongly suggest that Lys-42 plays an essential part in mediating oncogenic activities of K-Ras.
Figure 5.
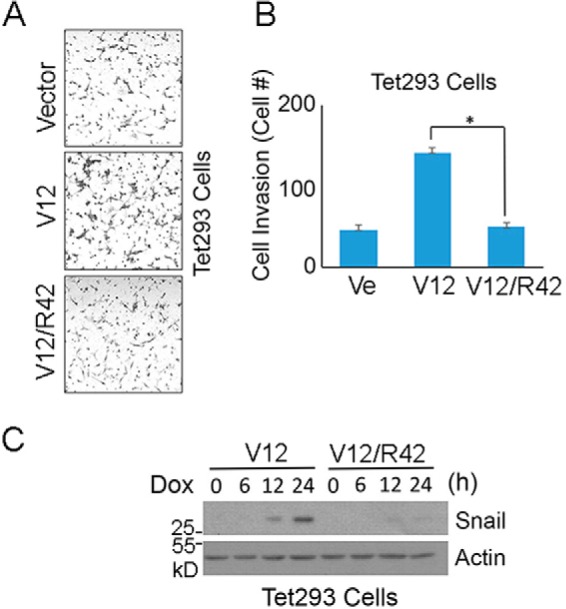
Lys-42 is essential for mediating invasion of transformed cells. A, Tet293 cells stably transfected with K-RasV12 or K-RasV12/R42 expression plasmid were cultured in the presence of Dox for 24 h, after which treated cells, along with parental cells, were used for transwell migration assays. Representative images are shown. B, cell invasiveness as described in A was quantified. Data are summarized from three independent experiments. Ve, vector-transfected control. C, Tet293 cells stably transfected with a plasmid construct expressing K-RasV12 or K-RasV12/R42 were cultured in the presence of Dox for various times as indicated, after which cells were collected and lysed. Equal amounts of cell lysates were blotted with antibodies to Snail and β-actin, respectively. Molecular weight markers are also indicated. *, p < 0.05. Error bars, S.D.
Figure 6.
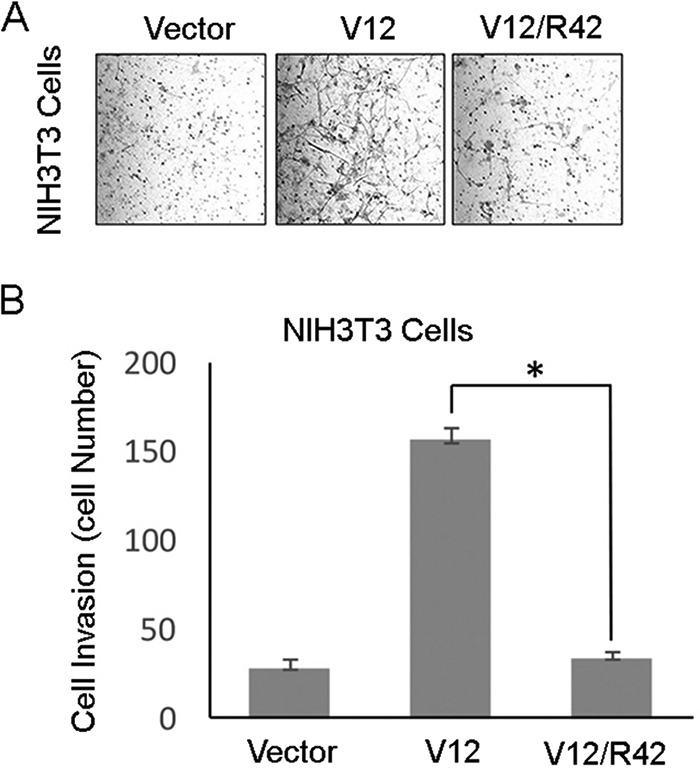
Expression of Lys-42 mutant of K-Ras compromises invasion of nontransformed cells. A, NIH3T3 cells were transfected with a plasmid construct expressing K-RasV12 or K-RasV12/R42 for 24 h, after which transfected cells, along with cells transfected with vector alone, were subjected to transwell migration assays. Representative images are shown. B, cell invasiveness as described in A was quantified. Data are summarized from three independent experiments. *, p < 0.05. Error bars, S.D.
