

Abstract
Upon repeated exposure to endotoxin or lipopolysaccharide (LPS), myeloid cells enter a refractory state called endotoxin tolerance as a homeostatic mechanism. In innate immune cells, LPS is recognized by co-receptors Toll-like receptor 4 (TLR4) and CD-14 to initiate an inflammatory response for subsequent cytokine production. One such cytokine, interleukin (IL)-27, is produced by myeloid cells in response to bacterial infection. In monocytes, IL-27 has proinflammatory functions such as up-regulating TLR4 expression for enhanced LPS-mediated cytokine production; alternatively, IL-27 induces inhibitory functions in activated macrophages. This study investigated the effects of IL-27 on the induction of endotoxin tolerance in models of human monocytes compared with macrophages. Our data demonstrate that IL-27 inhibits endotoxin tolerance by up-regulating cell surface TLR4 expression and soluble CD14 production to mediate stability of the surface LPS-TLR4-CD14 complex in THP-1 cells. In contrast, elevated basal expression of membrane-bound CD14 in phorbol 12-myristate 13-acetate (PMA)–THP-1 cells, primary monocytes, and primary macrophages may promote CD14-mediated endocytosis and be responsible for the preservation of an endotoxin-tolerized state in the presence of IL-27. Overall, the efficacy of IL-27 in inhibiting endotoxin tolerance in human THP-1 monocytes and PMA–THP-1 macrophages is affected by membrane-bound and soluble CD14 expression.
Keywords: inflammation, cytokine action, Toll-like receptor 4 (TLR4), lipopolysaccharide (LPS), NF-κB, CD14, endotoxin tolerance, interleukin-27 (IL-27), monocytes/macrophages
Introduction
Endotoxin or lipopolysaccharide (LPS)5 tolerance is defined as a refractory, anti-inflammatory state in response to a secondary lethal dose of LPS following a primary low-dose LPS exposure (1). The endotoxin-tolerant state is characterized by LPS hyporesponsiveness whereby proinflammatory cytokine production, such as tumor necrosis factor (TNF)-α, is decreased compared with cytokine levels after a single dose of LPS, as observed in vitro (2, 3), ex vivo (4, 5), and in vivo (5–7). Endotoxin tolerance is a control mechanism that myeloid cells implement to prevent tissue damage from overactive inflammatory responses, which can cause sepsis syndrome. However, it is important to note that the inability to fight secondary infection in an endotoxin-tolerized state can be detrimental to immunocompromised individuals.
Upon Gram-negative bacterial infection, LPS-binding protein (LBP) and cluster of differentiation (CD)-14 are required for LPS binding to Toll-like receptor 4 (TLR4) and scaffold protein myeloid differentiation protein (MD)-2 to initiate signal transduction (8–10). LPS signaling via TLR4-MD2-CD14 induces both the myeloid differentiation response gene 88(MyD88)–dependent and Toll/IL-1R domain–containing adaptor-inducing IFN-β (TRIF)–dependent pathways for downstream cytokine production (11–13). Immediately following MyD88 signaling, CD14-mediated endocytosis internalizes TLR4 to traffic to the endosome for subsequent TRIF-dependent signaling (14–17).
In myeloid cells, LPS ligation with TLR4 induces cytokine production, including interleukin (IL)-27 subunits IL-27p28 and Epstein-Barr virus–induced gene 3 (EBI3) (18, 19). IL-27 is part of the IL-12 family of heterodimeric cytokines including IL-12, IL-23, IL-30 (IL-27p28), and IL-35 (20–22). IL-27 receptor subunits WSX-1 and gp130 (glycoprotein-130) are expressed on monocytes and macrophages among other cells to signal using the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway (23–26).
Our laboratory has shown previously that IL-27 enhances TLR4 expression for the amplification of LPS-mediated signaling (27–29). Specifically, IL-27–treated monocytes up-regulate TLR4 expression (27–29) and TLR4-CD14 colocalization (27) for greater LPS responsiveness. IL-27–mediated increases in TLR4 expression have also been attributed to greater LPS-induced inflammasome activation in monocytes (28). There are discrepancies between the role of IL-27 in myeloid cells with proinflammatory functions in monocytes (27–30) and anti-inflammatory effects in macrophages (31–33). In this study, we examined whether IL-27 modulated endotoxin tolerance in both human monocytes and macrophages. We investigated the effects of IL-27 on TLR4/CD14 receptor expression, LPS binding, and signal transduction in THP-1 monocytes compared with PMA–THP-1 macrophages as well as downstream LPS-mediated cytokine production.
Upon IL-27 treatment, endotoxin-tolerized THP-1 cells show complete restoration of TNF-α production following LPS challenge, whereas PMA–THP-1 cells, primary monocytes, and primary macrophages show only moderate tolerance inhibition. The heightened effects of IL-27 on the induction of endotoxin tolerance in THP-1 cells compared with PMA–THP-1 cells may depend on CD14-mediated mechanisms for subsequent TLR4 signal transduction in the endosome. Understanding the mechanisms used by IL-27 to alter LPS responsiveness may reveal novel targets for prevention, diagnosis, or treatment of inflammatory illnesses affiliated with endotoxin tolerance, such as septic shock.
