

Abstract
Chemokines interact with glycosaminoglycans (GAGs) at the cellular surface and to specific cell-surface receptors to activate signaling pathways. The GAG interaction allows the formation of a chemotactic gradient of chemokine required for cell haptotaxis and chemokine oligomerization. Poxviruses encode secreted chemokine-binding proteins with no sequence similarity to their cellular counterparts to modulate the host immune system. The E163 protein from ectromelia virus, the causative agent of mousepox, binds chemokines through their GAG-binding domain. In addition, E163 interacts with GAGs to be anchored at the cell surface, but its ability to interfere with chemokine-GAG interactions has not been demonstrated. We report the identification of the GAG-binding regions in E163 and the generation of mutant forms deficient of GAG binding. Chemokine binding assays show that some of the E163 GAG-binding sites are also involved in the interaction with chemokines. By using recombinant GAG-binding mutant forms we demonstrate that E163 prevents the interaction of chemokines with cell-surface GAGs, providing mechanisms for the immunomodulatory activity of the viral chemokine-binding protein E163.
Keywords: chemokine, glycosaminoglycan, virus, poxvirus, inflammation, cell migration, immune evasion, viral chemokine binding protein
Introduction
The large DNA viruses, herpesviruses and poxviruses, have evolved many strategies to interfere with the immune system of their hosts (1–3). One of these strategies is the production of cytokine receptors that are secreted from the cell and bind with high affinity the cognate cytokines (4–6). Many of these secreted receptors share amino sequence similarity with cellular receptors, but some of them, such as the viral chemokine-binding proteins (vCKBPs)3 have unique structure and amino acid sequences unrelated to their cognate cytokine receptors (7, 8). This atypical topology is found in a range of poxvirus proteins that interact with immune mediators, and this structure has been named poxvirus immune evasion domain (6, 9). Some viral cytokine receptors interact with glycosaminoglycans (GAGs) as a mechanism to be retained at the cell surface. Some examples are the type I interferon-binding protein encoded by vaccinia virus (VACV) and the interleukin 18-binding protein from molluscum contagiosum virus and variola virus (10–17).
Chemokines are small (8–14 kDa) chemotactic cytokines and can be classified as inflammatory or homeostatic. The inflammatory chemokines can be induced after tissue damage or infection. One of the major roles of chemokines is to promote leukocyte recruitment to sites of infection (18–20). For this reason, the chemokine system is a relevant target for pathogenic viruses. Chemokines are classified in four classes, C, CC, CXC, and CX3C, according to the position of the first two conserved cysteines at the N terminus of the protein (19). The biological activity of chemokines depends of two sequential and essential interactions. First, an interaction with GAGs, and second, binding to cell-surface G protein-coupled receptors (GPCR), with seven transmembrane domains, that activate specific signaling pathways (21). The chemokine-GAG interaction permits the formation of a chemotactic gradient of chemokines required for cell haptotaxis and chemokine oligomerization, which are considered essential for their activity in vivo (22, 23).
The vCKBPs are secreted proteins with no amino acid sequence similarity to cellular proteins (4, 5, 8). These proteins interact with chemokines through their GAG or GPCR-binding domain. Also, some vCKBPs can interact with the cell surface, through their interaction with GAGs, and simultaneously interact with chemokines. A family of poxvirus proteins containing the smallpox virus-encoded chemokine receptor (SECRET) domain interact with a reduced number of CC and CXC chemokines through their GPCR-binding domain (24). The M3 protein from murine gammaherpesvirus 68 (MHV-68) binds C, CC, CXC, and CX3C chemokines through their GAG and GPCR domains (25–27) and it has been shown to displace chemokines previously bound to the cell surface, suggesting that M3 disrupts preformed chemokine gradients (27). Glycoprotein G from alphaherpesviruses infecting animals binds CC and CXC chemokines, and inhibits their activity (28). By contrast, glycoprotein G from herpes simplex virus 1 and 2, relevant human pathogens, binds CC and CXC chemokines through the GAG-binding domain of the chemokines and potentiates their activity (29–31). R17 from herpesvirus Peru interacts with C and CC chemokines through their GPCR domain and bind GAGs simultaneously (32). The poxvirus 35-kDa vCKBP binds through the GPCR-binding domain of the chemokines and abrogates cellular chemotaxis in vitro (33–36). The three-dimensional structure of some vCKBPs, alone or complexed with chemokines was solved and show unique structural foldings not found in host proteins (5, 6, 37–41).
The ectromelia virus (ECTV) E163 vCKBP and its orthologue A41 encoded by VACV bind a reduced set of CC and CXC chemokines with high affinity at the nanomolar range (42, 43). The three-dimensional structure of the A41 protein showed structural similarity to the poxvirus 35-kDa vCKBP and the SECRET domain (5, 6, 42), belonging to the poxvirus immune evasion domain (6, 9). The E163 vCKBP interacts with the GAG-binding region of the chemokines and does not inhibit cellular chemotaxis in vitro. Also, this vCKBP is capable of binding GAGs (heparin, heparan sulfate, chondroitin sulfate A, and chondroitin sulfate B) with different affinities and, consistently, it shows canonical GAG binding sequences (43). The biological consequences of the interaction of E163 with GAGs have not been elucidated and the GAG-binding region of this protein has not been characterized.
We report a further characterization of the interaction of ECTV E163 with GAGs and the construction of variant forms of E163 deficient in their GAG-binding capacity. We also show that E163 blocks the interaction of chemokines with cell-surface GAGs and propose a mechanism of action for this vCKBP.
Results
Expression of E163 protein and mutant versions deficient in GAG binding
The ECTV E163 protein is a GAG-binding protein. Two potential GAG-binding sites (152LKPRDFKT159 and 213RKILKKKF220) were previously identified (43) and we detected a third potential GAG-binding site (141KTKDFMK147) (Fig. 1, A and C). These regions of the E163 protein (A: BXBXXXB; B: BXBXXB; and C: BBXXBBB) closely fit to canonical GAG-binding motifs (BBXB and BBBXXB) (44). To identify GAG-binding sites of this vCKBP we generated six recombinant protein mutants in the GAG-binding regions, named E163-1 to E163-6 (Fig. 1B). Site-directed mutagenesis was performed to replace the basic residues, Arg or Lys, of these GAG-binding regions by A (Table 1), and the protein variants were expressed in the baculovirus system. Purification of a representative mutant protein, E163-2, is shown in Fig. 2A. The Western blot identified a higher molecular size band suggesting that the protein may form dimers (Fig. 2B).
Figure 1.

E163 mutant proteins deficient in putative GAG-binding sites. A, amino acid sequence of ECTV E163 and VACV A41 proteins showing the locations of the putative GAG-binding sites A (residues 141–147), B (residues 152–159), and C (residues 213–220). B, schematic representation of E163 mutant proteins. The putative GAG-binding sites A (red), B (green) and C (purple) are represented. C, three-dimensional structure of A41 VACV. The structure of the A41 (PDB 2VGA) protein was used to indicate the GAG-binding regions of the vCKBP E163. The β-sheet I (blue) and the β-sheet II (red) of the protein are indicated. The latter was proposed as the chemokine-binding region of this vCKBP. The localization of the mutations introduced in region A (yellow), region B (pink), and region C (orange) of the E163 protein are indicated.