SUMO inhibitor blocks cell migration in a K-Ras–dependent manner
Ras proteins were the first and remain the best-studied oncogene products. However, to date, no compounds that target Ras have been approved for cancer treatments in the clinic. Given that sumoylation plays a crucial role in activating Ras, we next determined whether 2-D08, a SUMO E2 inhibitor (25), would suppress tumor cell migration in a manner that depends on K-Ras activation. To this end, we selected MiaPaCa-2 and BxPA-3 cells for our studies, as MiaPaCa-2 cells harbor mutated K-RasV12, whereas BxPC-3 cells contain WT K-Ras. Wound-healing assays revealed that MiaPaCa-2 cells closed the wound gap much faster than BxPC-3 cells did, suggesting that expression of oncogenic K-Ras enhances cell migration (Fig. 7, A and B). Significantly, SUMO E2 inhibitor, 2-D08, inhibited MiaPaCa-2 cell migration in a concentration-dependent manner, whereas it had little effect on BxPC-3 cells.
Figure 7.
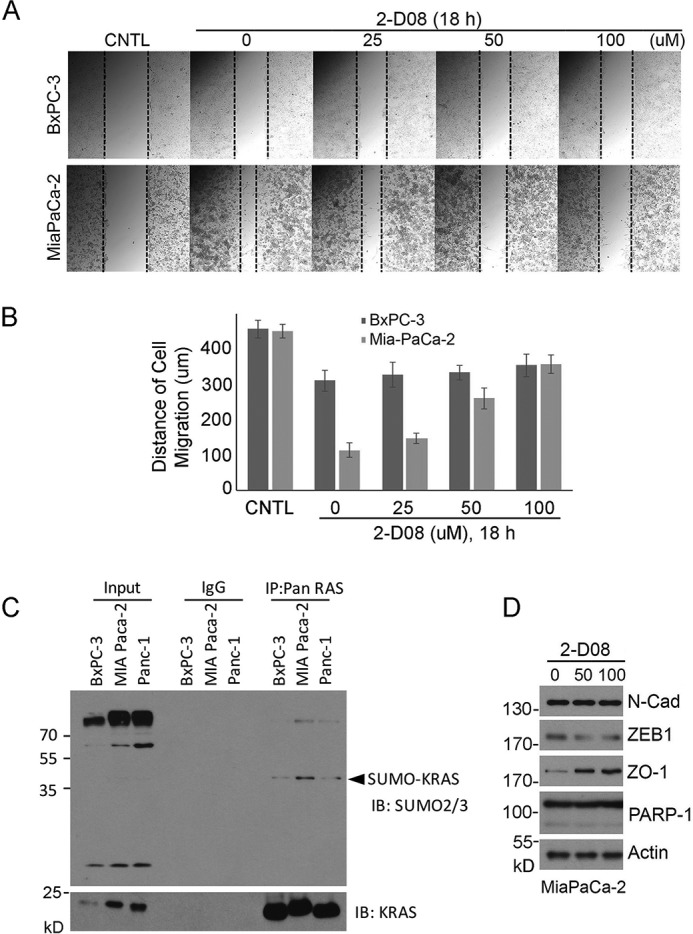
SUMO inhibitor blocks migration of pancreatic cancer cells with K-RasV12 mutation. A, both BxPC-3 (without K-Ras mutation) and MiaPaCa-2 (with K-RasV12 mutation) cells were employed for wound-healing assays. Cells with open scratches were treated with various concentrations of 2-D08 for 18 h. Representative images of cells before (CNTL) and after recovery in the presence of 2-D08 for 18 h are shown. B, cell migration of various treatments, as shown in A, was quantified. Data are summarized from three independent experiments. C, pancreatic cell lines as indicated were immunoprecipitated with the Ras antibody (Pan-Ras) or IgG as control. Immunoprecipitates, along with lysate controls, were blotted with antibodies to SUMO2/3 or to K-Ras. D, MiaPaCa-2 cells treated with or without 2-D08 were lysed, and equal amounts of cell lysates were blotted with antibodies to N-cadherin (N-Cad), ZEB1, ZO-1, PARP-1, and β-actin, respectively. Molecular weight markers are also indicated.