Results
IL-27 enhances TLR4 expression in monocytes and macrophages
We demonstrated previously that IL-27 enhances TLR4 expression in THP-1 cells and primary monocytes (27–29). To test whether IL-27 also modulates TLR4 expression in human macrophages, we used THP-1 monocytes, PMA-treated THP-1 macrophages (PMA–THP-1), primary monocytes, and primary macrophages as model systems. To establish monocyte and macrophage phenotypes in these cells, CD14 and CD16 expression were measured by flow cytometry (Fig. 1A). In agreement with of others (34, 35), we found that THP-1 cells had moderate CD14 and CD16 expression, whereas PMA treatment increased CD14 and decreased CD16 expression in PMA–THP-1 cells relative to THP-1 cells. Primary human monocytes exhibited three distinct populations: classical phagocytic monocytes (high CD14 and low CD16), intermediate proinflammatory monocytes (high CD14 and moderate CD16), and nonclassical patrolling monocytes (low CD14 and high CD16), as expected (36–38). Furthermore, primary macrophages showed moderate CD14 and higher CD16 compared with classical primary monocytes (Fig. 1A).
Figure 1.

IL-27 enhances TLR4 but not CD14 expression in classical monocytes and macrophages. A, resting THP-1 cells, PMA–THP-1 cells, primary human monocytes, and primary human macrophages were stained with anti-CD16 and anti-CD14 antibodies and acquired by flow cytometry. Autofluorescence (gray) and stained cells (black) were overlaid in contour plots. B, THP-1 cells, PMA–THP-1 cells, primary human monocytes, and primary human macrophages were stimulated with LPS (10 ng/ml), IL-27 (100 ng/ml), or LPS + IL-27 simultaneously for 24 h. Cells were stained with anti-TLR4 and anti-CD14 antibodies and acquired by flow cytometry. The -fold change of median fluorescence intensity was calculated relative to medium (med) controls. Graphs present the median ± S.D. of at least three independent experiments or at least three different blood donors.
To assess the effects of IL-27 on LPS co-receptors, we examined TLR4 and CD14 expression in response to LPS and IL-27 co-stimulation in all cell types (Fig. 1B). THP-1 cells, PMA–THP-1 cells, primary monocytes, and primary macrophages were stimulated with LPS (10 ng/ml), IL-27 (100 ng/ml), or both LPS + IL-27 for 24 h to measure surface TLR4 and CD14 expression. Compared with untreated cells (referred to as “med”), IL-27- and LPS + IL-27–treated THP-1 cells had significantly enhanced TLR4 expression, whereas LPS alone induced only a slight up-regulation of TLR4 expression. PMA–THP-1 cells, primary monocytes, and primary macrophages also displayed significantly increased TLR4 expression in response to IL-27 alone. PMA–THP-1 cells had a significant reduction in TLR4 expression in response to LPS or LPS + IL-27 compared with untreated or IL-27–treated cells, respectively, and significantly higher levels in response to LPS + IL-27 than LPS alone. There was a significant increase in CD14 expression in THP-1 cells in response to LPS, IL-27, and LPS + IL-27 stimulation relative to untreated cells. In PMA–THP-1 cells and primary macrophages, LPS and LPS + IL-27 stimulation down-regulated membrane CD14 expression relative to either unstimulated or IL-27–treated cells. Conversely, CD14 expression displayed no significant changes between the conditions in primary monocytes. Interestingly, THP-1 cells expressed the highest levels of TLR4 (data not shown) and the lowest CD14 expression (Fig. 1A) across all cell types, indicating that THP-1 cells may have an altered LPS responsiveness compared with the other cell types examined.
IL-27 inhibits induction of endotoxin tolerance in monocytes and macrophages
Because IL-27 modulates LPS responsiveness (27–30) and LPS receptor expression, we explored the effects of IL-27 on repeated LPS stimulation and induction of endotoxin tolerance in monocytes and macrophages. THP-1 cells, PMA–THP-1 cells, primary monocytes, and primary macrophages were stimulated with LPS (10 ng/ml), IL-27 (100 ng/ml), or both LPS + IL-27 for 24 h. Then cells were washed and challenged with or without a secondary dose of LPS (100 ng/ml) for 4 h, and TNF-α production was measured in cell-free supernatants. In THP-1 cells, LPS + IL-27 treatment induced significantly more TNF-α than IL-27 or LPS alone in unchallenged cells (2° medium) (Fig. S1A). As expected, following LPS challenge (2° LPS), LPS pretreated cells (tolerized cells) produced significantly less TNF-α compared with medium controls (untolerized cells) (Figs. 1A and 2A). IL-27–pretreated THP-1 cells (1° IL-27)6 followed by LPS challenge (2° LPS) had significantly increased TNF-α production compared with untolerized cells (Fig. 2A, 1° med), confirming results previously obtained in human monocytes (27, 29). However, the addition of IL-27 to the LPS tolerizing dose (1° LPS + IL-27) resulted in significantly enhanced TNF-α in response to LPS challenge (2° LPS) compared with cells tolerized with LPS alone (1° LPS) as well as untolerized cells (1° med), indicating that IL-27 effectively blocked induction of endotoxin tolerance and even enhanced TNF-α expression in tolerized THP-1 cells (Fig. 2A).
Figure 2.