Table 1.
Mutagenesis of putative GAG-binding sites in the E163 protein
| Protein | GAG-binding region | Mutations |
|---|---|---|
| E163-1 | 152LKPRDFKT159 (B) | K153A,R155A,K158A |
| E163-2 | 141KTKDFMK147 (A) | K141A,K143A,K147A |
| E163-3 | 213RKILKKKF220 (C) | R213A,K214A,K217A,K218A,K219A |
| E163-4 | A + B | |
| E163-5 | B + C | |
| E163-6 | A + C |
Figure 2.

Expression and purification of the GAG-binding mutant protein E163-2. A, different steps of the purification of recombinant E163-2 protein from supernatants of Hi5 cells infected with a recombinant baculovirus expressing the E163-2 protein. Protein samples were analyzed by SDS-PAGE and Coomassie Blue staining. 1–13, proteins eluted with increasing concentration of imidazole. Molecular size markers (kDa) and the position of the E163-2 protein are indicated. SN, supernatant; C, concentrated; UB, unbound material. B, Western blot of purified E163 and E163-2 proteins developed with a polyclonal anti-E163 antibody. The cowpox virus 35-kDa was used as negative control.
Characterization of the GAG-binding region of E163
We next assessed whether purified recombinant E163 and E163-1 to E163-6 proteins retained the ability to bind GAGs. First, we immobilized heparin onto a BIAcore CM5 sensor chip and tested the binding of E163 and E163-1 to E163-6 to the artificial GAG surface by surface plasmon resonance (SPR) (Fig. 3). The E163 protein bound to heparin, consistent with the previously described high affinity interaction of E163 with heparin (43). However, the E163-1 to E163-6 mutant proteins totally or partially lost their ability to bind heparin. The E163-1 (lacking the B region) and E163-3 (lacking the C region) proteins lost their capacity to bind heparin. The E163-2 protein (lacking the A region), E163-4 (lacking the A and B regions), E163-5 (lacking the B and C regions), and E163-6 (lacking the A and C regions) showed reduced binding to heparin. The double mutant proteins, E163-4, E163-5, and E163-6, showed low association and dissociated slowly. These results indicated that the A, B, and C regions of the E163 vCKBP are involved in the interaction with heparin.
Figure 3.
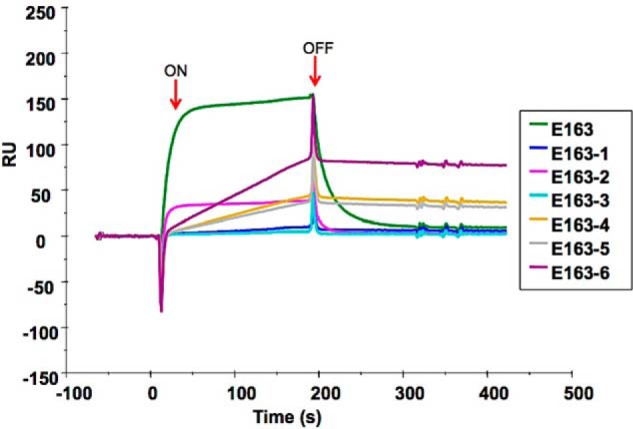
SPR analysis of the binding of WT and mutant E163 proteins to heparin. 55 RU of heparin was immobilized onto a BIAcore CM5 sensor chip. The indicated purified proteins (150 nm) were injected over both flow cells at a rate of 10 μl/min. Arrows indicate the time of the start (ON) and end (OFF) of the injection. The sensorgram shows binding of the E163 recombinant proteins to heparin.
To test the interaction of E163 with more complex GAGs present on the cellular surface we used CHO-K1 cells that express all types of GAGs and the mutant CHO-618 cells that do not express GAGs (45). We demonstrated by flow cytometry analysis that the recombinant E163 protein binds to the surface of CHO-K1 cells (Fig. 4, A and B). The VACV type I interferon-binding protein (B18), known to interact with cell-surface GAGs (10), and the vCKBP MHV-68 M3 (26) were used as positive and negative controls, respectively. We also showed that E163 secreted from ECTV-infected cells bound to CHO-K1 cells, whereas no positive signal was observed with supernatants from cells infected with a mutant virus lacking E163 expression (Fig. S2). Reduced binding to CHO-K1 cells was observed with all mutant E163 proteins, and the results from three independent experiments showed that mutant proteins E163-4 (lacking the A and B regions) and E163-5 (lacking the B and C regions) significantly lost their binding to the cellular surface compared with the WT protein (Fig. 4, A and B). Binding of the proteins to the surface of CHO-618 cells, deficient in GAG expression, showed nonspecific binding to cells, but in this case no significant differences were observed between the WT E163 protein and the mutant proteins (Fig. 4, C and D). Lack of binding of the VACV B18 protein to CHO-618 cells confirmed reduced GAGs at the surface of these cells (Fig. 4C). This experiment confirmed that the A, B, and C regions of the E163 vCKBP are specific GAG-binding sites. In summary, we observed that the B region of the E163 protein is essential for high affinity binding to GAGs but it needs the mutation of the A or C regions simultaneously to lose significant interaction of the protein to the cellular surface, as shown for the E163-4 and E163-5 proteins (Fig. 4B). The E163-6 protein did not lose binding to CHO-K1 cells because the B region was not mutagenized (Fig. 4B).
Figure 4.

Interaction of WT and mutant E163 proteins with the cellular surface. Cells were incubated with 250 nm of the indicated recombinant purified proteins and bound E163 protein was detected with a polyclonal anti-E163 antibody by flow cytometry. A, histograms show representative binding to CHO-K1 cells of the recombinant proteins: E163 (blue), E163-1 to E163-6 (green), negative control without protein (gray), B18 (pink), and M3 (cyan). B, scatter plots show the binding, mean (horizontal lines) and standard deviation (S.D., vertical lines), of the proteins to CHO-K1 cells in each case (n = 3). The asterisk indicates significant differences (p < 0.05) with respect to E163 protein binding. C, histograms show representative binding to CHO-618 cells of the recombinant proteins: E163 (blue), E163-1 to E163-6 (green), negative control without protein (gray), B18 (pink), and M3 (cyan). D, scatter plots show the binding, mean (horizontal lines) and S.D. (vertical lines), of the proteins to CHO-618 cells in each case (n = 3). Data shown are representative of three experiments.