Moreover, 2-D08 treatment suppressed expression of ZEB1, a gene product positively associated with cell migration and EMT (Fig. 7D). In contrast, it promoted expression of ZO-1, a gene product whose expression is negatively associated with cell proliferation and migration. Immunoblotting revealed that K-Ras sumoylation levels were lower in BxPC-3 cells than those in MiaPaCa-2 cells (Fig. 7D). These results are thus consistent with our early observation that cells expressing oncogenic Ras contain more sumoylated counterpart (20). These results also explain why MiaPaCa-2 cells are sensitive to treatment with 2-D08.
Based on our studies, we propose the following model to explain Lys-42 in mediating K-Ras biochemical and biological functions (Fig. 8). K-Ras is modified by sumoylation, which in turn regulates its physical interaction with downstream component c-Raf via the Ras-binding domain (RBD). Other domain structure(s) of Raf, such as the SUMO-interacting motif, may recognize the SUMO moiety, positively promoting the interaction between Ras and Raf. Lys-42 mutation significantly compromises its sumoylation and thus the physical interaction with Raf. Desumoylated K-Ras has a significantly reduced ability to activate downstream components owing to compromised interaction with c-Raf, leading to reduced biological/pathological functions, including cell migration and invasion.
Figure 8.
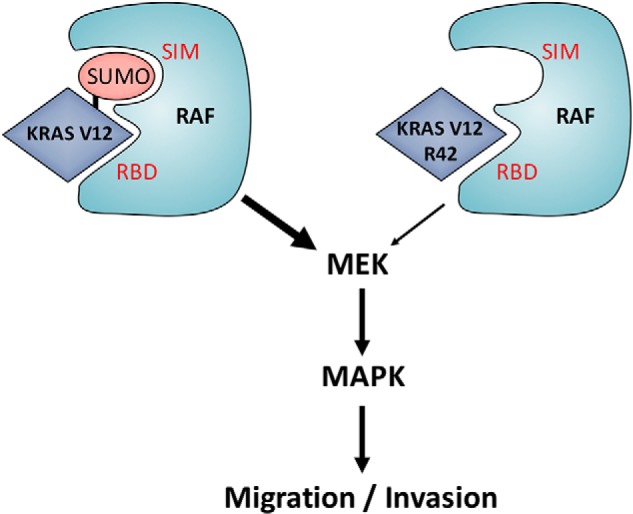
A model depicting sumoylation in regulating Ras signaling and functions. SIM, SUMO-interacting motif; RBD, Ras-binding domain.
Discussion
Posttranslational modifications play an essential role in regulating the subcellular localization, stability, and/or activity of numerous cellular proteins. Ras proteins are modified by almost all known posttranslational modifications (9, 26). Lysine residues in proteins are major targets for posttranslational modifications, including acetylation, methylation, ubiquitination, and sumoylation. In fact, extensive research in the past has demonstrated that many lysine residues in Ras proteins are modified (12, 14). We recently demonstrated that Lys-42 of Ras proteins is important for sumoylation, a previously undocumented posttranslational modification for the oncogene product (20). In the current study, we have further analyzed the functional consequence of K-RasR42 in mediating its biologic and oncogenic functions. We have demonstrated that inducible expression of K-RasV12/R42 mutant inhibits the activation of the RAF/MEK/ERK signaling axis. Moreover, expression of this mutant suppresses cell migration and invasion in multiple cell lines.
Profiling a kinase array reveals that expression of the K-RasV12/R42 mutant leads to changes in phosphorylation of protein kinases that mediate several distinct downstream pathways. Notably, we have observed that the phosphorylation level of FAK, a kinase functioning as a biosensor or integrator to control cell motility, is also significantly reduced in cells expressing K-RasV12/R42. This is consistent with our subsequent study showing that expression of K-RasV12/R42 leads to a weakened ability of cell migration and invasion.