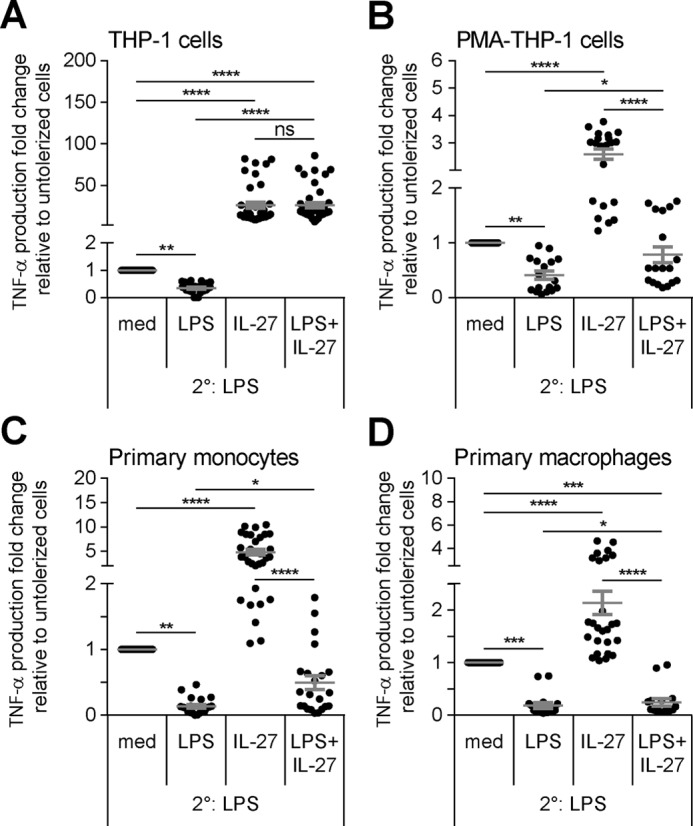
Co-treatment of IL-27 with a tolerizing dose of LPS inhibits induction of endotoxin tolerance in human monocytes and macrophages. THP-1 cells (A), PMA–THP-1 cells (B), primary human monocytes (C), and primary human macrophages (D) were stimulated with LPS (10 ng/ml), IL-27 (100 ng/ml), or LPS + IL-27 simultaneously for 24 h (1°). Cells were washed and challenged with LPS (100 ng/ml) for 4 h (2°). TNF-α production was measured in cell-free supernatants by ELISA. The -fold change of TNF-α production was calculated relative to untolerized (1° med/2° LPS) cells. Graphs present mean ± S.E. of at least six independent experiments or at least six different blood donors.
In PMA–THP-1 cells, primary monocytes, and primary macrophages, tolerized cells produced significantly less TNF-α relative to untolerized cells (Fig. 2, B–D), as expected. In all three cells types, pretreatment with IL-27 alone followed by LPS challenge significantly enhanced TNF-α production compared with untolerized cells, indicating that, in addition to monocytes, IL-27 enhances LPS responsiveness in human macrophages. LPS + IL-27–pretreated PMA–THP-1 cells, primary monocytes, and primary macrophages all showed significantly greater TNF-α production in response to 2° LPS compared with tolerized cells (Fig. 2, B–D), similarly observed were significantly elevated cytokine concentration in these cells (Fig. S1, B–D). Unlike in THP-1 cells, 2° LPS-induced TNF-α production in cells pretreated with 1° LPS + IL-27 remained lower than LPS-induced TNF-α produced from untolerized cells. Overall, IL-27 inhibited induction of endotoxin tolerance in both monocytes and macrophages, however, the magnitude of IL-27–mediated restoration of TNF-α production was greater in THP-1 cells compared with other cell types examined.
NF-κB/AP-1 activity is down-regulated in endotoxin-tolerized cells
LPS-TLR4 ligation results in immediate NF-κB p65/p50 activation and translocation from the cytoplasm to the nucleus (39). Although IL-27 is known to promote NF-κB signaling (26, 27), endotoxin-tolerized cells have been shown to reduce NF-κB activation relative to untolerized cells (40). To examine the effects of IL-27 on NF-κB signaling in endotoxin-tolerized cells, we compared NF-κB p65 and p50 nuclear translocation in response to LPS and/or IL-27 pretreatment in THP-1 and PMA–THP-1 cells. Cells were stimulated with LPS (10 ng/ml), IL-27 (100 ng/ml), or LPS + IL-27 for 24 h followed by a secondary dose of LPS (100 ng/ml) for 2 h. Cells were stained with antibodies against p65 and p50, and images were collected by confocal microscopy. Pixel intensity ratios of nuclear:cytoplasmic regions were calculated for each condition. In both THP-1 and PMA–THP-1 cells, LPS stimulation for 2 h (med/LPS) resulted in significantly enhanced p65 nuclear translocation compared with unstimulated controls (med/med) (Fig. 3, A and B). Although p50 nuclear translocation was significantly greater in LPS-treated THP-1 cells (med/LPS) relative to unstimulated cells, this was not observed in PMA–THP-1 cells (Figs. S2 and S3). Endotoxin-tolerized THP-1 and PMA–THP-1 cells showed reduced p65 nuclear translocation relative to untolerized cells; however, these changes only reached statistical significance in PMA–THP-1 cells (Fig. 3, A and B). The effect of IL-27 on NF-κB nuclear translocation was more prominent on the p65 subunit than the p50 (Figs. 3, S2, and S3). Finally, LPS + IL-27–pretreated PMA–THP-1 cells had comparable LPS-mediated p65 nuclear translocation to tolerized cells, indicating that the effects of IL-27 on inhibiting endotoxin tolerance may not be strictly dependent on NF-κB activation.
Figure 3.