Involvement of the GAG-binding regions of the E163 protein in chemokine binding
The three-dimensional structure of the 35-kDa vCKBP in complex with chemokine has been determined by NMR spectroscopy (41). The residues that are essential for the interaction of the vCKBP with the chemokine are localized at β-sheet II and at the 2–4 loop of the protein. Although the A41-chemokine complex has not been solved by crystallography, the structural modeling based on the crystal structure of the A41 protein suggested that the interaction of A41 and 35-kDa vCKBP with chemokines involves the same regions (42). We carried out a sequence alignment between A41 and E163 proteins (Fig. 1A) to localize the residues mutated in proteins E163-1 to E163-6 in the three-dimensional structure of the A41 protein (Fig. 1C). The B and C regions of the E163 protein are situated closely to the β-sheet I (Fig. 1C). To investigate whether the GAG- and chemokine-binding regions of the E163 proteins are independent or overlap we carried out binding assays by SPR immobilizing E163 and E163-1 to E163-5 proteins onto BIAcore chip and tested murine chemokines (mCxcl12α, mCxcl12β, mCxcl13, mCxcl14, mCxcl16, mCcl2, mCcl21, mCcl24, and mCcl27) that are known ligands of E163 (43) (Fig. 5). We used mCxcl16 and mCcl2 as negative controls. The E163-1 to E163-5 proteins interacted with all these chemokines but may be with different binding affinities (Fig. 5), also indicating that their folding was largely preserved after the introduction of mutations. We also performed kinetic binding analysis between E163 mutant proteins and some chemokines to know whether there are differences in chemokine binding affinity compared with the WT protein (Table 2). Representative examples of the SPR kinetics assays are shown in Fig. S1. We observed that the affinity constant (KD) of all these interactions were in the nanomolar range. Mutations in the A region (E163-2) did not affect the affinity for mCxcl12β and had a minor effect on mCcl21 binding. By contrast, mutations in the B region (E163-1) reduced the binding to the chemokines mCxcl12β and mCxcl13. Simultaneous mutations in the B and C regions (E163-5) reduced the binding affinity of mCxcl12β but had a minor effect on hCCL21 binding. These results suggested that the GAG interacting domain A of E163 does not contribute to chemokine binding, whereas the GAG interacting domains B and C contribute to chemokine binding, but they play a more important role in the binding of the CXC chemokines than that of the CC chemokines tested.
Figure 5.

Chemokine-binding assay of E163 and E163-1 to E163-5 proteins by SPR. Binding of the chemokines indicated at the bottom of the figure was tested on CM4 or CM5 chips coated with E163, and E163-1 to E163-5 proteins. The analytes (100–200 nm) were injected over both flow cells at a rate of 10 μl/min. The association phase (3 min) and the dissociation step (2 min) are shown.
Table 2.
Affinity constants of E163 and E163 mutant proteins for chemokines
| Chemokinea | Protein | Ka | Kd | KD |
|---|---|---|---|---|
| 1/ms | 1/s | nm | ||
| mCxcl12β | E163 | 1.96 × 106 | 4.10 × 10−3 | 2.00 |
| E163-1 | 3.22 × 106 | 9.55 × 10−2 | 29.65 | |
| E163-2 | 4.47 × 105 | 9.00 × 10−4 | 2.00 | |
| E163-5 | 2.41 × 106 | 9.65 × 10−2 | 40.03 | |
| mCcl21 | E163 | 2.22 × 106 | 1.88 × 10−3 | 0.85 |
| E163-2 | 5.30 × 106 | 2.26 × 10−2 | 4.26 | |
| E163 | 4.97 × 105 | 2.60 × 10−3 | 5.24 | |
| mCclx13 | E163-1 | 1.24 × 105 | 5.30 × 10−3 | 42.76 |
| E163 | 9.99 × 105 | 8.74 × 10−4 | 0.87 | |
| hCCL21 | E163-5 | 7.14 × 105 | 1.97 × 10−3 | 2.76 |
a m, mouse.
E163 has the capacity of inhibiting the interaction between chemokines and the cellular surface
E163 interacts with the GAG-binding domain of chemokines and it was proposed to block the binding of chemokines to cell-surface GAGs (43). However, this could not be tested in binding assays of chemokines to cells because E163 itself binds to the GAGs, and the E163-chemokine complex would remain bound to the cell surface in flow cytometry assays. However, the mutant versions of E163 with reduced GAG-binding activity allowed us to address whether E163 prevents the interaction of chemokines with GAGs. With this purpose, we tested the binding of hCCL21, a chemokine recognized by E163 (43), to the cell surface in the absence or presence of E163. First, we confirmed that hCCL21 binds to CHO-K1 cells but does not bind to the GAG-deficient cell line CHO-618 (Fig. 6A). The inhibition of the binding of hCCL21 to cells in the presence of heparin confirmed that interaction between hCCL21 and CHO cells is mediated by GAGs (Fig. 6A). Next, we incubated hCCL21 in the presence of the E163, E163-4, or E163-5 proteins (Fig. 6B). As predicted, when CCL21 was preincubated with WT E163 we did not find any difference with the chemokine alone in terms of CCL21 binding to cells. We observed a reduction in chemokine binding to cells, compared with CCL21 alone, in the presence of E163-4 and E163-5, which was significant in the latter case (Fig. 6, B and C).
Figure 6.

Interaction between CCL21 and the cellular surface in the presence of the E163 vCKBP. A, histograms show representative binding of the control (gray), CCL21 chemokine alone (blue), or CCL21 in the presence of heparin (H + CCL21, pink) to CHO-K1 and CHO-618 cells. B, histograms show representative binding of the control (gray), CCL21 chemokine alone (blue), CCL21 in the presence of the E163 protein (yellow), CCL21 in the presence of the E163-4 protein (green), or CCL21 in the presence of the E163-5 protein (red) to CHO-K1 cells. C, scatter plots show the binding, mean (horizontal line) and S.D. (vertical lines), of the proteins to CHO-K1 cells in each case (n = 5). The asterisk indicates significant differences (p < 0.05) between CCL21 alone and in the presence of E163, E163-4, or E163-5. Data shown are representative of three experiments.
The E163 protein can oligomerize
Western blot analysis suggested that the E163 protein may form dimers (Fig. 2B). To know whether E163 vCKBP can form oligomers we carried out size exclusion chromatography of the purified recombinant E163 protein. The purified protein eluted in two peaks (Fig. 7A). Analysis of the relevant fractions by SDS-PAGE and Western blot with a specific anti-E163 antibody showed that the first peak contained two bands of 33 and 66 kDa, corresponding to monomeric and dimeric forms, whereas the second peak contained a single band of 33 kDa (Fig. 7A). We also immobilized purified recombinant E163 protein onto CM5 chips and tested the binding of purified E163, or 35-kDa vCKBP and BSA as negative controls (Fig. 7B). The interaction of purified E163, but not the negative controls, with E163 protein covalently bound to the CM5 chip confirmed that this vCKBP is capable of interacting with itself.
Figure 7.