One intriguing observation is that there were no significant changes of physical interaction between K-RasV12/R42 and c-Raf compared with K-RasV12 and c-Raf; however, when SENP1 was expressed, the interaction between K-RasV12/R42 and c-Raf was greatly reduced (Fig. 1C). There are two possibilities to explain these results. 1) Lys-42 regulates K-Ras sumoylation but is not the residue accepting the SUMO moiety. In other words, the Arg-42 mutant reduces, but does not abolish, K-Ras sumoylation. Thus, further inhibition of K-Ras sumoylation by 2-D08 synergizes with Arg-42 mutation, leading to reduced physical interaction with c-Raf as well as compromised downstream signaling. 2) An alternative explanation is that sumoylation of another component(s) upstream of Ras is required for its full activation and efficient interaction with c-Raf. Supporting this, it has been reported that Grb2 is modified by sumoylation, which positively regulates ERK signaling through promoting its interaction with Sos1.
K-Ras is the most frequently mutated oncogene product in human malignancies. K-Ras mutations are frequently associated with tumor development, poor prognosis, and therapeutic resistances. Ras proteins have been generally considered druggable targets, given their enzymatic activities and roles as key components of signaling pathways for cell survival, proliferation, transformation, and differentiation (1, 9, 27). Despite extensive research in the past, no pharmacological agents are available for malignancies derived partly or primarily from K-Ras mutations. One aspect of K-Ras biology that has not been extensively studied is the role of lysine residues in mediating its oncogenic/pathologic activities because of their involvement in several types of posttranslational modifications. Our observations that demonstrate the importance of lysine residues in oncogenic functions open up new avenues for identifying druggable targets, including those essential for participating in ubiquitination, sumoylation, acetylation, and methylation. As a proof-of-principle, we show that SUMO E2 inhibitor 2-D08 is capable of blocking cell migration that is at least in part promoted by K-RasV12 mutation.
We realize that Lys-42 lies in close proximity to the so-called switch domain and that a mutation at Lys-42 could in theory cause a conformation change, leading to compromised interactions with upstream regulators and/or downstream effectors. On the other hand, available evidence suggests that the substitution of Lys with Arg at residue 42 does not appear to perturb its conformation: 1) K-RasV12/R42 regulates AKT signaling/activation in a manner similar to that of K-RasV12 (Fig. 1B); 2) K-RasV12/R42 interacts with Ral.GDS, Raf, and PI3K in the same manner as that of WT Ras or RasV12 mutant regardless of GTP loading (21). However, when the overall sumoylation status in the cells is compromised due to expression of SENP1, K-RasV12/R42 displays weakened interaction with c-Raf compared with K-RasV12 (Fig. 1C), supporting the notion that sumoylation plays an essential role in Ras downstream signaling.
Activated K-Ras functions to drive oncogenesis in major human malignancies. The K-RasV12 mutant is thought to be constitutively locked in the active, GTP-bound state, which makes it very challenging to inhibit its oncogenic activity through traditional drug design approaches. One approach is to disrupt signaling between K-Ras protein and its downstream effector(s) via a small molecular compound, thus blocking its activities (28). In this study, we demonstrate that SUMO inhibitor 2-D08 suppresses expression of genes involved in EMT and reduces the migration rate of pancreatic cancer cells with K-RasV12 in a concentration-dependent manner. Given that sumoylation closely resembles ubiquitination and that small molecular inhibitors for ubiquitin and the proteasome have been moved to the clinic for cancer treatments (29, 30), we believe that a better understanding of the role of Lys-42 in regulating K-Ras will open a new path to develop innovative anti-cancer therapies.
Experimental procedures
Cell culture and transfection
HEK293T (kidney carcinoma), MCF7 (mammary carcinoma), NIH3T3 (fibroblast), MiaPaCa-2, and BxPC-3 cell lines obtained from the American Type Culture Collection were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Invitrogen) and antibiotics (100 μg/ml penicillin and 50 μg/ml streptomycin sulfate; Invitrogen) at 37 °C under 5% CO2. Transfection of individual cell lines was achieved with either LF2000 (Invitrogen) or Fugene HD (Roche Diagnostics) following the manufacturers' protocol. Transfection efficiency was estimated to be between 80 and 100% in all cases through co-transfecting a GFP-expressing plasmid (data not shown).
Plasmids
HA-tagged SUMO3 and UBC9 expression plasmids were obtained from Addgene. K-Ras and its mutant cDNAs were obtained using the QuikChange site-directed mutagenesis kit (Agilent Technologies). All mutations were confirmed by DNA sequencing.
