

Abstract
A sensitization of inositol 1,4,5-trisphosphate receptor (IP3R)–mediated Ca2+ release is associated with oxidative stress in multiple cell types. These effects are thought to be mediated by alterations in the redox state of critical thiols in the IP3R, but this has not been directly demonstrated in intact cells. Here, we utilized a combination of gel-shift assays with MPEG-maleimides and LC–MS/MS to monitor the redox state of recombinant IP3R1 expressed in HEK293 cells. We found that under basal conditions, ∼5 of the 60 cysteines are oxidized in IP3R1. Cell treatment with 50 μm thimerosal altered gel shifts, indicating oxidation of ∼20 cysteines. By contrast, the shifts induced by 0.5 mm H2O2 or other oxidants were much smaller. Monitoring of biotin–maleimide attachment to IP3R1 by LC–MS/MS with 71% coverage of the receptor sequence revealed modification of two cytosolic (Cys-292 and Cys-1415) and two intraluminal cysteines (Cys-2496 and Cys-2533) under basal conditions. The thimerosal treatment modified an additional eleven cysteines, but only three (Cys-206, Cys-767, and Cys-1459) were consistently oxidized in multiple experiments. H2O2 also oxidized Cys-206 and additionally oxidized two residues not modified by thimerosal (Cys-214 and Cys-1397). Potentiation of IP3R channel function by oxidants was measured with cysteine variants transfected into a HEK293 IP3R triple-knockout cell line, indicating that the functionally relevant redox-sensitive cysteines are predominantly clustered within the N-terminal suppressor domain of IP3R. To our knowledge, this study is the first that has used proteomic methods to assess the redox state of individual thiols in IP3R in intact cells.
Keywords: redox regulation; inositol 1,4,5-trisphosphate (IP3); calcium intracellular release; calcium transport; endoplasmic reticulum (ER); calcium channel; inositol 1,4,5-trisphosphate receptor; oxidative stress; proteomics; redox-sensitive cysteine
Introduction
Inositol 1,4,5-trisphosphate receptor (IP3R)2 channels serve as the principal mechanism to mobilize Ca2+ from endoplasmic reticulum (ER) stores in response to cell stimulation by many hormones, growth factors, and neurotransmitters. The three isoforms of the IP3R family contain an N-terminal ligand-binding domain, a central regulatory domain, and a C-terminal channel domain. The ligand-binding domain contains a suppressor domain (SD; residues 1–223) that acts to inhibit IP3 binding to the core binding domain (residues 224–604). Channel opening is stimulated when IP3 levels are elevated during agonist stimulation. An alternative mode of channel opening results from regulatory modulation of IP3Rs, which sensitize channels to basal or modest changes in IP3 levels. A well-documented example of IP3R sensitization results from channel oxidation produced by exogenous or endogenous redox modulators. The first redox agent reported to sensitize IP3R channels was the organomercurial preservative thimerosal (reviewed in Ref. 1). Other compounds with similar effects include t-butylhydroperoxide (2), menadione (3), H2O2 (4), 2,2′-dithiodipyridine (5), diamide (6, 7), oxidized GSH (8, 9), and superoxide-generating systems such as xanthine/xanthine oxidase (10–13). In the case of thimerosal, the effects on purified reconstituted receptors suggest a direct reaction with thiol groups on the channel (14, 15). The recruitment of multiple IP3R channels in isolated patches has been shown to be regulated by the redox state of the receptor (16). The effects of prolonged ER stress and enhanced translocation of Ca2+ into mitochondria has also been linked to altered IP3R redox state as a result of hyperoxidation of the ER lumen promoting oxidation of critical intraluminal thiols of the IP3R (17–19).
Although there is substantial evidence for the regulation of IP3Rs by redox state, there is very little known regarding which IP3R thiols are involved, the types of thiol modifications occurring in the intact cell, or how the channel is sensitized by redox modifications. Although changes in IP3R redox state are presumed to occur, they have never been directly measured. The present study is the first to measure the redox state of IP3R in intact cells treated with various oxidants and to apply proteomic methods to interrogate the redox state of individual thiols in the IP3R1 isoform, which contains a total of 60 cysteines. These studies identify 4 oxidized thiols under basal conditions and an additional 11 or 4 thiols, which become oxidized in cells exposed to thimerosal or H2O2, respectively. The functional relevance of these oxidations was investigated using cysteine mutants transfected into a HEK293 IP3R triple-knockout cell line. The results show that the redox-sensitive cysteines required for IP3R1 channel potentiation are predominantly clustered within the N-terminal suppressor domain.
Results
We have adapted a method originally described by Leichert and Jakob (20, 21) to measure the redox state of IP3Rs in situ (Fig. 1A). The critical initial step is the lysis of cells directly in 10% TCA to prevent any alteration of thiol status. Precipitated proteins are solubilized under strongly denaturing conditions (0.5% SDS, 6 m urea), and free thiols are blocked irreversibly by reaction with 10 mm iodoacetamide (IAM). The samples are then reprecipitated with TCA, and any oxidized thiols are reduced with 10 mm DTT. A further TCA precipitation was used to remove the DTT, and free thiols on the receptor were reacted with PEG-maleimides of different sizes (2, 5, and 20 kDa). The resulting gel shifts can be measured on SDS–PAGE after immunoblotting for IP3R. HEK293 cells were used as an experimental system. These cells contain very low levels of IP3R1 and endogenously express IP3R2 and IP3R3 (22). Data from gel-shift assays are shown in Fig. 1 (B–E) for HEK293 cells transfected with IP3R1. Reproducible, small shifts are observed with both MPEG-2 and MPEG-5 under basal conditions (Fig. 1B). These shifts are not artifactual because they are blocked by inclusion of DTT during the MPEG reactions (Fig. 1B) or by DTT pretreatment of the intact cells prior to lysate preparation (Fig. 1C, lanes 1–3). The addition of the oxidants H2O2 (0.5 mm; Fig. 1C, lanes 7–9) or thimerosal (50 μm; Fig. 1C, lanes 10–12) enhanced the shift observed with both MPEG-2 and MPEG-5. Thimerosal produced larger changes than H2O2, and the effect of both oxidants were not additive (Fig. 1C, lanes 13–15). There are 60 cysteines in the primary sequence of the rat IP3R1. Lysates prepared in the absence of IAM treatment allow maximum reactivity to be measured, but shifted bands were retained on the gel only for the small MPEG-2 (Fig. 1D). In principle, given the molecular weight of the MPEGs, the total number of cysteines involved can be determined from the magnitude of the shift, but quantitation is subject to several inaccuracies and assumptions. One of these is the unusual hydrodynamic properties of MPEG-2 and MPEG-5 that produces gel shifts on SDS–PAGE corresponding to 5 and 15 kDa, respectively (23, 24). Taking this into account, the shifts with either MPEG-2 or MPEG-5 correspond to the presence of ∼5 oxidized cysteines/IP3R1 subunit under basal conditions and ∼10 and ∼20 oxidized cysteines/subunit in the presence of H2O2 and thimerosal, respectively (Fig. 1E). Previous studies have suggested that IP3Rs in hepatocytes are stimulated by GSSG (25), and IP3Rs are reported to be glutathionylated in response to diamide in endothelial cells (6, 7). Changes in the GSH redox status of HEK293 cells in response to H2O2 and thimerosal are shown in Fig. 2A. Both thimerosal and H2O2 caused similar increases in GSSG levels, but only thimerosal caused a large drop in total glutathione (GSH + GSSG). In contrast to H2O2, thimerosal forms an ethyl mercury adduct with reduced thiols and is known to deplete GSH in other systems (26). Preincubation of cells with buthionine sulfoximine (BSO, an inhibitor of GSH biosynthesis) reduced total GSH and GSSG levels but did not alter basal, H2O2, or thimerosal-induced gel shifts in IP3R (Fig. 2B). Diamide selectively oxidizes GSH to GSSG (27). An IP3R gel shift was observed at concentrations of diamide (50 μm) that did not produce detectable elevations of GSSG (Fig. 2C). Higher diamide concentrations (250 μm), which caused large elevations of GSSG, only produced a small additional gel shift (Fig. 2C). Diamide and other oxidants such as menadione and t-butylhydroperoxide were no more effective than H2O2 (not shown). Overall, the data do not show a clear correlation of IP3R oxidation with the GSH redox state. Rapid reversibility of both the total GSH levels (Fig. 2D) and the gel shifts were observed when DTT was added to the cells after the thimerosal (Fig. 2E).