IL-27 differentially affects NF-κB activity in tolerized THP-1 cells relative to PMA–THP-1 cells. THP-1 cells (A) and PMA–THP-1 (B) cells were stimulated with LPS (10 ng/ml), IL-27 (100 ng/ml), or LPS + IL-27 simultaneously for 24 h. Cells were washed and challenged with LPS (100 ng/ml) for 2 h. Cells were stained with anti-NF-κB p65 (red) and NucRed DNA stain (blue). The relative brightness of NF-κB p65 was measured in identically sized regions of the cell nucleus and cytoplasm. The images are representative of three independent experiments. DIC images are shown on the left of the panels. Merged images display overlays of NucRed and p65. A scale bar = 30 μm is displayed at the bottom left for each cell type. The nuclear/cytoplasmic ratios were calculated for each condition (bottom). Graphs present the mean ± S.E. of 18 cells/condition. Three representative images, including those presented here in A and B, also are depicted in Fig. S2, where they are denoted with an asterisk. THP-1 XBlue cells (C) and PMA–THP-1 XBlue cells (D) were stimulated with LPS (10 ng/ml), IL-27 (100 ng/ml), or LPS + IL-27 simultaneously for 24 h (1°). Cells were washed and challenged with LPS (100 ng/ml) for 24 h (2°) to allow for NF-κB–induced SEAP production and secretion. SEAP production was quantified using a colormetric QUANTI-BlueTM assay. Mean ± S.E. absorbance values are displayed. Data are representative of six independent experiments.
To further explore LPS-mediated transcription factor activity under endotoxin-tolerized conditions, we utilized THP-1 XBlue cells that express an NF-κB/AP-1–inducible secreted embryonic alkaline phosphatase (SEAP) reporter gene to quantify NF-κB/AP-1 activation. THP-1 XBlue cells and PMA–THP-1 XBlue cells were stimulated with either LPS (10 ng/ml) or IL-27 (100 ng/ml) or co-stimulated with LPS + IL-27 simultaneously for 24 h. Cells were then washed and stimulated with or without LPS (100 ng/ml) for 24 h to allow for NF-κB/AP-1–induced SEAP production and secretion. THP-1 XBlue cells pretreated with IL-27 and LPS + IL-27 followed by 2° medium showed increased activation of NF-κB/AP-1 compared with 1° medium controls, but LPS pretreatment had minimal effects (Fig. 3C). Upon 2° LPS challenge, untolerized (1° med) and IL-27–pretreated cells had significantly greater LPS-induced NF-κB/AP-1 activation compared with tolerized (1° LPS) and LPS + IL-27–pretreated THP-1 XBlue cells, respectively. In PMA–THP-1 XBlue cells, IL-27 stimulation alone activated NF-κB/AP-1 relative to medium control cells. However, following 2° LPS challenge, neither IL-27 nor LPS + IL-27 altered NF-κB/AP-1 signaling relative to medium or LPS-treated cells, respectively (Fig. 3D). Similar to THP-1 XBlue cells, following LPS challenge, LPS- or LPS + IL-27–pretreated PMA–THP-1 XBlue cells showed reduced NF-κB/AP-1 activity compared with cells without LPS pretreatment, showing significantly reduced NF-κB/AP-1 activation upon induction of endotoxin tolerance. However, endotoxin tolerance results in a greater decrease in NF-κB/AP-1 activity in PMA–THP-1 XBlue compared with THP-1 XBlue cells, which supports the notion that CD14high-expressing PMA–THP-1 cells are more sensitive to LPS stimulation than THP-1 cells.
IL-27 differentially impacts LPS binding in tolerized THP-1 cells compared with PMA–THP-1 cells, primary monocytes, and primary macrophages
Given that we detected lower CD14 expression levels in THP-1 cells than in the other cells examined (Fig. 1A) and that CD14 regulates LPS-TLR4 binding (8, 41), there may be differences in CD14-mediated LPS binding between cell types. Thus, we postulated that IL-27 may differentially affect LPS binding in THP-1 cells compared with PMA–THP-1 cells, primary monocytes, and primary macrophages and that this may be dependent on CD14 expression. In addition to the membrane-bound form, CD14 exists in soluble form to enhance LPS responsiveness in cells lacking membrane-bound CD14 while inhibiting LPS binding in CD14high-expressing cells (42, 43). Using LPS conjugated to Alexa Fluor 488 (LPS-Alexa), we investigated the effects of IL-27 and LPS on secondary LPS-Alexa binding by flow cytometry; and to investigate the requirement for CD14, we used the matrix metalloproteinase inhibitor batimastat (BB-94) to block cleavage membrane CD14 (44–48). Resting PMA–THP-1 cells, primary monocytes, and primary macrophages exhibited significantly greater LPS-Alexa positive cells compared with THP-1 cells (Fig. S4), which correlated with levels of CD14 expression in these cell types (Fig. 1A). To investigate the effects of IL-27 on LPS-Alexa binding, THP-1 cells, PMA–THP-1 cells, primary monocytes, and primary macrophages were treated with LPS (10 ng/ml), IL-27 (100 ng/ml), or LPS + IL-27 for 24 h in the presence or absence of BB-94 (25 μm), as indicated, and then washed and incubated with LPS-Alexa (100 ng/ml) for 1 h. In THP-1 cells, pretreatment with LPS, IL-27, and LPS + IL-27 significantly increased LPS-Alexa binding relative to medium controls (Fig. 4A, black dots). In PMA–THP-1 cells, primary monocytes, and primary macrophages, LPS-tolerized and LPS + IL-27–pretreated cells had reduced LPS-Alexa positive cells, whereas pretreatment with IL-27 did not have any notable effect (Fig. 4, B–D, black dots). The addition of BB-94 significantly enhanced LPS-Alexa binding in THP-1 cells but not in PMA–THP-1 cells, primary monocytes, or primary macrophages (Fig. 4, A–D, gray dots), suggesting that cleavage of CD14 may contribute to the enhanced LPS binding in THP-1 cells. Interestingly, although minimally altering LPS-Alexa binding, BB-94 treatment abolished all LPS-induced TNF-α production following LPS and/or IL-27 pretreatment in the four cell types examined (data not shown).