Protein extracts and immunoblotting
Total cell lysates were prepared in a buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1% IGEPAL, 0.1% SDS, and 0.5% sodium deoxycholate) supplemented with a mixture of protease and phosphatase inhibitors. HEK293T Tet-on cells were engineered to express K-RasV12 or K-RasV12/R42 mutants in the presence of doxycycline. At various times after the addition of doxycycline, cells were collected and lysed in the same buffer as above. Protein concentrations were measured using the bicinchoninic acid protein assay reagent kit (Pierce). Equal amounts of protein lysates from various samples were used for SDS-PAGE analysis followed by immunoblotting. Antibodies to HA, FLAG, phospho-cRaf, Raf1, Ral.GDS, phospho-MEK, MEK, phospho-ERK42/44, ERK1/2, Snail, Claudin-1, N-Cad, ZEB1, ZO-1, PARP-1, and actin were purchased from Cell Signaling Technology. Specific signals on immunoblots (polyvinylidene difluoride) were visualized using enhanced chemiluminescence (Super-Signal, Pierce).
Immunoprecipitation
Cells were lysed in TBSN buffer (20 mm Tris-Cl (pH 8.0), 150 mm NaCl, 0.5% Nonidet P-40, 5 mm EGTA, 1.5 mm EDTA, 0.5 mm Na3VO4, and 20 mm β-glycerol phosphate). The cell lysates were clarified by centrifugation at 15,000 × g for 20 min at 4 °C. Cleared lysates (1 mg) were added to FLAG M2 agarose (Sigma) followed by incubation in the TBSN buffer for 1 h at 4 °C. After incubation, proteins bound to each resin were washed extensively with the binding buffer, eluted in the SDS-PAGE sample buffer, and analyzed by SDS-PAGE.
Wound-healing assay
NIH3T3 fibroblast cells were transiently transfected with plasmid vector alone or pcDNA-K-RasV12 or pcDNA-K-RasV12/R42 for 24 h before they were used for the wound-healing assay. Transfected cells were serum-starved for 18 h and then wounded by manual scraping with a pipette tip. Plates were washed several times to remove nonadherent cells, and cells were cultured in the same medium containing 1% fetal bovine serum. Assessment of cell migration into the wounded area was performed under a light microscope after an additional 18 h of incubation.
Phosphokinase array
For phosphokinase array analysis, cellular extracts (500 mg) were incubated with the Phospho-Kinase Array Kit (Proteome Profiler; R&D Systems, Abingdon, UK) following the manufacturer's instructions. Densitometry values were estimated by the ImageJ software and were expressed as arbitrary units. Average signal of the pair of duplicate spots, representing each phosphorylated kinase protein, was calculated after subtraction of background values (pixel density) from negative control spots and normalization to average values from positive control spots.
Cell invasion assays
HEK293T and NIH3T3 cells were transiently transfected with pcDNA plasmid vector (control), pcDNA-K-RasV12, or pcDNA-K-RasV12/R42. Twenty-four h posttransfection, cells (4 × 104 cells/well) were seeded onto transwell inserts and incubated at 37 °C for 12 h. Nonmigrated cells were removed from the upper face of the transwell insert using a cotton swab. Cells that migrated through membranes were stained. Cell density in each treatment was recorded under a light microscope.
Statistical analysis
Each experiment was performed at least three times. The data were plotted as the mean ± S.D. Student's t test was used for all comparisons. A p value of <0.05 was considered statistically significant.
Author contributions
B. H. C., M. R. P., and W. D. conceptualization; B. H. C. data curation; B. H. C. and W. D. formal analysis; B. H. C. investigation; B. H. C., M. R. P., Y. C., and L. L. methodology; B. H. C., M. R. P., Y. C., and L. L. writing-review and editing; Y. C. and L. L. resources; W. D. supervision; W. D. funding acquisition; W. D. writing-original draft; W. D. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Feng Wu and Ziyue Kou in the laboratory for assistance during the study. The New York University Center was supported by NIEHS, National Institutes of Health, Center Grant ES000260.