Figure 1.

Gel-shift assays to measure IP3R redox state. A, cartoon showing the sequential treatments employed in the two main assays used in this study. B and C, TCA precipitates solubilized from HEK293 cells transfected with IP3R1 were sequentially treated with IAM and DTT and then reacted for 1 h with 0.5 mm MPEG-2 or MPEG-5. Gel shifts in IP3R were assessed by immunoblotting. Treatment of cells was for 10 min with H2O2 (0.5 mm) and thimerosal (Thim, 50 μm). In B, DTT (10 mm) was added during the MPEG reaction, and in C (lanes 1–3), the cells were pretreated for 30 min with DTT before lysis. Lanes 4–15 are from the same blot, but a higher exposure of lanes 10–12 is shown for better visualization of the bands. D, IAM (10 mm) treatment of the lysates was omitted as a control. E, the number of reactive cysteines was calculated from the magnitude of the gel shift relative to molecular weight markers as described under “Materials and methods.” The data shown are the means ± S.E. for three to five measurements. For additional details see text.
Figure 2.

Additional characterization of IP3R redox responses. A, total GSH and oxidized GSH was measured after treatment for 10 min with H2O2 (0.5 mm), thimerosal (50 μm), thapsigargin (1 μm), the indicated concentrations of diamide, and 16 h preincubation with 0.25 mm BSO. B, gel shift assays after BSO treatment. C, gel-shift dose responses for thimerosal and diamide. D and E, rapid DTT (10 mm) reversibility of thimerosal-induced changes in total GSH (D) and IP3R (E) gel shifts. In the last two lanes, the DTT was added before the thimerosal.
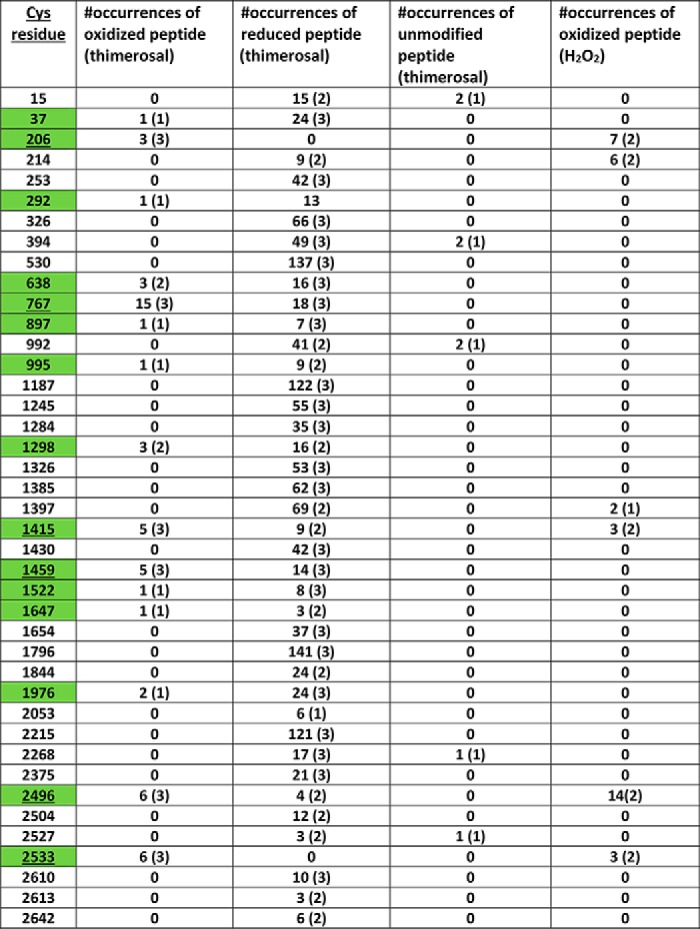
To get a comprehensive picture of the status of individual IP3R cysteines, we adopted a proteomic approach in which the denatured cell lysates prepared after sequential iodoacetamide and DTT treatment were incubated with biotin–maleimide instead of PEG-maleimide (Fig. 1A). The modified IP3R1 was then immunoprecipitated from the lysate, separated on SDS–PAGE, and silver-stained. The excised band was subjected to in-gel digestion with trypsin and processed for LC–MS/MS. The mass spectra were interrogated for cysteine containing peptides that were either modified with iodoacetamide (corresponding to reduced thiols) or modified with biotin–maleimide (corresponding to oxidized thiols). The compiled results from three independent experiments derived from control and thimerosal-treated cells are shown in Fig. 3 and Table 1. The mean sequence coverage was 71 ± 2% and resulted in the detection of 41 of 60 total cysteines when trypsin was used for cleavage. We observed four cysteines that were oxidized under basal conditions: Cys-292, Cys-1415, Cys-2496, and Cys-2533. Two of these cysteines are cytosol-facing (Cys-292 and Cys-1415), and two are intraluminal (Cys-2496 and Cys-2533). Thus of the five intraluminal cysteines, only two were oxidized. In the presence of thimerosal, the cysteines oxidized under basal conditions remained oxidized, and an additional 11 cytosol-facing cysteines were identified as being oxidized. These were Cys-37, Cys-206, Cys-638, Cys-767, Cys-897, Cys-995, Cys-1298, Cys-1459, Cys-1522, Cys-1647, and Cys-1976. It should be noted, however, that only six of these cysteines were consistently oxidized in all three trials (boxed in Fig. 3 and underlined in Table 1). In the case of Cys-206 and Cys-2533, we were able to identify only the oxidized form of the peptides in the spectra from thimerosal-treated cells. However, with the other redox-sensitive cysteines, both the oxidized and reduced forms of the peptides were evident (Table 1). For several cysteines the observed oxidation events were rare and in some cases observed only once in the three trials. However, the frequency of oxidation events was much higher (35–100%) for the six cysteines that were reproducibly oxidized. Lysates from H2O2-treated cells were analyzed in two experiments (Fig. 3, yellow boxes, and Table 1). A total of six cysteines were oxidized in both trials with three residues found under basal conditions (Cys-1415, Cys-2496, and Cys-2533), and an additional three residues oxidized upon H2O2 addition (Cys-206, Cys-214, and Cys-1397). The residues repeatedly oxidized by thimerosal and H2O2 were conserved in all three IP3R isoforms, with the exception of the thimerosal-sensitive residue Cys-1459, which was present only in the IP3R1 isoform (Fig. 3).
Figure 3.

Identification of redox-sensitive thiols in IP3R1 using LC–MS/MS. Redox lysates were prepared from transfected HEK293 cells treated in the presence and absence of thimerosal (50 μm) and processed with IAM to block free thiols. Oxidized residues were liberated with DTT, and the lysate was TCA-precipitated again and reacted with biotin–maleimide (0.5 mm, 5 h). The lysates were processed on a Sephadex G25 column to exchange the sample into a buffer suitable for immunoprecipitation, which was carried out with a C-terminal Ab. The immunoprecipitates were processed on 5% SDS–PAGE, and the silver-stained IP3R was excised and processed for LC–MS/MS. The spectra were analyzed with the SEQUEST search engine for tryptic peptides containing cysteines modified with IAM (reduced) or biotin–maleimide (oxidized). The boxed residues were identified in each of three independent trials. Oxidized residues observed in H2O2 (0.5 mm)–treated cells are indicated by yellow rectangles. Cysteine residues located in the ER lumen are underlined. All oxidized residues are conserved in all three isoforms with the exceptions shown in filled blue circles.
Table 1.
The cysteine residues oxidized by thimerosal in three independent trials are shown in green
The numbers of occurrences of biotin maleimide-modified peptides (oxidized), carbidomethylated peptides (reduced), or unmodified peptides in these samples are tabulated with the number of experiments shown in parentheses. The underlined residues are where at least one occurrence of oxidized peptide was observed in all three trials. In three trials the cysteine residues that were oxidized under basal conditions were Cys-292, Cys-1415, Cys-2496, and Cys-2533. The numbers of occurrences of peptides containing these oxidized residues were 10 (2), 16 (3), 18 (3), and 6 (3) with the number of experiments shown in parentheses. The oxidized peptides in H2O2-treated cells were also tabulated for two independent trials.