Figure 4.

IL-27 partially affects LPS binding in tolerized THP-1 cells but not in PMA–THP-1 cells, primary monocytes, or primary macrophages. THP-1 cells (A), PMA–THP-1 cells (B), primary monocytes (C), and primary macrophages (D) were stimulated with LPS (10 ng/ml), IL-27 (100 ng/ml), or LPS + IL-27 simultaneously for 24 h with or without BB-94 (25 μm). Cells were subsequently incubated with LPS-Alexa (100 ng/ml) for 1 h at 37 °C. Bound LPS was measured by flow cytometry. The -fold changes were calculated for LPS-, IL-27-, and LPS + IL-27–treated cells relative to medium controls. Graphs present data from at least three independent experiments.
IL-27 increases production of soluble CD14 in THP-1 cells but not in PMA–THP-1 cells, primary monocytes, and primary macrophages
Soluble CD14 (sCD14) and LBP are required for LPS binding (8), and therefore, we sought to determine whether IL-27 directly affects sCD14 or LBP production in monocytes and macrophages. THP-1 cells, PMA–THP-1 cells, primary monocytes, and primary macrophages were stimulated with LPS (10 ng/ml), IL-27 (100 ng/ml), or LPS + IL-27 for 24 h. Soluble LBP and sCD14 production was measured in cell-free supernatants by ELISA. Soluble LBP production was undetectable across all conditions in THP-1 cells and PMA–THP-1 cells (data not shown). In THP-1 cells, IL-27 and LPS + IL-27 treatment alone increased sCD14 production compared with medium- or LPS-stimulated controls (Fig. 5A, black dots). In PMA–THP-1 cells, LPS or LPS + IL-27 stimulation decreased the amount of sCD14 produced relative to unstimulated controls (Fig. 5B, black dots). Soluble CD14 production was not altered by the addition of IL-27 in PMA–THP-1 cells. In primary human monocytes, IL-27 significantly down-regulated sCD14 expression (Fig. 5C, black dots), whereas in primary macrophages, neither IL-27 nor LPS had a significant effect on its production (Fig. 5D, black dots). It is important to note that unstimulated PMA–THP-1 cells, primary monocytes, and primary macrophages produced at least 10-fold more sCD14 than unstimulated THP-1 cells (Fig. 5, A and B, black dots). This suggests that for PMA–THP-1 cells, primary monocytes, and primary macrophages, the higher levels of surface CD14 expression result in higher levels of sCD14 generation compared with THP-1 cells. Using BB-94 to block the cleavage of membrane CD14 (44–48), sCD14 was significantly reduced in THP-1 cells, PMA–THP-1 cells, and primary monocytes treated with BB-94, relative to solvent controls across all conditions (Fig. 5, A–C, gray dots). BB-94 did not significantly alter sCD14 production in primary macrophages (Fig. 5D, gray dots). Overall, IL-27–induced sCD14 production in THP-1 cells may contribute to differences in LPS responsiveness in PMA–THP-1 cells and primary cells compared with THP-1 cells.
Figure 5.

IL-27 induces soluble CD14 in THP-1 cells but not in PMA–THP-1 cells. THP-1 cells (A), PMA–THP-1 cells (B), primary monocytes (C), and primary macrophages (D) were stimulated with LPS (10 ng/ml), IL-27 (100 ng/ml), or LPS + IL-27 simultaneously for 24 h (1°) in the absence or presence of BB-94 inhibitor (25 μm) or DMSO solvent control. Soluble CD14 production was measured in cell-free supernatants. Graphs present mean ± S.E. of at least three independent experiments.