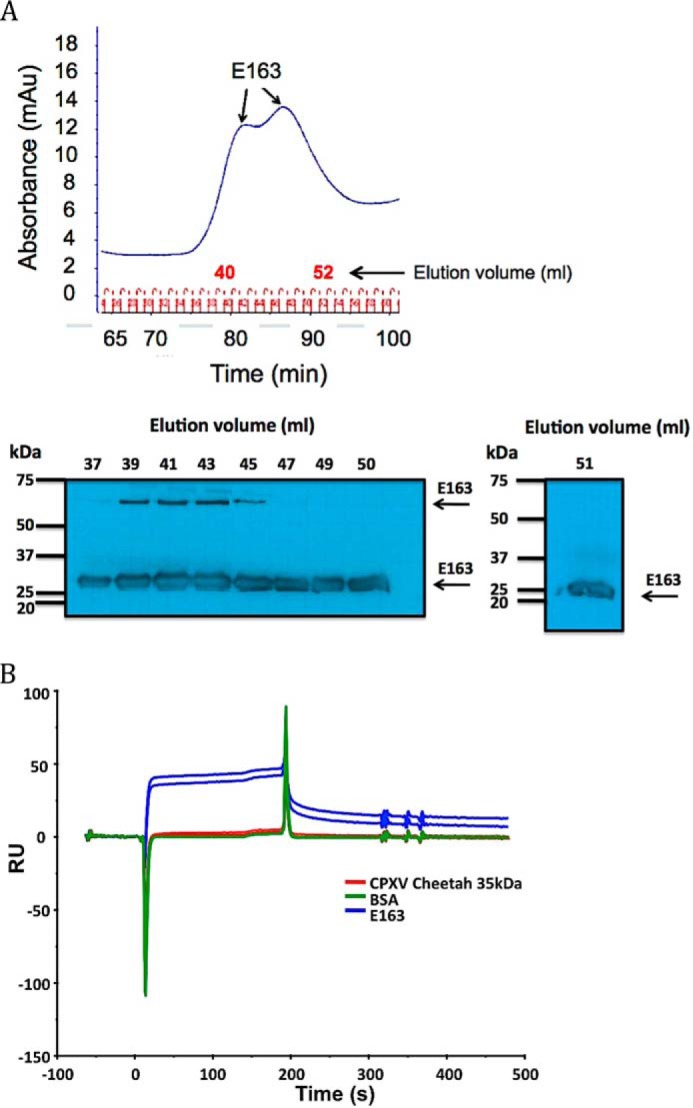
Oligomeric state of purified E163 protein. A, analysis of purified E163 protein by size exclusion chromatography. Recombinant E163 expressed in the baculovirus system and purified with Ni2+-nitrilotriacetic acid columns was analyzed by gel filtration chromatography. The arrows indicate the two peaks in the chromatogram and the elution volume of the fractions is indicated in red. The lower panel shows Western blot analysis with anti-E163 specific antibodies of selected fractions containing E163. The elution volume of the fractions analyzed by Western blot, molecular size in kDa, and arrows marking the position of the E163 protein are indicated. B, homotypic interaction of E163 in SPR-binding assays. The E163 protein was immobilized onto a BIAcore chip and the interaction with 2 different concentrations (250 and 500 nm) of E163 (blue), cowpox virus 35-kDa strain Cheetah (red), and BSA (green) was tested. The flow rate was 10 μl/min and the association (3 min) and the dissociation phases were monitored. A sensorgram is shown.
Discussion
A variety of CKBPs have been identified in viruses and other pathogens (7, 8). These immune modulatory molecules bind chemokines with high affinity but their mechanism of action to modulate chemokine activity is varied. Here we have characterized further the properties of the vCKBP E163 and its mechanism of action.
The interaction of chemokines with GAGs confers to the chemokines the capacity of being immobilized on the cell surface and extracellular matrix to generate a concentration gradient to guide immune cells to sites of inflammation and infection (46, 47). It has been proposed that interruption of the chemokine-GAG interaction may decrease the activity of chemokines in vivo (22, 23). Chemokine mutants lacking GAG binding capacity are able to trigger cellular chemotaxis in vitro but lose chemotactic functions in vivo (22). Transgenic mice carrying a Cxcl12 gene mutation that expresses a CXCL12 that cannot bind to GAGs but interacts with its specific CXCR4 receptor failed to induce both homeostasis and physiological functions in vivo (23).
vCKBPs have distinct mechanisms of action to modulate the chemokine system. Some vCKBPs, such as the VACV 35-kDa protein, prevent the interaction of chemokines with specific chemokine receptors and consequently prevent the activation of chemokine-mediated intracellular pathways and cell migration (33). Alternatively, vCKBPs, such as the VACV A41 protein, interact with the GAG-binding domain of the chemokine and do not inhibit chemokine-induced cell migration in vitro. It was proposed that these vCKBPs block the presentation of the chemokine at the cell surface and the formation of a chemokine gradient required for their function in vivo (42, 43). This hypothesis was supported by the ability of VACV A41 to modulate lymphocyte migration to sites of infection in animal models (48). MHV-68 M3 protein has been shown to have both functions, preventing the binding of chemokines to cell receptors and the interaction of chemokines with GAGs in vitro (25, 27).
In addition, there are vCKBPs, such as ECTV E163 and the 35-kDa homologue (M-T1) from myxoma virus, that simultaneously bind chemokines and GAGs by itself (43, 49). This would retain the secreted vCKBP in the vicinity of the infected tissues and may also interfere with the interaction of chemokines with GAGs. GAG-mediated interaction with the cell surface has been described for other poxvirus cytokine decoy receptors, such as binding proteins for type 1 interferon and interleukin 18 (10, 13, 14).
The GAG-binding regions of ECTV E163 or its orthologue VACV A41 had not been solved to date and the functional implications of this interaction remained unclear. We have identified here three GAG-binding regions in the E163 protein: 141KTKDFMK147 (A), 152LKPRDFKT159 (B), and 213RKILKKKF220 (C). The discrepancies between the results of the binding to heparin by SPR and binding to cells may reflect the expression of complex GAGs at the surface of the cell, also expressing heparan sulfate or chondroitin sulfate. Also, characterization of the interaction of the E163 mutants to the cell surface is closer to the physiological conditions that occur in the infected host. Mutations in regions A+B or B+C had a major effect on cell binding, whereas the mutation of regions B or C alone was sufficient to lose binding to heparin. Cardin and Weintraub (44) defined canonical sequences involved in GAG binding, and other factors such as the existence of an Arg residue near these canonical sequences or secondary structure can influence the affinity (50). The B and C regions of E163 include an Arg residue that may contribute to the heparin binding that is lost after mutagenesis, which is not the case of the A region. Mutation of the B region was necessary, together with simultaneous mutations in either region A or region C, to abrogate binding to cells. Accordingly, simultaneous mutations of the A and C regions were not sufficient to prevent E163 binding to cells. These results suggest that the A, B, and C regions work cooperatively to interact with GAGs on the cellular surface.
These GAG-binding regions are located in areas of the E163 protein exposed and available to interact with other proteins or components (Fig. 1C). As described for the 35-kDa vCKBP, the ECTV E163 orthologue in VACV has been proposed to interact with chemokines through the β-sheet II (41, 42). We show that mutations in the GAG-binding regions do not abrogate chemokine binding but may affect the chemokine binding affinity.
Mutation of the A region had a minor effect on chemokine binding, which is consistent with its location far from both β-sheets of E163 and suggests that this region does not contribute to chemokine binding. By contrast, mutations in the B and C regions reduced the binding affinity for CXC chemokines. Since these regions are close to β-sheet I, our results suggest that these sites may contribute to some extent to chemokine binding or that mutations in these regions may affect the overall protein folding and the structure of β-sheet II, the proposed chemokine-binding domain of E163. It should be noticed that mutations in the B and C regions had a minor effect on the interaction of hCCL21, suggesting that the interaction of CXC chemokines with E163 may be different to that of CC chemokines.
The E163 protein may form oligomers, likely dimers of 66 kDa. The Western blot results suggest that dimers may be formed through disulfide bonds. The crystal structure of VACV A41 showed that the 9 cysteine residues present in the protein were involved in the formation of 4 intramolecular disulfide bonds, leaving one cysteine residue available for the potential formation of an intermolecular bond to generate a dimer (42). Whether or not disulfide bonds are involved, SPR analysis showed that E163 interacts with itself with the potential to form larger complexes. Dimerization of the herpesvirus M3 protein is required for its correct function (37). The oligomerization of 35-kDa vCKBP homologues encoded by orf virus or myxoma virus (M-T1) has been demonstrated or suggested, but whether this is required for chemokine binding has not been addressed (8, 49).