To use a mutagenesis approach to assess the functional role of individual cysteines in redox-modulation of the IP3R channel, we chose to employ a recently created human HEK293 cell line from which all three IP3R isoforms have been deleted by CRISPR (28). These 3KO cells do not elicit agonist or IP3-mediated cytosolic Ca2+ signals (28, 29) as illustrated in Fig. 4A for stimulation with carbachol in the presence of extracellular Ca2+. By contrast, robust carbachol responses are observed in WT cells, and Ca2+ signals can be restored in 3KO cells by transfection with IP3R1 DNA (Fig. 4B). Comparable gel shifts in response to thimerosal (50 μm) were observed in IP3R1 transfected WT or 3KO cells (not shown). To assay for redox potentiation of IP3R1, we pretreated the cells for 2 min with thimerosal. In initial trials, we observed that the use of concentrations of thimerosal of >20 μm induced by itself a delayed increase of cytosolic Ca2+ (Fig. 4C). This presumably reflects sensitization of IP3R1 to endogenous IP3 because the response to thimerosal was greatly blunted in untransfected 3KO cells (Fig. 4D). To be able to measure redox potentiation of carbachol responses, we chose to use a low concentration of thimerosal (10 μm), which allowed a stable baseline to be maintained during thimerosal pretreatment prior to stimulation by carbachol. The carbachol addition was combined with EGTA to chelate extracellular Ca2+ and ensured that only intracellular Ca2+mobilization was being measured. Under these conditions a thimerosal-induced potentiation of the Ca2+ signal was observed for WT IP3R1 at low subsaturating concentrations of carbachol (Fig. 4E) but not at a maximal dose of carbachol (Fig. 4F). IP3R1 in which the N-terminal 12 cysteines have been mutated to alanine (Cys-less) has been shown to be a functional channel that is insensitive to a stimulatory effect of thimerosal on [3H]IP3 binding (30, 31). In agreement with these studies, the Cys-less mutant failed to show thimerosal-induced potentiation of IP3R function in 3KO cells (Fig. 4, G and H). Thimerosal at higher concentrations has been found to inhibit IP3-mediated Ca2+ release (32). It should be noted that in our studies, using 10–50 μm thimerosal, only stimulatory effects were observed.
Figure 4.

Measurements of carbachol-mediated cytosolic [Ca2+] in HEK293 IP3R 3KO cells transfected with IP3R1. Cytosolic free [Ca2+] changes were measured with Fluo-8 in 96-well plates on a Flex station apparatus as described under “Materials and methods.” A, representative traces showing responses of WT and IP3R 3KO HEK293 cells to a maximal dose of carbachol (Cch) in the presence of extracellular Ca2+. B, changes in cytosolic Ca2+ of 3KO cells transfected with IP3R1 and stimulated with the indicated concentrations of carbachol. EGTA (1.3 mm) was added together with the carbachol to chelate extracellular Ca2+. C, thimerosal (10 or 50 μm) was added at 18 s to 3KO cells transfected with IP3R1. Cystosolic Ca2+ changes are shown as the means ± S.D. of Fluo-8 fluorescence for four wells of a representative experiment. D, as in C but using untransfected 3KO cells. E and G, 3KO cells transfected with IP3R1 were stimulated with a submaximal (E) or maximal dose (G) of carbachol in the absence of extracellular Ca2+ under control conditions (black symbols) or after pretreatment with 10 μm thimerosal (red symbols) for 2 min. F and H, as in E and G but with 3KO cells transfected with a cysteine-less IP3R1 construct in which the first 12 N-terminal cysteines are mutated to alanine (31). The data shown for E–H are the mean ± S.D. of four wells from a representative experiment. Pooled data from independent experiments are shown in Fig. 5.
A selected group of cysteine mutants were further analyzed for redox potentiation. These mutants included the individual residues within the N-terminal Cys-less construct, as well as three residues that were reproducibly oxidized in the MS assays (C767S, C1415S, and C1459S), and a cysteine found only in the SI(+) alternatively spliced form of IP3R1 (C326S). Several of these mutants have been used in our previous studies (33). The expression of new mutants in 3KO cells was verified by Western blotting (Fig. 5A). To quantitate potentiation we measured the ratio of Ca2+ responses in the presence and absence of thimerosal pretreatment using 0.25 μm carbachol as in Fig. 4C. The maximal response to 20 μm carbachol was also measured and normalized to the maximal response observed with WT IP3R1 DNA. The potentiation ratio and maximal responses for each of the mutants are shown in Fig. 5 (B and C), respectively. Five of the eleven mutants showed diminished potentiation by thimerosal. Of these, four showed a partial inhibition (C37S, C56S/C61S, C214S, and C326S), whereas C15S was almost as effective as the Cys-less in removing the potentiating effect of thimerosal. The majority of the mutants showed maximal carbachol responses that were comparable with the WT IP3R1, although several had significantly lower (e.g. C37S) or higher (e.g. C15S) responses (Fig. 5C). The variation in maximal responses may reflect different levels of functional receptors in the ER stores that may not be evident at the level of immunoblots. It should be noted, however, that the maximal responses did not correlate with diminished redox potentiation.
Figure 5.
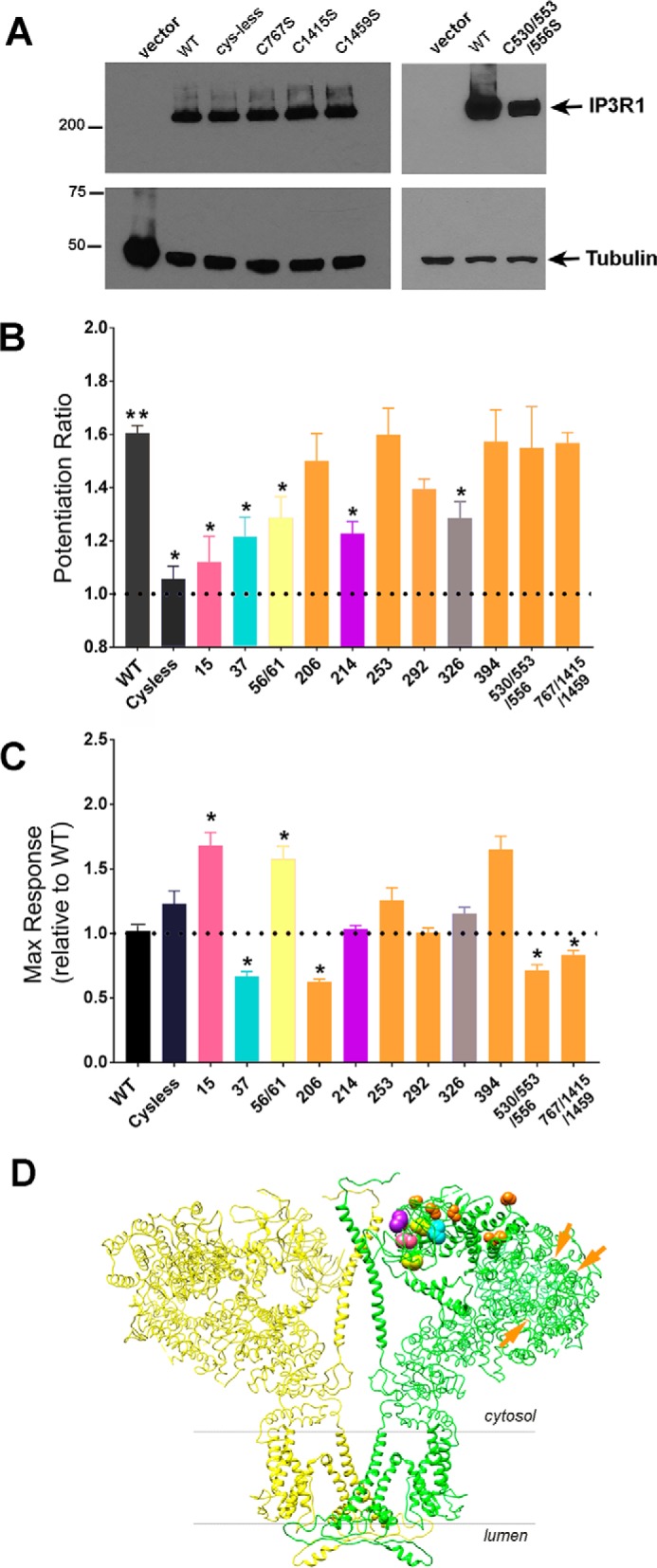
Quantitation of thimerosal potentiation of carbachol-mediated intracellular Ca2+release for specific cysteine mutants of IP3R1. A, expression of selected cysteine mutants in 3KO cells detected by immunoblotting with IP3R1 Ab. The expression of the remaining cysteine mutants used in this study has been verified previously (33). B, the amplitude of the Ca2+ response to 0.25 μm carbachol after thimerosal treatment was expressed as a ratio to the control response using data obtained as shown in Fig. 4E. C, the response to a maximal dose of carbachol (20 μm) was quantitated for each mutant and expressed as a ratio of WT IP3R1 responses. All data are the means ± S.E. of n = 3–6 independent experiments. *, p < 0.001 significantly different from WT; **, p < 0.0001 significantly different from 1 (one sample t test). D, two opposing subunits of the tetrameric structure of apo-IP3R1 determined by cryo-EM (Protein Data Bank (PDB) ID 3JAV) are shown in a side view. The redox-sensitive cysteine residues are color coded according to the data in B. Redox-insensitive residues are colored orange. Side chains for Cys-767, Cys-1415, and Cys-1459 were not resolved in the structure and are indicated by orange arrows. Cys-326, which is located in the SI splice site, was also not present in the structure.