IL-27 enhances LPS-induced IFN-β production in PMA–THP-1 cells, primary monocytes, and primary macrophages
LPS-TLR4-CD14 signaling induces MyD88-dependent TNF-α production, and subsequent CD14-mediated endocytosis of the LPS-TLR4 complex allows for endosomal TRIF-dependent type I IFN production (49). As IL-27 promotes sCD14 expression in THP-1 cells (Fig. 5A), we used IFN-β production as a readout for CD14-dependent internalization of the LPS-TLR4 complex. THP-1 cells, PMA–THP-1 cells, primary monocytes, and primary macrophages were stimulated with LPS (10 ng/ml), IL-27 (100 ng/ml), or LPS + IL-27 for 24 h and subsequently washed and challenged with or without LPS (100 ng/ml) for 4 h. IFN-β production was measured in cell-free supernatants by ELISA. THP-1 cells did not produce detectable amounts of IFN-β in response to LPS, IL-27, or LPS + IL-27 (Fig. 6A, black dots); however, PMA–THP-1 cells produced IFN-β in response to LPS treatment (Fig. 6A, gray dots) with similar trends to primary monocytes and primary macrophages (Fig. 6B). Specifically, PMA–THP-1 cells co-stimulated with LPS + IL-27 resulted in significantly higher levels of IFN-β compared with cells that were unstimulated or those treated with LPS or IL-27 alone (Fig. 6A, 1° only and 2° medium, gray dots). Upon examination of IFN-β production in our model of tolerized PMA–THP-1 cells, primary monocytes, and primary macrophages, we observed a similar expression pattern as that for TNF-α (Figs. 6, A and B, and 2, B–D). In response to 2° LPS, tolerized cells produced significantly less IFN-β production than untolerized cells, whereas IL-27 priming significantly enhanced LPS-induced IFN-β production compared with untolerized cells. Interestingly, pretreatment of PMA–THP-1 cells with LPS + IL-27 enhanced IFN-β production following 2° LPS challenge relative to tolerized cells, although these treatments did not reach statistically significant differences in primary monocytes or macrophages. These results indicate that LPS-mediated induction of IFN-β, like TNF-α, is sensitive to endotoxin tolerance. Furthermore, in CD14high-expressing cells, but not in THP-1 cells, the LPS-TLR4-CD14 complex is efficiently internalized to induce TRIF-mediated signaling. In PMA–THP-1 cells, IL-27–mediated inhibition of endotoxin tolerance translated to enhanced IFN-β production relative to tolerized cells, similar to TNF-α; however, IFN-β production in LPS- or LPS + IL-27–tolerized PMA–THP-1 cells, primary monocytes, and primary macrophages was below the sensitivity range of the ELISA. Overall, surface CD14 expression correlated with IFN-β production in response to LPS stimulation and IL-27–enhanced IFN-β in untolerized PMA–THP-1 cells, primary monocytes, and primary macrophages.
Figure 6.
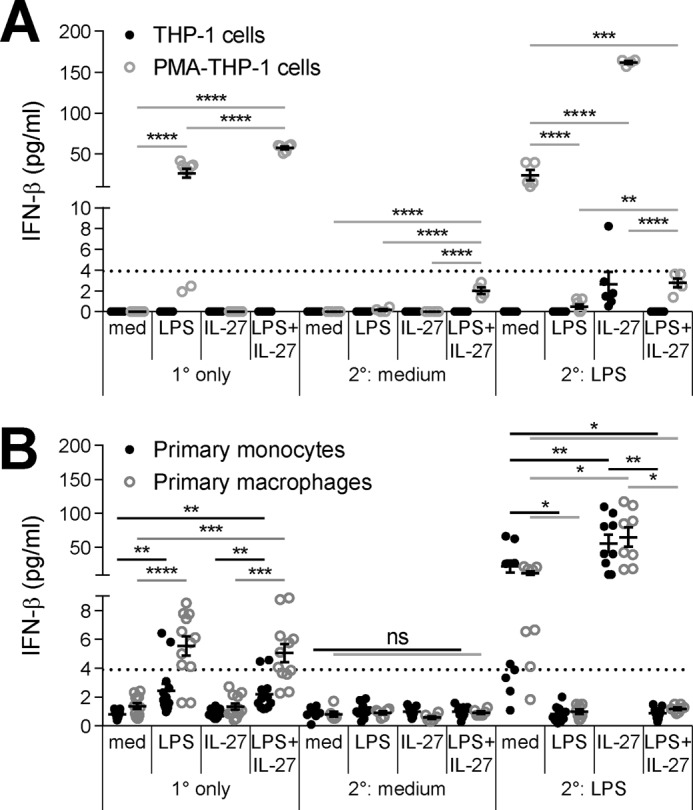
IL-27 up-regulates IFN-β production in tolerized PMA–THP-1 cells, primary monocytes, and primary macrophages. THP-1 and PMA–THP-1 cells (A) as well as primary monocytes and macrophages (B) were stimulated with LPS (10 ng/ml), IL-27 (100 ng/ml), or LPS + IL-27 simultaneously for 24 h (1°). Cells were washed and challenged with LPS (100 ng/ml) for 4 h (2°). Human IFN-β production was measured in cell-free supernatants. The graphs present mean ± S.E. of at least three independent experiments or at least three independent blood donors. The dashed line depicts the lower sensitivity of the human IFN-β ELISA kit at 3.90625 pg/ml.