The interaction of E163 with the GAG-binding domain of chemokines was demonstrated previously, but the ability of E163 to block the interaction of chemokines with GAGs has not been shown before. This was difficult to demonstrate because E163 also interacts with GAGs and the E163-chemokine complex would remain bound to the cell surface. We took advantage of mutant forms of E163 that do not interact with GAGs to show that the protein blocks the interaction of chemokines with GAGs at the surface of cells. Structural predictions suggest that E163 should be able to bind GAGs and chemokines simultaneously, as demonstrated for myxoma virus M-T1 (49). The fact that a GAG-binding mutant E163, but not WT E163, inhibits the binding of CCL21 to cells supports the notion that E163 simultaneously interacts with chemokines while bound to the cell surface through GAGs.
The results presented in this report suggest that ECTV E163 interacts with the GAG-binding domain of chemokines and prevents the attachment of chemokines to GAGs at the cell surface and the formation of a chemokine gradient. E163 itself interacts with GAGs and the E163-chemokine complex is retained at the cell surface. In this way E163 may disrupt the correct presentation of chemokines to leukocytes, maybe by changing the three-dimensional structure of the chemokine or preventing the formation of larger, active chemokine clusters on the surface of cells, which have been proposed to be formed as a result of their interaction with cell-surface GAGs (51). Alternatively, as suggested for the TSG-6 human CKBP, E163 may inhibit the transport of chemokines across the endothelial cell monolayer to be presented to the leukocytes (52, 53). The important role of VACV A41 in virulence (48, 54) suggests that the ability of E163 and its A41 orthologue to sequester chemokines and alter their structure to allow these viruses to modulate the host immune response.
Experimental procedures
Cells, reagents, and viruses
Chinese hamster ovary K1 (CHO-K1) and proteoglycan-deficient variant CHO-618 cells (CHO-618) were kindly provided by Dr. F. Arenzana-Seisdedos (Institute Pasteur, Paris, France) and were grown in 10% fetal calf serum (FCS) containing Dulbecco's modified Eagle's medium (Gibco)/Ham's F-12 (1:1) (Gibco) medium. Hi5 insect cells were cultured in TC-100 medium supplemented with 10% FCS. BSC-1 cells were grown in medium supplemented with 2.5% FCS. Recombinant baculoviruses were grown in adherent Hi5 insect cells in 10% FCS TC-100 medium or suspension Hi5 cells maintained in Express Five (Life Technologies) medium for the generation of recombinant baculoviruses or protein expression, respectively. Recombinant cytokines were obtained from R&D Systems and Peprotech Inc. Biotin-conjugated heparin (from bovine intestinal mucosa) was from Calbiochem. ECTV strain Naval was grown in BSC-1 cells (55). A mutant ECTV lacking the EVN163 gene (E163ΔECTV) was used as a control and will be described elsewhere.4
Cloning and expression of recombinant proteins
To generate plasmid pHH01 for the expression of a tagged E163-C protein, the sequences coding for amino acid residues 23–223 from the EVN163 gene from ECTV strain Naval (55) were amplified by PCR using primers containing EcoRI and XhoI restriction sites at the 5′ and 3′ termini, respectively. The PCR-amplified fragment was cloned into pAL7 (14), a pFastBac1 vector bearing the honeybee melittin signal peptide at the 5′ termini and a C-terminal V5-His6 tag. Plasmids pHH02 to pHH07, bearing mutations in the EVN163 gene, were engineered by using the QuikChange II site-directed mutagenesis kit (Stratagene, Cedar Creek, TX). Plasmids were sequenced to confirm the presence of the desired mutations and the absence of unwanted mutations. The resulting constructions were named pHH01 (E163), pHH02 (E163-1), pHH03 (E163-2), pHH04 (E163-3), pHH05 (E163-4), pHH06 (E163-5), and pHH07 (E163-6) (Table 1). Recombinant baculoviruses were generated using the Bac-to-Bac system (Life Technologies) (24). Viral stocks were amplified by infecting Hi5 cells at low multiplicity of infection (0.1–0.01 pfu/cell) to obtain high-titer baculovirus stocks for protein production. We also used as controls recombinant proteins B18 from VACV (56), M3 from murine gammaherpesvirus 68 (MHV-68) (26), and the 35-kDa vCKBP from cowpox virus strain Cheetah (33).
Protein expression and purification
Recombinant His-tagged proteins were purified with Ni2+-nitrilotriacetic acid columns (Qiagen) from supernatants of Hi5 cells infected at high multiplicity of infection (43). Protein stocks were dialyzed against phosphate-buffered saline (PBS), quantified by BCA assay (Pierce Biotechnology) and gel densitometry, and stored at −80 °C. The purified protein was used to generate a polyclonal rabbit anti-E163 serum. The analysis of the molecular size of the purified proteins was done in an AKTA-FLP purifier (GE Healthcare, Sweden). We detected the protein at 280 nm of absorbance and used molecular mass standards of 12–200 kDa (Sigma). The fractions containing E163 were concentrated using a 10-kDa cutoff ultracentrifuge tube and further purified by gel filtration chromatography through a SuperdexTM 200 HiLoad 16/60 prepacked column in 0.02 m Hepes buffer (pH 7.0) containing 0.15 m NaCl and 0.001% sodium azide. The purified E163 protein was tested by SDS-PAGE (SDS-PAGE) and Western blotting.
Characterization of protein-protein interactions by SPR
Chemokine binding and affinity constants were determined by SPR using a BIAcore X100 biosensor (GE Healthcare). For ligand screening experiments, purified recombinant E163, E163-1 to E163-5 proteins were amine coupled to a CM5 or CM4 sensor chips (BIAcore, Inc.) to a level of ∼300–800 response units (RU) (300–800 pg/mm2) as described (24). Recombinant chemokines were injected at 100–200 nm into HBS-EP buffer (10 mm Hepes, 150 mm NaCl, 3 mm EDTA, 0.005% (v/v) surfactant P20 (pH 7.4)) at a flow rate of 10 μl/min during 3 min and association and dissociation were monitored. The surface was regenerated after each injection by using 10 mm glycine-HCl (pH 2.0). For kinetic analysis, the recombinant protein was immobilized at a low density (Rmax, 200 RU) to minimize the effects of mass transfer. We used different concentrations of the corresponding chemokine at a flow rate of 30 μl/min over a 2-min period and allowed to dissociate for an additional 5–15 min. Bulk refractive index changes were removed by subtracting the reference flow cell responses and the average response to a blank injection was subtracted from all analyte sensorgrams. Five different concentrations at single cycle kinetics assays and at least 7 different concentrations at multiple cycle kinetics of cytokines (in the range 2–100 nm) were injected at 30 μl/min in HBS-EP and the collected sensorgrams were aligned and fitted to a 1:1 Langmuir model using the Biacore X100 Evaluation software. The kinetic constants were determined from fittings containing no less than 5 concentration sensorgrams.