This work was supported in part by National Institutes of Health, NCI Grant CA216987 (to W. D. and Y. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
- MEK
- mitogen-activated protein kinase/extracellular signal–regulated kinase kinase
- ERK
- extracellular signal–regulated kinase
- FAK
- focal adhesion kinase
- MAPK
- mitogen-activated protein kinase
- Dox
- doxycycline
- PI3K
- phosphatidylinositol 3-kinase
- SUMO
- small ubiquitin-like modifier
- EMT
- epithelial and mesenchymal transition.
References
- 1. Ahearn I. M., Haigis K., Bar-Sagi D., and Philips M. R. (2011) Regulating the regulator: post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 13, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shaw R. J., and Cantley L. C. (2006) Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441, 424–430 10.1038/nature04869 [DOI] [PubMed] [Google Scholar]
- 3. Hall A. (1990) The cellular functions of small GTP-binding proteins. Science 249, 635–640 10.1126/science.2116664 [DOI] [PubMed] [Google Scholar]
- 4. Boguski M. S., and McCormick F. (1993) Proteins regulating Ras and its relatives. Nature 366, 643–654 10.1038/366643a0 [DOI] [PubMed] [Google Scholar]
- 5. Yaswen P., and Campisi J. (2007) Oncogene-induced senescence pathways weave an intricate tapestry. Cell 128, 233–234 10.1016/j.cell.2007.01.005 [DOI] [PubMed] [Google Scholar]
- 6. Sternberg P. W., and Alberola-Ila J. (1998) Conspiracy theory: RAS and RAF do not act alone. Cell 95, 447–450 10.1016/S0092-8674(00)81612-8 [DOI] [PubMed] [Google Scholar]
- 7. Lynch S. J., Snitkin H., Gumper I., Philips M. R., Sabatini D., and Pellicer A. (2015) The differential palmitoylation states of N-Ras and H-Ras determine their distinct Golgi subcompartment localizations. J. Cell Physiol. 230, 610–619 10.1002/jcp.24779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sung P. J., Tsai F. D., Vais H., Court H., Yang J., Fehrenbacher N., Foskett J. K., and Philips M. R.. Phosphorylated K-Ras limits cell survival by blocking Bcl-xL sensitization of inositol trisphosphate receptors. Proc. Natl. Acad. Sci. U.S.A. 110, 20593–20598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Philips M. R., and Cox A. D. (2007) Geranylgeranyltransferase I as a target for anti-cancer drugs. J. Clin. Invest. 117, 1223–1225 10.1172/JCI32108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bivona T. G., Quatela S. E., Bodemann B. O., Ahearn I. M., Soskis M. J., Mor A., Miura J., Wiener H. H., Wright L., Saba S. G., Yim D., Fein A., Pérez de Castro I., Li C., Thompson C. B., et al. (2006) PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell 21, 481–493 10.1016/j.molcel.2006.01.012 [DOI] [PubMed] [Google Scholar]
- 11. Bar-Sagi D., and Hall A. (2000) Ras and Rho GTPases: a family reunion. Cell 103, 227–238 10.1016/S0092-8674(00)00115-X [DOI] [PubMed] [Google Scholar]
- 12. Jura N., Scotto-Lavino E., Sobczyk A., and Bar-Sagi D. (2006) Differential modification of Ras proteins by ubiquitination. Mol. Cell 21, 679–687 10.1016/j.molcel.2006.02.011 [DOI] [PubMed] [Google Scholar]
- 13. Xu L., Lubkov V., Taylor L. J., and Bar-Sagi D. (2010) Feedback regulation of Ras signaling by Rabex-5-mediated ubiquitination. Curr. Biol. 20, 1372–1377 10.1016/j.cub.2010.06.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang M. H., Nickerson S., Kim E. T., Liot C., Laurent G., Spang R., Philips M. R., Shan Y., Shaw D. E., Bar-Sagi D., Haigis M. C., and Haigis K. M.. Regulation of RAS oncogenicity by acetylation. Proc. Natl. Acad. Sci. U.S.A. 109, 10843–10848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Henis Y. I., Hancock J. F., and Prior I. A. (2009) Ras acylation, compartmentalization and signaling nanoclusters (Review). Mol. Membr. Biol. 26, 80–92 10.1080/09687680802649582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Herrero A., Pinto A., Colón-Bolea P., Casar B., Jones M., Agudo-Ibáñez L., Vidal R., Tenbaum S. P., Nuciforo P., Valdizán E. M., Horvath Z., Orfi L., Pineda-Lucena A., Bony E., Keri G., et al. (2015) Small molecule inhibition of ERK dimerization prevents tumorigenesis by RAS-ERK pathway oncogenes. Cancer Cell 28, 170–182 10.1016/j.ccell.2015.07.001 [DOI] [PubMed] [Google Scholar]