The residues having no effects (colored orange in Fig. 5) include Cys-767, Cys-1415, and Cys-1459, which were all residues that were reproducibly oxidized in MS assays after thimerosal treatment, with Cys-1459 being constitutively oxidized even under basal conditions (Fig. 3). Three residues that are included in the Cys-less mutant but did not appear in the mass spectra (Cys-530, Cys-553, and Cys-556) were also without effect on redox potentiation. The location of the redox-sensitive and -insensitive residues, color-coded as in Fig. 5B, have been mapped to the cryo-EM structure of IP3R1 (34) in Fig. 5D. The main conclusion is that the redox-sensitive residues are clustered within the first 225 residues. This region has variously been referred to as the suppressor domain (or β-trefoil domain-1) and has been shown to be critical for channel gating (35–37). The structural implications of these findings are discussed in more detail below.
In the final series of experiments, we have examined the potentiating effects of H2O2 and also tested the redox sensitivity of the IP3R2 and IP3R3 isoforms (Fig. 6). A maximal dose of carbachol (25 μm) elicited similar Ca2+ responses from IP3R1 and IP3R2 transfected cells and a 20% smaller response in IP3R3 transfected cells (Fig. 6A). The dose response to carbachol was examined for all three isoforms (Fig. 6B). The EC50 for carbachol for IP3R1 (0.62 ± 0.05 μm) and IP3R2 (0.66 ± 0.07 μm) were similar, although IP3R3 (1.7 ± 0.12 μm) was somewhat less sensitive. This can be compared with the rank order of sensitivity IP3R2 > IP3R1 >IP3R3 found in direct measurements with IP3 in permeabilized DT40 3KO cells stably expressing different receptor isoforms (13, 38, 39). All of the cysteine mutants also showed similar IP3 sensitivities as the WT IP3R1 (not shown). This is illustrated for the C15S mutant (EC50 = 0.76 ± 0.02 μm) in Fig. 6B. An N-terminal fusion protein containing the Cys-less mutant has also been shown to have a comparable IP3-binding affinity as WT receptors (30). Thus the pronounced loss of redox potentiation seen with the C15S and Cys-less mutants (Fig. 5B) is unlikely to be related to an intrinsically different IP3 sensitivity of the channel.
Figure 6.

Redox potentiation by thimerosal and H2O2 of IP3R1, IP3R2, and IP3R3. A, the three different IP3R isoforms were transfected into HEK293 3KO cells (10 μg of DNA/60-cm plate) and the maximal Ca2+ response obtained with 25 μm carbachol in each experiment was quantitated relative to the maximal response for IP3R1. B, intracellular Ca2+ release was measured using different concentrations of carbachol. Also shown are the responses of the C15S IP3R1 mutant. The amplitude of the Ca2+ response was normalized to the maximum and is shown as means ± S.E. (n = 3). The lines drawn through the data points are fits to the equation y = ax/(b + x), where b corresponds to the EC50 values (given in the text). C, the 3KO cells transfected with the three different isoforms were pretreated for 2 min with either 10 μm thimerosal (Thi) or 100 μm H2O2, and the potentiation of responses to 0.25 μm carbachol or 0.5 μm carbachol (IP3R3) was measured as described in Fig. 5B. The data shown are means ± S.E. of three to five independent experiments. *, p < 0.05 statistical significant from thimerosal for each isoform.
Thimerosal potentiated the carbachol response approximately equally with IP3R1 and IP3R2 isoforms but was much less effective with IP3R3 (Fig. 6C). A similar isoform preference was observed when H2O2 (100 μm) was used to potentiate carbachol responses. The lack of redox sensitivity of the IP3R3 isoform is in agreement with previous findings in DT40 cells (13, 31). As observed with thimerosal, the C15S IP3R1 mutant was insensitive to potentiation by H2O2.
Discussion
Although IP3Rs have been established to be redox-sensitive channels, the present study is the first to directly measure changes in the redox state of the IP3R protein in intact cells. To do this, we have utilized gel-shift assays and MS. Although the gel-shift assay is convenient and provides qualitative information, the method is also insensitive, because heterogeneous small shifts in a large protein such as IP3Rs can be easily missed. Many membrane proteins are known to retain folded structures even in the presence of SDS and urea (40–42). It is therefore possible that not all thiols are freely accessible to the MPEG gel-shift reagent even in the “denaturing” conditions used in our experiments. The MS approach is more sensitive and provides more information. However, this method is limited by incomplete sequence coverage, which resulted in 19 cysteines not being observed in the mass spectrum when the sample was digested with trypsin. Interestingly, the same cysteines were missing in each attempt, including Cys-556 and a cluster of six cysteines located prior to the first transmembrane domain (Fig. 3). To increase coverage, a single trial was conducted with chymotrypsin as the cleavage enzyme, but the same cysteines were also absent in this analysis (not shown). Several recent studies have shown that both ryanodine receptors (43) and IP3Rs (44) are subject to S-palmitoylation. The attachment of palmitic acid is known to interfere with the chromatography of trypsin peptides, making it difficult to identify modified residues by LC–MS/MS (45). Using MALDI–TOF, Cys-56 and Cys-849 were identified as sites of S-palmitoylation (44), providing a possible explanation for why peptides containing these sites were absent from the LC–MS/MS analysis. Overall, the limitations in coverage make it likely that the number of redox-sensitive thiols identified by MS is underestimated.
The redox state of the ER lumen is considerably more oxidized than the cytosol, based on the GSH redox couple (46). Therefore, it may be anticipated that the intraluminal thiols of the IP3R would be predominantly oxidized. However, it is now understood that conditions favoring oxidative protein folding can occur under more reducing conditions and that the reactive thiols of key ER luminal oxidoreductases are predominantly in the reduced state (47–49). Indeed, under basal conditions only Cys-2496 and Cys-2533 of the five intraluminal IP3R1 cysteines were found to be oxidized. The pair of intraluminal cysteines at Cys-2496 and Cys-2504 have previously been suggested to form a disulfide bond based on NMR studies of a fusion protein encoding the intraluminal loop (50). However, in our experiments Cys-2504 was consistently reduced, and therefore a disulfide bond between these residues is unlikely to occur in an intact cell under basal conditions. Both Cys-2496 and Cys-2504 have been implicated as important for association with ERp44, an intraluminal chaperone of the thioredoxin family, which selectively associates with the reduced form of IP3R1 and has an inhibitory effect on channel function (51). Our results would suggest that ERp44 would preferentially associate with the Cys-2504 residue under basal conditions. The Cys-2533 residue is located at the intraluminal end of a pore helix, and mutation of this residue has previously been shown to inactivate channel function (52). A constitutively oxidized Cys-2533 residue may stabilize the intraluminal structure of the pore, possibly by formation of disulfide bonds with other ER luminal proteins. Neither thimerosal nor H2O2 changed the redox state of IP3R intraluminal thiols.
Despite the reducing environment in the cytosol, a significant number of cysteines in key cytosolic signaling proteins are in the oxidized state (53). In the case of IP3R1, two cytosol-facing cysteines (Cys-292 and Cys-1415) were found to be oxidized under basal conditions. This could indicate that these thiols have a sufficiently low pKa to allow the formation of thiolate anions that can form disulfides with GSH or with other redox-sensitive proteins. It is also possible that IP3Rs are present in a local environment that is more oxidizing than the bulk redox potential of the cytosol (54).