Heparin binding analysis by SPR
We generated an artificial GAG surface onto CM5 sensor chips (BIAcore, Inc.). First, 2000 RU of streptavidin (Sigma) was immobilized by amine coupling in 10 mm acetate (pH 4.0). Then, we added 1 m ethanolamine (pH 8.5) and later passed 5 μg/ml of biotinylated heparin diluted in 300 mm NaCl HBS-EP over one of the two flow cells, the other flow cell was used as specific binding control. Then we captured ∼55 RU of biotinylated heparin. E163, E163-1 to E163-6 proteins were injected at a concentration of 150 nm over both flow cells at a rate of 30 μl/min. Following the association phase, HBS-EP was allowed to flow over both cells to monitor the dissociation phase. The surface was regenerated with 1 m NaCl during 1 min. All BIAcore sensorgrams were analyzed using BIAcore X100 Evaluation software. Bulk refractive index changes were removed by subtracting the reference flow cell responses and the average response of a blank injection was subtracted from all analyte sensorgrams to remove systematic artifacts.
Sequence alignments and three-dimensional structure
Sequence alignments of A41 from VACV strain Western Reserve (GenBank accession number YP_233082) and E163 from ECTV strain Naval (EVN163) (55) were performed with ClustalW and ESPript 3.0 Bioinformatics tools to represent the three-dimensional structure of the A41 protein using PyMOL Bioinformatics tools (Delano Scientific LLC).
Analysis of protein binding to cells by flow cytometry
CHO-K1 and CHO-618 adherent cells were harvested with 2 mm EDTA in PBS and incubated on ice with recombinants proteins (E163, E163-1 to E163-6, M3 and B18) for 30 min in a total volume of 50 μl, in triplicate. To analyze the interaction of the E163 protein naturally secreted from infected cells, 100 μl of supernatant was incubated with CHO-K1 cells during 1 h at 4 °C, in quadruplicate. Supernatants were harvested from BSC-1 cell cultures infected at low multiplicity of infection (0.05 pfu/cell) with ECTV or E163ΔECTV, inactivated with psoralen and UV treatment (57), and concentrated 35 times with Vivaspin 500 centrifugal concentrators (Sartorius) with 10-kDa cut-off. After incubation, cells were washed with 1% bovine serum albumin (BSA) and 1% FCS in PBS, and protein binding was assessed by flow cytometry using a polyclonal rabbit anti-E163 antibody followed by Alexa Fluor 488 anti-rabbit IgG secondary antibody. The data were collected on a FACSCalibur (BD Biosciences, San Jose, CA) and analyzed using FlowJo 8.8.6 software (Tree star, Ashland, OR).
Chemokine binding to cells by flow cytometry
To analyze the capacity of E163 to interfere with the interaction between CCL21 and CHO-K1 or mutant CHO-618 cells, we used the biotinylated human CCL21/6Ckine kit (R&D Systems Inc.) following the manufacturer's instructions. Biotinylated CCL21 was incubated with the cell suspension in the absence or presence of 500 nm E163, E163-4, E163-5 or control biotinylated protein at the same time as the chemokine, in quintuplicate. To show specificity of binding of the chemokine to GAGs of the cellular surface, excess heparin (1 mg/ml; Sigma) was added to the cells at the same time as the labeled chemokine.
Statistics analysis
Statistical analysis we performed with IBM SPSS Statistics 21 (IBM Software). We used the Student's t test for independent samples to evaluate the significance differences (p value<0.05).
Author contributions
H. H. and A. A. formal analysis; H. H. and A. A. investigation; H. H. and A. A. methodology; H. H. writing-original draft; H. H. and A. A. writing-review and editing; A. A. conceptualization; A. A. supervision; A. A. funding acquisition; A. A. validation; A. A. project administration.
Supplementary Material
Acknowledgments
We thank J. M. Alonso-Lobo for helpful discussions and Rocío Martín, M. Carmen Férnandez, and Carolina Sánchez for excellent technical support.
This work was supported Spanish Ministry of Economy and Competitiveness and European Union (European Regional Development's Funds, FEDER) Grants SAF2012-38957 and SAF2015-67485-R. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1 and S2.
H. Heidarieh and A. Alcami, unpublished results.
- vCKBP
- viral chemokine-binding proteins
- ECTV
- ectromelia virus
- E163ΔECTV
- ECTV lacking the EVN163 gene
- FCS
- fetal calf serum
- GAGs
- glycominoglycans
- MHV-68
- murine gammaherpesvirus 68
- RU
- response units
- SECRET
- smallpox virus-encoded chemokine receptor
- GPCR
- G protein-coupled receptors
- SPR
- surface plasmon resonance
- VACV
- vaccinia virus
- HBS
- Hepes-buffered saline.
References
- 1. Finlay B. B., and McFadden G. (2006) Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 124, 767–782 10.1016/j.cell.2006.01.034 [DOI] [PubMed] [Google Scholar]
- 2. Seet B. T., Johnston J. B., Brunetti C. R., Barrett J. W., Everett H., Cameron C., Sypula J., Nazarian S. H., Lucas A., and McFadden G. (2003) Poxviruses and immune evasion. Annu. Rev. Immunol. 21, 377–423 10.1146/annurev.immunol.21.120601.141049 [DOI] [PubMed] [Google Scholar]
- 3. Smith G. L., Benfield C. T., Maluquer de Motes C., Mazzon M., Ember S. W., Ferguson B. J., and Sumner R. P. (2013) Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J. Gen. Virol. 94, 2367–2392 10.1099/vir.0.055921-0 [DOI] [PubMed] [Google Scholar]
- 4. Alcami A. (2003) Viral mimicry of cytokines, chemokines and their receptors. Nat. Rev. Immunol. 3, 36–50 10.1038/nri980 [DOI] [PubMed] [Google Scholar]
- 5. Epperson M. L., Lee C. A., and Fremont D. H. (2012) Subversion of cytokine networks by virally encoded decoy receptors. Immunol. Rev. 250, 199–215 10.1111/imr.12009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Felix J., and Savvides S. N. (2017) Mechanisms of immunomodulation by mammalian and viral decoy receptors: insights from structures. Nat. Rev. Immunol. 17, 112–129 10.1038/nri.2016.134 [DOI] [PubMed] [Google Scholar]
- 7. González-Motos V., Kropp K. A., and Viejo-Borbolla A. (2016) Chemokine binding proteins: an immunomodulatory strategy going viral. Cytokine Growth Factor Rev. 30, 71–80 10.1016/j.cytogfr.2016.02.007 [DOI] [PubMed] [Google Scholar]
- 8. Heidarieh H., Hernáez B., and Alcamí A. (2015) Immune modulation by virus-encoded secreted chemokine binding proteins. Virus Res. 209, 67–75 10.1016/j.virusres.2015.02.028 [DOI] [PubMed] [Google Scholar]