Overexpression of CD14 alters the effects of IL-27 on LPS responsiveness
To further investigate the role of CD14 in IL-27–modulated endotoxin tolerance we used THP-1 cells stably transfected with a CD14-expressing plasmid (CD14-THP-1 cells) to model CD14high THP-1 cells. CD14-THP-1 cells have TLR4 and CD14 expression comparable to primary monocytes (50). To assess how IL-27 affected endotoxin tolerance in this cell model, CD14-THP-1 and PMA-treated CD14-THP-1 (PMA–CD14-THP-1) cells were stimulated with LPS (10 ng/ml), IL-27 (100 ng/ml), or LPS + IL-27 for 24 h and subsequently washed and challenged with or without LPS (100 ng/ml) for 4 h, as described above. CD14-THP-1 cells produced a lower amount of TNF-α in response to 1° LPS + IL-27 after the 24-h stimulation without LPS challenge (2° medium) relative to other cell types examined (Figs. S5A and S1). However, in response to the 2° LPS challenge, untolerized CD14-THP-1 cells (1° medium) produced more TNF-α than the untolerized THP-1 cells (Fig. 7A and Fig. S1A), and TNF-α levels were comparable to PMA–THP-1 cells and primary monocytes (Figs. S5A and S1, B and C). As expected, TNF-α production in response to 2° LPS challenge was decreased in tolerized CD14-THP-1 cells (1° LPS) compared with untolerized cells (1° med) (Fig. 7B). Interestingly, similar to THP-1 cells, IL-27 or LPS + IL-27 pretreatment of CD14-THP-1 cells resulted in LPS-induced TNF-α production that surpassed levels produced by untolerized cells. Upon PMA treatment of CD14-THP-1 cells, PMA–CD14-THP-1 cells presented similar trends to PMA–THP-1 cells (Figs. 7B and 2B); tolerized cells had reduced TNF-α production and IL-27–enhanced TNF-α production relative to untolerized cells. PMA–CD14-THP-1 cells had significantly enhanced LPS-induced TNF-α production upon 1° LPS + IL-27 pretreatment relative to tolerized cells (Fig. 7B), although the increased cytokine concentration did not reach statistical significance (Fig. S5B).
Figure 7.
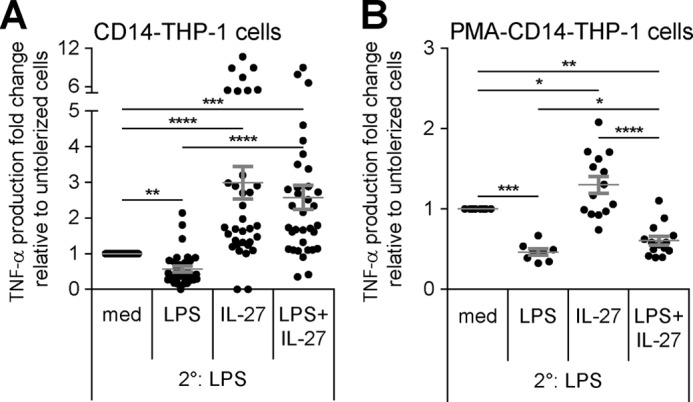
Overexpression of CD14 in THP-1 cells does not alter IL-27–mediated inhibition of endotoxin tolerance. CD14-THP-1 cells (A) and PMA–CD14-THP-1 cells (B) were stimulated with LPS (10 ng/ml), IL-27 (100 ng/ml), or LPS + IL-27 simultaneously for 24 h (1°). Cells were washed and challenged with LPS (100 ng/ml) for 4 h (2°). TNF-α production was measured in cell-free supernatants by ELISA. The -fold change of TNF-α production was calculated relative to untolerized (1° med/2° LPS) cells. Graphs present mean ± S.E. of at least six independent experiments.
Because CD14-THP-1 cells express high levels of stable CD14 and respond similarly to IL-27–treated THP-1 cells in endotoxin-tolerized conditions, we examined whether the addition of exogenous sCD14 to THP-1 cells would affect LPS- and/or IL-27–induced TNF-α in tolerized and untolerized cells. THP-1 cells were stimulated with 1° LPS (10 ng/ml), IL-27 (100 ng/ml), or LPS + IL-27 for 24 h and subsequently washed and challenged with or without 2° LPS (100 ng/ml) and/or recombinant sCD14 (500 ng/ml) for 4 h. Interestingly, the addition of recombinant sCD14 enhanced TNF-α production in THP-1 cells in all conditions where they received IL-27, regardless of the presence or absence of LPS (Fig. 8A), suggesting an association between IL-27 and sCD14 for enhanced TNF-α production. Recombinant CD14 did not have a statistically significant effect on TNF-α production in PMA–THP-1 cells in our model (Fig. 8B). In addition to up-regulating TLR4 expression, IL-27 may interact with CD14 or mediate CD14 production mechanisms in CD14low-expressing cells to further contribute to LPS responsiveness.
Figure 8.

The addition of recombinant CD14 with IL-27 enhances TNF-α production in tolerized THP-1 cells but not in PMA–THP-1 cells. THP-1 cells (A) and PMA–THP-1 cells (B) were stimulated with LPS (10 ng/ml), IL-27 (100 ng/ml), or LPS + IL-27 simultaneously for 24 h (1°). Cells were washed and challenged with LPS (100 ng/ml) for 4 h (2°) with recombinant soluble CD14 (500 ng/ml) added 30 min prior to 2° medium or LPS. TNF-α production was measured in cell-free supernatants. Graphs present mean ± S.E. of at least five independent experiments.
Discussion
This study focused on the effects of IL-27 on LPS-mediated endotoxin tolerance and found that IL-27 differentially inhibited the induction of endotoxin tolerance in human myeloid cells dependent on both membrane and soluble CD14 expression. In CD14high-expressing primary monocytes, PMA–THP-1 cells, and primary macrophages, IL-27 treatment allowed for a partial restoration of LPS responsiveness while completely inhibiting endotoxin tolerance induction in CD14low-expressing THP-1 cells. We suggest that expression levels of membrane or soluble CD14 may determine the efficacy of IL-27 in restoring cytokine production in our endotoxin tolerance model.
Gram-negative bacteria infection results in exposure to LPS, initiating an immune response that causes symptoms such as fever and, in severe cases, sepsis (51–53). In endotoxin tolerance, myeloid cell signaling is reprogrammed to prevent excessive inflammation and to protect against tissue damage and septic shock. On the other hand, the lack of responsiveness in endotoxin-tolerized cells may impede clearance of bacteria (54–56).