Mass spectroscopy indicated a total of 11 cytosolic cysteines that were modified in the presence of thimerosal. However, there was considerable variability in the results with some of the cysteines being modified in only one of three experiments. The reason for this variability is not clear but could be related to the experimental methodology and/or heterogeneity in the redox state of specific cysteines in the IP3R. Only three cysteines were consistently oxidized by thimerosal in all three experiments, and they were Cys-206, Cys-767, and Cys-1459. In the case of H2O2, there were also three cysteines that were oxidized in each of two experiments. They were Cys-206, Cys-214, and Cys-1397. Cys-206 and Cys-214 are located in the SD, whereas Cys-767, Cys-1397, and Cys-1459 are located in the regulatory domain. To assess the functional relevance of these residues, we have used an assay in which cysteine mutants were transfected into HEK293 IP3R 3KO cells. Redox potentiation was measured by preincubating cells for 2 min with oxidant prior to stimulation of intracellular Ca2+ mobilization with a subsaturating concentration of carbachol. These studies revealed that redox sensitivity was lost in an IP3R1 mutant lacking the N-terminal 12 cysteines. Further mutagenesis of the individual cysteines indicates that multiple cysteines confer redox sensitivity to thimerosal. With the exception of Cys-326, all the redox-sensitive residues are clustered in the SD (Fig. 5D).
The SD (also referred to as the β-trefoil domain 1 (34)) plays a critical role in channel gating (35–37). This domain makes contacts with two adjacent downstream domains called the β-trefoil domain-2 and the armadillo solenoid fold-1 domain, which together constitute the inner binding core that specifically binds IP3 (30). The cryo-EM structures of apo-IP3R1 (34) and multiple liganded form of IP3R3 (55) have been reported. Long-range conformational changes resulting from IP3 binding can be transmitted to the channel domain by several mechanisms (34, 55). This includes changes in the relative position of a loop variously referred to as a “hot spot” or “gating loop” within the SD that contains the Tyr-167 residue. Mutations within the Tyr-167 loop disrupt the coupling between IP3 binding and channel opening (56). We suggest that the oxidation of specific cysteines in the SD may enhance the coupling efficiency of the mechanisms by which the SD transduces the ligand-binding signal to the channel domain. This is consistent with earlier studies that found an interaction between recombinant SD and inner binding core proteins that was strengthened by adding thimerosal (32). A more detailed understanding of the molecular mechanism of redox potentiation of the IP3R channel awaits high resolution structures of the open state of the IP3R1 channel. Oxidation of key thiols could also influence channel activity indirectly by modifying interactions of the receptor with its many binding partners. For example, one of the interaction sites of Bcl-2 encompasses the H2O2-sensitive Cys-1397 (57). Interestingly, an R36C mutation within the SD has been identified in three affected individuals with spinocerebellar ataxia (58). This mutation would place a cysteine immediately adjacent to the redox-sensitive Cys-37 residue. Unlike other inactivating IP3R1 mutations linked to inherited neurodegenerative diseases, this mutation results in a gain of channel function. The redox regulation properties of the R36C mutant remain to be investigated.
An additional finding in the present study was that the results from MS and functional assays were not always well-correlated. For example, several residues were reproducibly oxidized by thimerosal in the MS measurements but played no role when tested in functional assays (e.g. Cys-206, Cys-767, Cys-1415, and Cys-1459). Cys-206 has previously been shown to be a highly accessible cysteine (33) and was oxidized by both thimerosal and H2O2. However, robust oxidation of specific thiols may simply reflect their higher accessibility/reactivity and may not necessarily result in altered channel function. There were also several cysteine residues that contributed to redox potentiation in functional measurements that were not detected as being oxidized in the MS assay (e.g. Cys-15 and Cys-326). A functional effect obtained from mutating a particular cysteine residue does not prove that this residue is the physiological target of oxidants in the WT receptor. Although there may be limitations regarding sensitivity and coverage, the MS analysis does accurately identify the redox-sensitive thiols of the IP3R in situ.
Although this study focused primarily on IP3R1, we have also made some functional measurements on IP3R2 and IP3R3 expressed in HEK293 3KO cells. In line with previous studies on DT40 cells (31, 32), we observed reduced potentiation of IP3R3 by thimerosal and H2O2 relative to IP3R1 or IP3R2 (Fig. 6). Five of the six cysteines present in the SD of IP3R1 are highly conserved in all three isoforms; only Cys-15 is unique to IP3R1, and its mutation markedly reduces redox potentiation of the IP3R1 channel by either H2O2 or thimerosal. Although the remaining five cysteines are conserved, it is also apparent from IP3-binding studies with chimeric receptors that the SD of IP3R3 is unique and is not interchangeable with the other two isoforms (35, 59). Thus differences in the precise intra- and intermolecular contacts made by the SD may determine differences in redox regulation of the three isoforms. We also cannot exclude the possibility that other nonconserved cysteine residues outside of the SD may play a role in redox regulation of IP3R2 and IP3R3. Clearly other cell and species–specific regulatory factors can also play a role because previous studies on DT40 cells have noted that the endogenous chicken IP3R3 and stably expressed rat IP3R3 differ in their redox sensitivity (13). Different redox microenvironments (54) or variable antioxidant defenses could also contribute to differential sensitivity of IP3R isoforms in different cell types.
Redox regulation of IP3R-mediated Ca2+ signaling has been implicated in ER stress and apoptosis (19, 60–63). The redox-insensitive mutants identified in this study may be potentially useful in examining the contribution of IP3R redox regulation in different models of cellular injury.
Materials and methods
Gel-shift assays for measurements of IP3R redox state
The method used was a modification of the procedure described by Leichert and Jacob (20, 21). The key step is the quenching of the cellular redox state by rapid acidification of the cells with 10% TCA (Fig. 1A). The cells were scraped from the plates and centrifuged. The protein precipitate was dissolved in a strongly denaturing buffer (DB) containing 200 mm Tris-HCl (pH 8.5), 10 mm EDTA, 0.5% SDS, and 6 m urea. The lysate was incubated in this buffer supplemented with 10 mm iodoacetamide for 45 min to block all available free thiols. The lysate was then reprecipitated with 10% TCA and resuspended in DB buffer containing 10 mm DTT and incubated for a further 45 min. After reprecipitation with 10% TCA, the pellet was resuspended in 0.25 ml of DB buffer and adjusted to pH 7.0 with NaOH. Lysate protein (15 μg) was incubated with 0.5 mm PEG-maleimides of various sizes (2, 5, and 20 kDa) for 1 h. After SDS–PAGE on 5% gels for 2.5 h, the samples were transferred to nitrocellulose and immunoblotted with a C-terminal Ab specific for IP3R1 raised to the C-terminal 18 amino acids (64). The size of the MPEG-shifted bands was estimated by comparing the relative mobility of the bands to standards retained on the 5% gels including the unmodified IP3R1 subunit (320 kDa), a lysate from a stable HEK293 cell expressing a concatenated dimer of IP3R1 (640 kDa) (65), prestained myosin (211 kDa), and prestained β-gal (114 kDa).
GSH assays
Total GSH was measured in the supernatant of TCA-quenched cell lysates using an enzymatic recycling assay utilizing 5,5′-dithiobis(nitrobenzoic acid), NADPH, and GSH reductase as originally described (66). Oxidized GSH was measured with a lumiglo kit obtained from Promega (Madison, WI). HEK293 cells (5 × 104) were grown for 24 h in 96-well plates and treated with various oxidants. The cells were lysed on the plate using a supplied buffer containing NEM to block GSH. Residual GSSG was converted to GSH and measured using a luciferin probe as described by the manufacturer.