- 9. Nelson C. A., Epperson M. L., Singh S., Elliott J. I., and Fremont D. H. (2015) Structural conservation and functional diversity of the poxvirus immune evasion (PIE) domain superfamily. Viruses 7, 4878–4898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alcamí A., Symons J. A., and Smith G. L. (2000) The vaccinia virus soluble α/β interferon (IFN) receptor binds to the cell surface and protects cells from the antiviral effects of IFN. J. Virol. 74, 11230–11239 10.1128/JVI.74.23.11230-11239.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Born T. L., Morrison L. A., Esteban D. J., VandenBos T., Thebeau L. G., Chen N., Spriggs M. K., Sims J. E., and Buller R. M. (2000) A poxvirus protein that binds to and inactivates IL-18, and inhibits NK cell response. J. Immunol. 164, 3246–3254 10.4049/jimmunol.164.6.3246 [DOI] [PubMed] [Google Scholar]
- 12. Born T. L., Smith D. E., Garka K. E., Renshaw B. R., Bertles J. S., and Sims J. E. (2000) Identification and characterization of two members of a novel class of the interleukin-1 receptor (IL-1R) family: delineation of a new class of IL-1R-related proteins based on signaling. J. Biol. Chem. 275, 29946–29954 10.1074/jbc.M004077200 [DOI] [PubMed] [Google Scholar]
- 13. Esteban D. J., Nuara A. A., and Buller R. M. (2004) Interleukin-18 and glycosaminoglycan binding by a protein encoded by Variola virus. J. Gen. Virol. 85, 1291–1299 10.1099/vir.0.79902-0 [DOI] [PubMed] [Google Scholar]
- 14. Montanuy I., Alejo A., and Alcami A. (2011) Glycosaminoglycans mediate retention of the poxvirus type I interferon binding protein at the cell surface to locally block interferon antiviral responses. FASEB J. 25, 1960–1971 10.1096/fj.10-177188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smith V. P., and Alcami A. (2000) Expression of secreted cytokine and chemokine inhibitors by ectromelia virus. J. Virol. 74, 8460–8471 10.1128/JVI.74.18.8460-8471.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Symons J. A., Alcamí A., and Smith G. L. (1995) Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell 81, 551–560 10.1016/0092-8674(95)90076-4 [DOI] [PubMed] [Google Scholar]
- 17. Xiang Y., and Moss B. (2003) Molluscum contagiosum virus interleukin-18 (IL-18) binding protein is secreted as a full-length form that binds cell surface glycosaminoglycans through the C-terminal tail and a furin-cleaved form with only the IL-18 binding domain. J. Virol. 77, 2623–2630 10.1128/JVI.77.4.2623-2630.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lau E. K., Allen S., Hsu A. R., and Handel T. M. (2004) Chemokine-receptor interactions: GPCRs, glycosaminoglycans and viral chemokine binding proteins. Adv. Protein Chem. 68, 351–391 10.1016/S0065-3233(04)68010-7 [DOI] [PubMed] [Google Scholar]
- 19. Zlotnik A., and Yoshie O. (2000) Chemokines: a new classification system and their role in immunity. Immunity 12, 121–127 10.1016/S1074-7613(00)80165-X [DOI] [PubMed] [Google Scholar]
- 20. Zlotnik A., Yoshie O., and Nomiyama H. (2006) The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 7, 243 10.1186/gb-2006-7-12-243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mellado M., Rodríguez-Frade J. M., Mañes S., and Martínez A. C. (2001) Chemokine signaling and functional responses: the role of receptor dimerization and TK pathway activation. Annu. Rev. Immunol. 19, 397–421 10.1146/annurev.immunol.19.1.397 [DOI] [PubMed] [Google Scholar]
- 22. Proudfoot A. E., Handel T. M., Johnson Z., Lau E. K., LiWang P., Clark-Lewis I., Borlat F., Wells T. N., and Kosco-Vilbois M. H. (2003) Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. U.S.A. 100, 1885–1890 10.1073/pnas.0334864100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rueda P., Richart A., Récalde A., Gasse P., Vilar J., Guérin C., Lortat-Jacob H., Vieira P., Baleux F., Chretien F., Arenzana-Seisdedos F., and Silvestre J. S. (2012) Homeostatic and tissue reparation defaults in mice carrying selective genetic invalidation of CXCL12/proteoglycan interactions. Circulation 126, 1882–1895 10.1161/CIRCULATIONAHA.112.113290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alejo A., Ruiz-Argüello M. B., Ho Y., Smith V. P., Saraiva M., and Alcami A. (2006) A chemokine-binding domain in the tumor necrosis factor receptor from variola (smallpox) virus. Proc. Natl. Acad. Sci. U.S.A. 103, 5995–6000 10.1073/pnas.0510462103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alexander-Brett J. M., and Fremont D. H. (2007) Dual GPCR and GAG mimicry by the M3 chemokine decoy receptor. J. Exp. Med. 204, 3157–3172 10.1084/jem.20071677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parry C. M., Simas J. P., Smith V. P., Stewart C. A., Minson A. C., Efstathiou S., and Alcami A. (2000) A broad spectrum secreted chemokine binding protein encoded by a herpesvirus. J. Exp. Med. 191, 573–578 10.1084/jem.191.3.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Webb L. M., Smith V. P., and Alcami A. (2004) The gammaherpesvirus chemokine binding protein can inhibit the interaction of chemokines with glycosaminoglycans. FASEB J. 18, 571–573 10.1096/fj.03-0485fje [DOI] [PubMed] [Google Scholar]
- 28. Bryant N. A., Davis-Poynter N., Vanderplasschen A., and Alcami A. (2003) Glycoprotein G isoforms from some alphaherpesviruses function as broad-spectrum chemokine binding proteins. EMBO J. 22, 833–846 10.1093/emboj/cdg092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Martínez-Martín N., Viejo-Borbolla A., and Alcami A. (2016) Herpes simplex virus particles interact with chemokines and enhance cell migration. J. Gen. Virol. 97, 3007–3016 10.1099/jgv.0.000616 [DOI] [PubMed] [Google Scholar]
- 30. Martinez-Martin N., Viejo-Borbolla A., Martín R., Blanco S., Benovic J. L., Thelen M., and Alcamí A. (2015) Herpes simplex virus enhances chemokine function through modulation of receptor trafficking and oligomerization. Nat. Commun. 6, 6163 10.1038/ncomms7163 [DOI] [PubMed] [Google Scholar]
- 31. Viejo-Borbolla A., Martinez-Martín N., Nel H. J., Rueda P., Martin R., Blanco S., Arenzana-Seisdedos F., Thelen M., Fallon P. G., and Alcamí A. (2012) Enhancement of chemokine function as an immunomodulatory strategy employed by human herpesviruses. PLoS Pathog. 8, e1002497 10.1371/journal.ppat.1002497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lubman O. Y., Cella M., Wang X., Monte K., Lenschow D. J., Huang Y. H., and Fremont D. H. (2014) Rodent herpesvirus Peru encodes a secreted chemokine decoy receptor. J. Virol. 88, 538–546 10.1128/JVI.02729-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alcamí A., Symons J. A., Collins P. D., Williams T. J., and Smith G. L. (1998) Blockade of chemokine activity by a soluble chemokine binding protein from vaccinia virus. J. Immunol. 160, 624–633 [PubMed] [Google Scholar]