- 17. LeBleu V. S., Taduri G., O'Connell J., Teng Y., Cooke V. G., Woda C., Sugimoto H., and Kalluri R. (2013) Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 19, 1047–1053 10.1038/nm.3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsai F. D., Lopes M. S., Zhou M., Court H., Ponce O., Fiordalisi J. J., Gierut J. J., Cox A. D., Haigis K. M., and Philips M. R. (2015) K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc. Natl. Acad. Sci. U.S.A. 112, 779–784 10.1073/pnas.1412811112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu B., Swatkoski S., Holly A., Lee L. C., Giroux V., Lee C. S., Hsu D., Smith J. L., Yuen G., Yue J., Ann D. K., Simpson R. M., Creighton C. J., Figg W. D., Gucek M., and Luo J.. Oncogenesis driven by the Ras/Raf pathway requires the SUMO E2 ligase Ubc9. Proc. Natl. Acad. Sci. U.S.A. 112, E1724–E1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Choi B. H., Chen C., Philips M., and Dai W. (2018) RAS GTPases are modified by SUMOylation. Oncotarget 9, 4440–4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodriguez-Viciana P., Warne P. H., Khwaja A., Marte B. M., Pappin D., Das P., Waterfield M. D., Ridley A., and Downward J. (1997) Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell 89, 457–467 10.1016/S0092-8674(00)80226-3 [DOI] [PubMed] [Google Scholar]
- 22. Shih J. Y., and Yang P. C. (2011) The EMT regulator slug and lung carcinogenesis. Carcinogenesis 32, 1299–1304 10.1093/carcin/bgr110 [DOI] [PubMed] [Google Scholar]
- 23. Shen L., Kim S. H., and Chen C. Y. (2012) Sensitization of human pancreatic cancer cells harboring mutated K-ras to apoptosis. PLoS One 7, e40435 10.1371/journal.pone.0040435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mezencev R., Matyunina L. V., Jabbari N., and McDonald J. F. (2016) Snail-induced epithelial-to-mesenchymal transition of MCF-7 breast cancer cells: systems analysis of molecular changes and their effect on radiation and drug sensitivity. BMC Cancer 16, 236 10.1186/s12885-016-2274-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim Y. S., Keyser S. G., and Schneekloth J. S. Jr. (2014) Synthesis of 2′,3′,4′-trihydroxyflavone (2-D08), an inhibitor of protein sumoylation. Bioorg Med Chem. Lett. 24, 1094–1097 10.1016/j.bmcl.2014.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mor A., and Philips M. R. (2006) Compartmentalized Ras/MAPK signaling. Annu. Rev. Immunol. 24, 771–800 10.1146/annurev.immunol.24.021605.090723 [DOI] [PubMed] [Google Scholar]
- 27. Cox A. D., Der C. J., and Philips M. R. (2015) Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin. Cancer Res. 21, 1819–1827 10.1158/1078-0432.CCR-14-3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Athuluri-Divakar S. K., Vasquez-Del Carpio R., Dutta K., Baker S. J., Cosenza S. C., Basu I., Gupta Y. K., Reddy M. V., Ueno L., Hart J. R., Vogt P. K., Mulholland D., Guha C., Aggarwal A. K., and Reddy E. P. (2016) A small molecule RAS-mimetic disrupts RAS association with effector proteins to block signaling. Cell 165, 643–655 10.1016/j.cell.2016.03.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Landré V., Rotblat B., Melino S., Bernassola F., and Melino G. (2014) Screening for E3-ubiquitin ligase inhibitors: challenges and opportunities. Oncotarget 5, 7988–8013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niewerth D., Dingjan I., Cloos J., Jansen G., and Kaspers G. (2013) Proteasome inhibitors in acute leukemia. Expert Rev. Anticancer Ther. 13, 327–337 10.1586/era.13.4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.