Mass spectroscopy
HEK293 cells were transfected with plasmid encoding rat IP3R1 for 48 h. The cells were treated in the presence or absence of 50 μm thimerosal for 10 min. For each condition two 150-mm plates were used. After removal of the medium, each plate was quenched in 2.5 ml of 10% TCA. The precipitated protein was sequentially treated with iodoacetamide and DTT as described above for the gel-shift assays. The final DTT-treated lysate was solubilized in DB buffer and adjusted to a pH of 7.0. The lysate (1 mg of protein) was incubated with 0.5 mm biotin–maleimide (Sigma) for 16 h at room temperature in the dark. The reaction was terminated with 10 mm DTT, and the entire reaction was loaded on a PD-10 desalting column (GE Healthcare) equilibrated in a buffer containing 150 mm NaCl, 20 mm NaMOPS (pH 7.0), 0.5 mm EDTA, 0.2 mm DTT, 0.1% deoxycholate, and 0.2% Triton X-100. This buffer exchange step was carried out to reduce the SDS and urea concentrations to allow more efficient immunoprecipitation. The eluted protein was incubated with Ab to the C terminus of IP3R1 and protein A–Sepharose overnight. Immunocomplexes were processed on 5% SDS–PAGE and stained with silver (SilverSNAP stain; Thermo Fisher Scientific) or with colloidal blue (Novex). The excised band above the myosin marker was processed for in-gel trypsin digestion and LC—MS/MS using a Thermo Q Exactive Plus mass spectrometer at the Wistar Proteomics facility (Wistar Institute, Philadelphia, PA). Peptide identification was achieved by searching against the rat IP3R1 (NP_001007236.2) and the nonredundant human database from the National Center for Biotechnology Information. Mass accuracy was limited to 10 ppm for precursor ions and 0.6 Da for product ions, with tryptic enzyme specificity and up to two missed cleavages. Variable modifications included cysteine alkylation by iodoacetamide (+57.02 Da) and reaction with biotin–maleimide (+451.54 Da) (67).
Cytosolic [Ca2+] measurement in HEK293 cells
The cells were grown in 60-cm plates to ∼70% confluence and were transfected with 10 μg of IP3R DNA using Lipofectamine 3000 according to the manufacturer's instructions (Thermo Fisher Scientific). After 24 h, the cells were removed with trypsin and seeded at 50,000 cells/well in a 96-well transparent black plate. The culture medium was removed after a further 24 h and replaced with 90 μl of Hanks' buffered salt solution, 10 mm HEPES (pH 7.2) (HBSS–HEPES) for 30 min. An additional 100 μl of HBSS–HEPES containing 5 μm Fluo-8 AM, 0.1 mm sulfinpyrazone, and 500 μm Brilliant Black (to quench extracellular dye fluorescence) was added to the plates, which were incubated for 1 h at 37 °C. Changes in cytosolic Ca2+ were monitored using a Flex Station II plate reader in the fluorescence mode. The plate was maintained at 37 °C, and measurements were made using excitation and emission wavelengths of 485 and 525 nm, respectively. The various additions of thimerosal, H2O2, and carbachol were made utilizing the fluidics addition system of the Flex station II. Data acquisition and analysis was carried out with SoftMax Pro and Excel.
DNA constructs and cysteine mutagenesis
The cDNA encoding the IP3R1 isoform was a gift of Dr. Greg Mignery (Loyola University, Chicago, IL) and encoded the splice variant SI(+), SII(+), SIII(−). All amino acid numbering is with reference to the rat IP3R1 (68). The cDNA encoding mouse IP3R2 was a gift of Dr. Katsuhiko Mikoshiba (69), and rat IP3R3 was a gift of Dr. Graeme Bell (70). The Cys-less construct was a kind gift of Dr. Colin Taylor and contains mutations of the first 12 N-terminal cysteines modified to alanine in IP3R1 (SI(−)). The Cys-less construct has previously been shown to retain channel function (30, 31). Individual mutation of each of the 12 N-terminal cysteines to serine was described previously and included an additional cysteine located within the SI splice site (C326S) (33). The C530A/C553A/C556A mutant was produced by excising the BamHI/KpnI fragment from the Cys-less mutant and replacing it with the BamHI/KpnI PCR-amplified product derived from WT IP3R1. The three amino acids Cys-767, Cys-1415, and Cys-1459 were changed to serine using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies; catalog no. 200521) and the primer design method developed by Zheng et al. (71).
Antibodies and reagents
MPEG-2 and MPEG-20 was purchased from Nektar (Huntsville, AL). MPEG-5, sulfinpyrazone, and Brilliant Black were obtained from Sigma. Fluo-8AM was from AbCam (Cambridge, MA).
Author contributions
S. K. J. conceptualization; S. K. J. funding acquisition; S. K. J. writing-original draft; S. K. J. project administration; S. K. J. writing-review and editing; M. Y., D. I. Y., M. A., D. M. B., and G. H. investigation; K. A. methodology.
Acknowledgment
We thank Colin Taylor for the Cys-less mutant.
This work was supported by National Institutes of Health Grants RO1 DK103558 (to S. K. J. and G. H.) and RO1 DK051526 (to G. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- IP3R
- inositol 1,4,5-trisphosphate receptor
- ER
- endoplasmic reticulum
- SD
- suppressor domain
- IAM
- iodoacetamide
- BSO
- buthionine sulfoximine
- 3KO
- triple-knockout
- DB
- denaturing buffer.
References
- 1. Elferink J. G. (1999) Thimerosal: a versatile sulfhydryl reagent, calcium mobilizer, and cell function-modulating agent. Gen. Pharmacol. 33, 1–6 10.1016/S0306-3623(98)00258-4 [DOI] [PubMed] [Google Scholar]
- 2. Bird G. S., Burgess G. M., and Putney J. W. Jr. (1993) Sulfhydryl reagents and cAMP-dependent kinase increase the sensitivity of the inositol 1,4,5-trisphosphate receptor in hepatocytes. J. Biol. Chem. 268, 17917–17923 [PubMed] [Google Scholar]
- 3. Pruijn F. B., Sibeijn J. P., and Bast A. (1990) Changes in inositol-1,4,5-trisphosphate binding to hepatic plasma membranes caused by temperature, N-ethylmaleimide and menadione. Biochem. Pharmacol. 40, 1947–1952 10.1016/0006-2952(90)90223-8 [DOI] [PubMed] [Google Scholar]
- 4. Redondo P. C., Salido G. M., Rosado J. A., and Pariente J. A. (2004) Effect of hydrogen peroxide on Ca2+ mobilisation in human platelets through sulphydryl oxidation dependent and independent mechanisms. Biochem. Pharmacol. 67, 491–502 10.1016/j.bcp.2003.09.031 [DOI] [PubMed] [Google Scholar]
- 5. Islam M. S., Kindmark H., Larsson O., and Berggren P. O. (1997) Thiol oxidation by 2,2′-dithiodipyridine causes a reversible increase in cytoplasmic free Ca2+ concentration in pancreatic beta-cells: role for inositol 1,4,5-trisphosphate-sensitive Ca2+ stores. Biochem. J. 321, 347–354 10.1042/bj3210347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lock J. T., Sinkins W. G., and Schilling W. P. (2011) Effect of protein S-glutathionylation on Ca2+ homeostasis in cultured aortic endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 300, H493–H506 10.1152/ajpheart.01073.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lock J. T., Sinkins W. G., and Schilling W. P. (2012) Protein S-glutathionylation enhances Ca2+-induced Ca2+ release via the IP3 receptor in cultured aortic endothelial cells. J. Physiol. 590, 3431–3447 10.1113/jphysiol.2012.230656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Henschke P. N., and Elliott S. J. (1995) Oxidized glutathione decreases luminal Ca2+ content of the endothelial cell Ins(1,4,5)P3-sensitive Ca2+ store. Biochem. J. 312, 485–489 10.1042/bj3120485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Renard D. C., Seitz M. B., and Thomas A. P. (1992) Oxidized glutathione causes sensitization of calcium release to inositol 1,4,5-trisphosphate in permeabilized hepatocytes. Biochem. J. 284, 507–512 10.1042/bj2840507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Madesh M., Hawkins B. J., Milovanova T., Bhanumathy C. D., Joseph S. K., Ramachandrarao S. P., Sharma K., Kurosaki T., and Fisher A. B. (2005) Selective role for superoxide in InsP3 receptor-mediated mitochondrial dysfunction and endothelial apoptosis. J. Cell Biol. 170, 1079–1090 10.1083/jcb.200505022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suzuki Y. J., and Ford G. D. (1992) Superoxide stimulates IP3-induced Ca2+ release from vascular smooth muscle sarcoplasmic reticulum. Am. J. Physiol. 262, H114–H116 [DOI] [PubMed] [Google Scholar]