- 34. Graham K. A., Lalani A. S., Macen J. L., Ness T. L., Barry M., Liu L. Y., Lucas A., Clark-Lewis I., Moyer R. W., and McFadden G. (1997) The T1/35kDa family of poxvirus-secreted proteins bind chemokines and modulate leukocyte influx into virus-infected tissues. Virology 229, 12–24 10.1006/viro.1996.8423 [DOI] [PubMed] [Google Scholar]
- 35. Lalani A. S., Ness T. L., Singh R., Harrison J. K., Seet B. T., Kelvin D. J., McFadden G., and Moyer R. W. (1998) Functional comparisons among members of the poxvirus T1/35kDa family of soluble CC-chemokine inhibitor glycoproteins. Virology 250, 173–184 10.1006/viro.1998.9340 [DOI] [PubMed] [Google Scholar]
- 36. Smith C. A., Smith T. D., Smolak P. J., Friend D., Hagen H., Gerhart M., Park L., Pickup D. J., Torrance D., Mohler K., Schooley K., and Goodwin R. G. (1997) Poxvirus genomes encode a secreted, soluble protein that preferentially inhibits β chemokine activity yet lacks sequence homology to known chemokine receptors. Virology 236, 316–327 10.1006/viro.1997.8730 [DOI] [PubMed] [Google Scholar]
- 37. Alexander J. M., Nelson C. A., van Berkel V., Lau E. K., Studts J. M., Brett T. J., Speck S. H., Handel T. M., Virgin H. W., and Fremont D. H. (2002) Structural basis of chemokine sequestration by a herpesvirus decoy receptor. Cell 111, 343–356 10.1016/S0092-8674(02)01007-3 [DOI] [PubMed] [Google Scholar]
- 38. Arnold P. L., and Fremont D. H. (2006) Structural determinants of chemokine binding by an ectromelia virus-encoded decoy receptor. J. Virol. 80, 7439–7449 10.1128/JVI.00576-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Carfí A., Smith C. A., Smolak P. J., McGrew J., and Wiley D. C. (1999) Structure of a soluble secreted chemokine inhibitor vCCI (p35) from cowpox virus. Proc. Natl. Acad. Sci. U.S.A. 96, 12379–12383 10.1073/pnas.96.22.12379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xue X., Lu Q., Wei H., Wang D., Chen D., He G., Huang L., Wang H., and Wang X. (2011) Structural basis of chemokine sequestration by CrmD, a poxvirus-encoded tumor necrosis factor receptor. PLoS Pathog. 7, e1002162 10.1371/journal.ppat.1002162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang L., Derider M., McCornack M. A., Jao S. C., Isern N., Ness T., Moyer R., and LiWang P. J. (2006) Solution structure of the complex between poxvirus-encoded CC chemokine inhibitor vCCI and human MIP-1β. Proc. Natl. Acad. Sci. U.S.A. 103, 13985–13990 10.1073/pnas.0602142103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bahar M. W., Kenyon J. C., Putz M. M., Abrescia N. G., Pease J. E., Wise E. L., Stuart D. I., Smith G. L., and Grimes J. M. (2008) Structure and function of A41, a vaccinia virus chemokine binding protein. PLoS Pathog. 4, e5 10.1371/journal.ppat.0040005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ruiz-Argüello M. B., Smith V. P., Campanella G. S., Baleux F., Arenzana-Seisdedos F., Luster A. D., and Alcami A. (2008) An ectromelia virus protein that interacts with chemokines through their glycosaminoglycan binding domain. J. Virol. 82, 917–926 10.1128/JVI.02111-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cardin A. D., and Weintraub H. J. (1989) Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 9, 21–32 10.1161/01.ATV.9.1.21 [DOI] [PubMed] [Google Scholar]
- 45. Zhang L., Lawrence R., Frazier B. A., and Esko J. D. (2006) CHO glycosylation mutants: proteoglycans. Methods Enzymol. 416, 205–221 10.1016/S0076-6879(06)16013-9 [DOI] [PubMed] [Google Scholar]
- 46. Handel T. M., Johnson Z., Crown S. E., Lau E. K., and Proudfoot A. E. (2005) Regulation of protein function by glycosaminoglycans: as exemplified by chemokines. Annu. Rev. Biochem. 74, 385–410 10.1146/annurev.biochem.72.121801.161747 [DOI] [PubMed] [Google Scholar]
- 47. Laguri C., Arenzana-Seisdedos F., and Lortat-Jacob H. (2008) Relationships between glycosaminoglycan and receptor binding sites in chemokines-the CXCL12 example. Carbohydr. Res. 343, 2018–2023 10.1016/j.carres.2008.01.047 [DOI] [PubMed] [Google Scholar]
- 48. Ng A., Tscharke D. C., Reading P. C., and Smith G. L. (2001) The vaccinia virus A41L protein is a soluble 30 kDa glycoprotein that affects virus virulence. J. Gen. Virol. 82, 2095–2105 10.1099/0022-1317-82-9-2095 [DOI] [PubMed] [Google Scholar]
- 49. Seet B. T., Barrett J., Robichaud J., Shilton B., Singh R., and McFadden G. (2001) Glycosaminoglycan binding properties of the myxoma virus CC-chemokine inhibitor, M-T1. J. Biol. Chem. 276, 30504–30513 10.1074/jbc.M011401200 [DOI] [PubMed] [Google Scholar]
- 50. Hileman R. E., Fromm J. R., Weiler J. M., and Linhardt R. J. (1998) Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. Bioessays 20, 156–167 10.1002/(SICI)1521-1878(199802)20:2%3C156::AID-BIES8%3E3.0.CO%3B2-R [DOI] [PubMed] [Google Scholar]
- 51. Proudfoot A. E. I., Johnson Z., Bonvin P., and Handel T. M. (2017) Glycosaminoglycan interactions with chemokines add complexity to a complex system. Pharmaceuticals (Basel) 10, pii: E70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dyer D. P., Salanga C. L., Johns S. C., Valdambrini E., Fuster M. M., Milner C. M., Day A. J., and Handel T. M. (2016) The anti-inflammatory protein TSG-6 regulates chemokine function by inhibiting chemokine/glycosaminoglycan interactions. J. Biol. Chem. 291, 12627–12640 10.1074/jbc.M116.720953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dyer D. P., Thomson J. M., Hermant A., Jowitt T. A., Handel T. M., Proudfoot A. E., Day A. J., and Milner C. M. (2014) TSG-6 inhibits neutrophil migration via direct interaction with the chemokine CXCL8. J. Immunol. 192, 2177–2185 10.4049/jimmunol.1300194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Clark R. H., Kenyon J. C., Bartlett N. W., Tscharke D. C., and Smith G. L. (2006) Deletion of gene A41L enhances vaccinia virus immunogenicity and vaccine efficacy. J. Gen. Virol. 87, 29–38 10.1099/vir.0.81417-0 [DOI] [PubMed] [Google Scholar]
- 55. Mavian C., Lopez-Bueno A., Bryant N. A., Seeger K., Quail M. A., Harris D., Barrell B., and Alcami A. (2014) The genome sequence of ectromelia virus Naval and Cornell isolates from outbreaks in North America. Virology 462–463, 218–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fernández de Marco Mdel M., Alejo A., Hudson P., Damon I. K., and Alcami A. (2010) The highly virulent variola and monkeypox viruses express secreted inhibitors of type I interferon. FASEB J. 24, 1479–1488 10.1096/fj.09-144733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tsung K., Yim J. H., Marti W., Buller R. M., and Norton J. A. (1996) Gene expression and cytopathic effect of vaccinia virus inactivated by psoralen and long-wave UV light. J. Virol. 70, 165–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.