- 12. Wesson D. E., and Elliott S. J. (1995) The H2O2-generating enzyme, xanthine oxidase, decreases luminal Ca2+ content of the IP3-sensitive Ca2+ store in vascular endothelial cells. Microcirculation 2, 195–203 10.3109/10739689509146767 [DOI] [PubMed] [Google Scholar]
- 13. Bansaghi S., Golenár T., Madesh M., Csordás G., RamachandraRao S., Sharma K., Yule D. I., Joseph S. K., and Hajnóczky G. (2014) Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J. Biol. Chem. 289, 8170–8181 10.1074/jbc.M113.504159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kaplin A. I., Ferris C. D., Voglmaier S. M., and Snyder S. H. (1994) Purified reconstituted inositol 1,4,5-trisphosphate receptors: thiol reagents act directly on receptor protein. J. Biol. Chem. 269, 28972–28978 [PubMed] [Google Scholar]
- 15. Thrower E. C., Duclohier H., Lea E. J., Molle G., and Dawson A. P. (1996) The inositol 1,4,5-trisphosphate-gated Ca2+ channel: effect of the protein thiol reagent thimerosal on channel activity. Biochem. J. 318, 61–66 10.1042/bj3180061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vais H., Siebert A. P., Ma Z., Fernández-Mongil M., Foskett J. K., and Mak D. O. (2010) Redox-regulated heterogeneous thresholds for ligand recruitment among InsP3R Ca2+-release channels. Biophys. J. 99, 407–416 10.1016/j.bpj.2010.04.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li G., Mongillo M., Chin K. T., Harding H., Ron D., Marks A. R., and Tabas I. (2009) Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 186, 783–792 10.1083/jcb.200904060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anelli T., Bergamelli L., Margittai E., Rimessi A., Fagioli C., Malgaroli A., Pinton P., Ripamonti M., Rizzuto R., and Sitia R. (2012) Ero1α regulates Ca2+ fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid. Redox Signal. 16, 1077–1087 10.1089/ars.2011.4004 [DOI] [PubMed] [Google Scholar]
- 19. Kiviluoto S., Vervliet T., Ivanova H., Decuypere J. P., De Smedt H., Missiaen L., Bultynck G., and Parys J. B. (2013) Regulation of inositol 1,4,5-trisphosphate receptors during endoplasmic reticulum stress. Biochim. Biophys. Acta 1833, 1612–1624 10.1016/j.bbamcr.2013.01.026 [DOI] [PubMed] [Google Scholar]
- 20. Leichert L. I., and Jakob U. (2004) Protein thiol modifications visualized in vivo. PLoS Biol. 2, e333 10.1371/journal.pbio.0020333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leichert L. I., and Jakob U. (2006) Global methods to monitor the thiol-disulfide state of proteins in vivo. Antioxid. Redox Signal. 8, 763–772 10.1089/ars.2006.8.763 [DOI] [PubMed] [Google Scholar]
- 22. Wojcikiewicz R. J. (1995) Type I, II, and III inositol 1,4,5-trisphosphate receptors are unequally susceptible to down-regulation and are expressed in markedly different proportions in different cell types. J. Biol. Chem. 270, 11678–11683 10.1074/jbc.270.19.11678 [DOI] [PubMed] [Google Scholar]
- 23. Makmura L., Hamann M., Areopagita A., Furuta S., Muñoz A., and Momand J. (2001) Development of a sensitive assay to detect reversibly oxidized protein cysteine sulfhydryl groups. Antioxid. Redox Signal. 3, 1105–1118 10.1089/152308601317203611 [DOI] [PubMed] [Google Scholar]
- 24. Schwaller M., Wilkinson B., and Gilbert H. F. (2003) Reduction-reoxidation cycles contribute to catalysis of disulfide isomerization by protein-disulfide isomerase. J. Biol. Chem. 278, 7154–7159 10.1074/jbc.M211036200 [DOI] [PubMed] [Google Scholar]
- 25. Renard-Rooney D. C., Joseph S. K., Seitz M. B., and Thomas A. P. (1995) Effect of oxidized glutathione and temperature on inositol 1,4,5-trisphosphate binding in permeabilized hepatocytes. Biochem. J. 310, 185–192 10.1042/bj3100185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. James S. J., Slikker W. 3rd, Melnyk S., New E., Pogribna M., and Jernigan S. (2005) Thimerosal neurotoxicity is associated with glutathione depletion: protection with glutathione precursors. Neurotoxicology 26, 1–8 10.1016/j.neuro.2004.07.012 [DOI] [PubMed] [Google Scholar]
- 27. Kosower N. S., Kosower E. M., Wertheim B., and Correa W. S. (1969) Diamide, a new reagent for the intracellular oxidation of glutathione to the disulfide. Biochem. Biophys. Res. Commun. 37, 593–596 10.1016/0006-291X(69)90850-X [DOI] [PubMed] [Google Scholar]
- 28. Alzayady K. J., Wang L., Chandrasekhar R., Wagner L. E. 2nd, Van Petegem F., and Yule D. I. (2016) Defining the stoichiometry of inositol 1,4,5-trisphosphate binding required to initiate Ca2+ release. Sci. Signal. 9, ra35 10.1126/scisignal.aad6281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mataragka S., and Taylor C. W. (2018) All three IP3 receptor subtypes generate Ca2+ puffs, the universal building blocks of IP3-evoked Ca2+ signals. J. Cell Sci. 131, jcs220848 10.1242/jcs.220848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seo M. D., Velamakanni S., Ishiyama N., Stathopulos P. B., Rossi A. M., Khan S. A., Dale P., Li C., Ames J. B., Ikura M., and Taylor C. W. (2012) Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature 483, 108–112 10.1038/nature10751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khan S. A., Rossi A. M., Riley A. M., Potter B. V., and Taylor C. W. (2013) Subtype-selective regulation of IP3 receptors by thimerosal via cysteine residues within the IP3-binding core and suppressor domain. Biochem. J. 451, 177–184 10.1042/BJ20121600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bultynck G., Szlufcik K., Kasri N. N., Assefa Z., Callewaert G., Missiaen L., Parys J. B., and De Smedt H. (2004) Thimerosal stimulates Ca2+ flux through inositol 1,4,5-trisphosphate receptor type 1, but not type 3, via modulation of an isoform-specific Ca2+-dependent intramolecular interaction. Biochem. J. 381, 87–96 10.1042/BJ20040072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Joseph S. K., Nakao S. K., and Sukumvanich S. (2006) Reactivity of free thiol groups in type-I inositol trisphosphate receptors. Biochem. J. 393, 575–582 10.1042/BJ20050889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fan G., Baker M. L., Wang Z., Baker M. R., Sinyagovskiy P. A., Chiu W., Ludtke S. J., and Serysheva I. I. (2015) Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature 527, 336–341 10.1038/nature15249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Iwai M., Michikawa T., Bosanac I., Ikura M., and Mikoshiba K. (2007) Molecular basis of the isoform-specific ligand-binding affinity of inositol 1,4,5-trisphosphate receptors. J. Biol. Chem. 282, 12755–12764 10.1074/jbc.M609833200 [DOI] [PubMed] [Google Scholar]
- 36. Rossi A. M., Riley A. M., Tovey S. C., Rahman T., Dellis O., Taylor E. J., Veresov V. G., Potter B. V., and Taylor C. W. (2009) Synthetic partial agonists reveal key steps in IP3 receptor activation. Nat. Chem. Biol. 5, 631–639 10.1038/nchembio.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chan J., Yamazaki H., Ishiyama N., Seo M. D., Mal T. K., Michikawa T., Mikoshiba K., and Ikura M. (2010) Structural studies of inositol 1,4,5-trisphosphate receptor: coupling ligand binding to channel gating. J. Biol. Chem. 285, 36092–36099 10.1074/jbc.M110.140160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miyakawa T., Maeda A., Yamazawa T., Hirose K., Kurosaki T., and Iino M. (1999) Encoding of calcium signals by differential expression of IP3 receptor subtypes. EMBO J. 18, 1303–1308 10.1093/emboj/18.5.1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chandrasekhar R., Alzayady K. J., Wagner L. E. 2nd, and Yule D. I. (2016) Unique regulatory properties of heterotetrameric inositol 1,4,5-trisphosphate receptors revealed by studying concatenated receptor constructs. J. Biol. Chem. 291, 4846–4860 10.1074/jbc.M115.705301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dutta A., Tirupula K. C., Alexiev U., and Klein-Seetharaman J. (2010) Characterization of membrane protein non-native states: 1. Extent of unfolding and aggregation of rhodopsin in the presence of chemical denaturants. Biochemistry 49, 6317–6328 10.1021/bi100338e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Borgnia M. J., Kozono D., Calamita G., Maloney P. C., and Agre P. (1999) Functional reconstitution and characterization of AqpZ, the E. coli water channel protein. J. Mol. Biol. 291, 1169–1179 10.1006/jmbi.1999.3032 [DOI] [PubMed] [Google Scholar]
- 42. Heinz C., Engelhardt H., and Niederweis M. (2003) The core of the tetrameric mycobacterial porin MspA is an extremely stable β-sheet domain. J. Biol. Chem. 278, 8678–8685 10.1074/jbc.M212280200 [DOI] [PubMed] [Google Scholar]
- 43. Chaube R., Hess D. T., Wang Y. J., Plummer B., Sun Q. A., Laurita K., and Stamler J. S. (2014) Regulation of the skeletal muscle ryanodine receptor/Ca2+-release channel RyR1 by S-palmitoylation. J. Biol. Chem. 289, 8612–8619 10.1074/jbc.M114.548925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fredericks G. J., Hoffmann F. W., Rose A. H., Osterheld H. J., Hess F. M., Mercier F., and Hoffmann P. R. (2014) Stable expression and function of the inositol 1,4,5-triphosphate receptor requires palmitoylation by a DHHC6/selenoprotein K complex. Proc. Natl. Acad. Sci. U.S.A. 111, 16478–16483 10.1073/pnas.1417176111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ji Y., Leymarie N., Haeussler D. J., Bachschmid M. M., Costello C. E., and Lin C. (2013) Direct detection of S-palmitoylation by mass spectrometry. Anal Chem 85, 11952–11959 10.1021/ac402850s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hwang C., Sinskey A. J., and Lodish H. F. (1992) Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257, 1496–1502 10.1126/science.1523409 [DOI] [PubMed] [Google Scholar]
- 47. Chakravarthi S., Jessop C. E., and Bulleid N. J. (2006) The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Reports 7, 271–275 10.1038/sj.embor.7400645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Appenzeller-Herzog C. (2011) Glutathione- and non-glutathione-based oxidant control in the endoplasmic reticulum. J. Cell Sci. 124, 847–855 10.1242/jcs.080895 [DOI] [PubMed] [Google Scholar]
- 49. Hudson D. A., Gannon S. A., and Thorpe C. (2015) Oxidative protein folding: from thiol-disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radic. Biol. Med. 80, 171–182 10.1016/j.freeradbiomed.2014.07.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kang S., Kang J., Kwon H., Frueh D., Yoo S. H., Wagner G., and Park S. (2008) Effects of redox potential and Ca2+ on Inositol 1,4,5-trisphosphate receptor L3–1 loop region: implications for receptor regulation. J. Biol. Chem. 283, 25567–25575 10.1074/jbc.M803321200 [DOI] [PubMed] [Google Scholar]
- 51. Higo T., Hattori M., Nakamura T., Natsume T., Michikawa T., and Mikoshiba K. (2005) Subtype-specific and ER lumenal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell 120, 85–98 10.1016/j.cell.2004.11.048 [DOI] [PubMed] [Google Scholar]
- 52. Schug Z. T., da Fonseca P. C., Bhanumathy C. D., Wagner L. 2nd, Zhang X., Bailey B., Morris E. P., Yule D. I., and Joseph S. K. (2008) Molecular characterization of the inositol 1,4,5-trisphosphate pore-forming segment. J. Biol. Chem. 283, 2939–2948 10.1074/jbc.M706645200 [DOI] [PubMed] [Google Scholar]
- 53. Go Y. M., Duong D. M., Peng J., and Jones D. P. (2011) Protein cysteines map to functional networks according to steady-state level of oxidation. J. Proteomics Bioinform. 4, 196–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Booth D. M., Enyedi B., Geiszt M., Várnai P., and Hajnóczky G. (2016) Redox nanodomains are induced by and control calcium signaling at the ER–mitochondrial interface. Mol. Cell 63, 240–248 10.1016/j.molcel.2016.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Paknejad N., and Hite R. K. (2018) Structural basis for the regulation of inositol trisphosphate receptors by Ca2+ and IP3. Nat. Struct. Mol. Biol. 25, 660–668 10.1038/s41594-018-0089-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yamazaki H., Chan J., Ikura M., Michikawa T., and Mikoshiba K. (2010) Tyr-167/Trp-168 in type1/3 inositol 1,4,5-trisphosphate receptor mediates functional coupling between ligand binding and channel opening. J. Biol. Chem. 285, 36081–36091 10.1074/jbc.M110.140129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rong Y. P., Barr P., Yee V. C., and Distelhorst C. W. (2009) Targeting Bcl-2 based on the interaction of its BH4 domain with the inositol 1,4,5-trisphosphate receptor. Biochim. Biophys. Acta 1793, 971–978 10.1016/j.bbamcr.2008.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Casey J. P., Hirouchi T., Hisatsune C., Lynch B., Murphy R., Dunne A. M., Miyamoto A., Ennis S., van der Spek N., O'Hici B., Mikoshiba K., and Lynch S. A. (2017) A novel gain-of-function mutation in the ITPR1 suppressor domain causes spinocerebellar ataxia with altered Ca2+ signal patterns. J. Neurol. 264, 1444–1453 10.1007/s00415-017-8545-5 [DOI] [PubMed] [Google Scholar]
- 59. Szlufcik K., Bultynck G., Callewaert G., Missiaen L., Parys J. B., and De Smedt H. (2006) The suppressor domain of inositol 1,4,5-trisphosphate receptor plays an essential role in the protection against apoptosis. Cell Calcium 39, 325–336 10.1016/j.ceca.2005.11.007 [DOI] [PubMed] [Google Scholar]
- 60. Back S. H., and Kaufman R. J. (2012) Endoplasmic reticulum stress and type 2 diabetes. Annu. Rev. Biochem. 81, 767–793 10.1146/annurev-biochem-072909-095555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Simmen T., Lynes E. M., Gesson K., and Thomas G. (2010) Oxidative protein folding in the endoplasmic reticulum: tight links to the mitochondria-associated membrane (MAM). Biochim. Biophys. Acta 1798, 1465–1473 10.1016/j.bbamem.2010.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mekahli D., Bultynck G., Parys J. B., De Smedt H., and Missiaen L. (2011) Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 3, a004317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ivanova H., Vervliet T., Missiaen L., Parys J. B., De Smedt H., and Bultynck G. (2014) Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim. Biophys. Acta 1843, 2164–2183 10.1016/j.bbamcr.2014.03.007 [DOI] [PubMed] [Google Scholar]
- 64. Joseph S. K., and Samanta S. (1993) Detergent solubility of the inositol trisphosphate receptor in rat brain membranes: evidence for association of the receptor with ankyrin. J. Biol. Chem. 268, 6477–6486 [PubMed] [Google Scholar]
- 65. Alzayady K. J., Wagner L. E. 2nd, Chandrasekhar R., Monteagudo A., Godiska R., Tall G. G., Joseph S. K., and Yule D. I. (2013) Functional inositol 1,4,5-trisphosphate receptors assembled from concatenated homo- and heteromeric subunits. J. Biol. Chem. 288, 29772–29784 10.1074/jbc.M113.502203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tietze F. (1969) Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal. Biochem. 27, 502–522 10.1016/0003-2697(69)90064-5 [DOI] [PubMed] [Google Scholar]
- 67. Muthuramalingam M., Matros A., Scheibe R., Mock H. P., and Dietz K. J. (2013) The hydrogen peroxide-sensitive proteome of the chloroplast in vitro and in vivo. Front. Plant Sci. 4, 54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mignery G. A., Newton C. L., Archer B. T. 3rd, and Südhof T. C. (1990) Structure and expression of the rat inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 265, 12679–12685 [PubMed] [Google Scholar]
- 69. Iwai M., Tateishi Y., Hattori M., Mizutani A., Nakamura T., Futatsugi A., Inoue T., Furuichi T., Michikawa T., and Mikoshiba K. (2005) Molecular cloning of mouse type 2 and type 3 inositol 1,4,5-trisphosphate receptors and identification of a novel type 2 receptor splice variant. J. Biol. Chem. 280, 10305–10317 10.1074/jbc.M413824200 [DOI] [PubMed] [Google Scholar]
- 70. Blondel O., Takeda J., Janssen H., Seino S., and Bell G. (1993) Sequence and functional characterization of a third inositol trisphosphate receptor subtype, IP3R-3, expressed in pancreatic islets, kidney, gastrointestinal tract, and other tissues. J. Biol. Chem. 268, 11356–11363 [PubMed] [Google Scholar]
- 71. Zheng L., Baumann U., and Reymond J. L. (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115 10.1093/nar/gnh110 [DOI] [PMC free article] [PubMed] [Google Scholar]