IL-27 is a biomarker for sepsis disease severity (57–60), and although it is protective against bacteria-induced inflammatory damage, IL-27 also inhibits antibacterial immunity, resulting in increased bacterial growth and survival (31, 61). Thus, the exploration of specific mechanisms utilized by IL-27 to modulate LPS signaling could present novel targets for disease prevention and treatment.
NF-κB p50 homodimers are implicated as a mechanism for reduced cytokine production in endotoxin-tolerized monocytes by blocking NF-κB p65/p50 from binding to promoter regions (62, 63). Although the NF-κB p65 protein is not induced in response to LPS, expression of the p50 precursor, called p105, is up-regulated (62). Cleavage of p105 gives rise to p50 and p50 homodimers for the inhibition of LPS-mediated TNF-α production following an accumulation of p50 (64, 65). How IL-27 affects p50 homodimer formation in endotoxin-tolerized cells is unknown; however, our data suggest that IL-27 impacts LPS-mediated p50 nuclear localization in THP-1 cells.
Previously, IL-27 stimulation was shown to enhance NF-κB p65 and p50 DNA binding in monocytes (27). This supports our observation that IL-27 significantly enhanced LPS-mediated p65 nuclear translocation relative to unstimulated cells in both THP-1 and PMA–THP-1 cells. On the other hand, the same conditions did not alter p50 nuclear translocation in PMA–THP-1 cells and even decreased the nuclear/cytoplasmic ratios of p50 in THP-1 cells. In turn, IL-27 may promote the up-regulation of p50 expression, resulting in greater cytoplasmic p50 protein levels. LPS + IL-27 did not significantly alter p65 activity in tolerized THP-1 and PMA–THP-1 cells. This could be attributed to other signaling molecules involved in endotoxin tolerance, such as IRAK-M and SHIP-1 (4, 7, 66–69), which also contribute to the regulation of NF-κB activation (70).
Similar to IL-27, IFN-γ pretreatment inhibits endotoxin tolerance induction in monocytes (71). It is interesting to note that with a primary dose of LPS at 10 ng/ml, TNF-α production was restored by IFN-γ pretreatment but not up to the level of untolerized cells in primary monocytes. These results are comparable with the effects of IL-27 on TNF-α production in tolerized primary monocytes. IFN-γ did not dramatically affect the NF-κB signal transduction pathway in tolerized monocytes (71), which is similar to the effects of IL-27 in tolerized THP-1 cells and PMA–THP-1 cells in our study. Chen and Ivashkiv (71) did not explore links between CD14 and IFN-γ but rather determined that IFN-γ-mediated inhibition of endotoxin tolerance occurs because of chromatin modifications for restored transcription factor binding to TNF and IL6 promoters. Our data do not rule out changes in chromatin remodeling as an additional mechanism for IL-27–mediated inhibition of endotoxin tolerance, and it is likely that multiple mechanisms exist to control such responses.
Interestingly, Park et al. (72) examined TNF-α–mediated induction of an endotoxin refractory state using exogenous TNF-α in the absence of primary LPS followed by a secondary LPS stimulation; TNF-α treatment inhibited NF-κB signaling following LPS challenge and led to a tolerized state in human macrophages. TNF-mediated tolerance (termed “TNF tolerance”) has been reviewed recently (73), and it has been suggested that TNF tolerance contributes to negative immune regulation similar to endotoxin tolerance. In our model, we observed differential TNF-α induction between THP-1 and PMA–THP-1 cells, where THP-1 cells exhibited less TNF-α induction compared with PMA–THP-1 cells. This could provide an explanation for the differential responses to IL-27; the higher levels of TNF-α produced by PMA–THP-1 cells potentially contribute to a more stable tolerized state such that IL-27 treatment could not block the formation of endotoxin tolerance to the same extent as in THP-1 cells.
Macrophages and monocytes exhibit different mechanisms to control inflammation (74, 75). Here, IL-27 primed both monocytes and macrophages for enhanced proinflammatory responses; however, others have shown anti-inflammatory functions whereby IL-27 suppresses TNF-α and IL-1β responsiveness in human macrophages (76) and induces the anti-inflammatory cytokine IL-10 in murine macrophages (33). These discrepancies may be explained by the various subcategories of monocytes and macrophages that reflect their specific functions. Specifically, monocyte subsets include proinflammatory, phagocytic, and patrolling monocytes, which are distinguished by CD14 and CD16 expression as well as chemokine receptors CCR2 and CX3CR1 (36, 37). Investigating the effects of IL-27 on purified subpopulations of monocytes and macrophages could clarify some of the observed differences.
Upon activation, phagocytic and inflammatory monocytes differentiate into classically activated M1 macrophages (77, 78). In contrast, patrolling monocytes differentiate into alternatively activated M2 macrophages (77, 78). The phenotype of THP-1 cells is closely related to classical phagocytic monocytes, and thus they may exhibit properties that allow LPS + IL-27 co-stimulation to completely restore cytokine production in tolerized THP-1 cells. In addition, endotoxin-tolerant macrophages express negative regulatory genes similar to alternatively activated M2 macrophages (79–81). The comparison between M2 macrophages and endotoxin-tolerized cells is a controversial topic. Porta et al. (81) aligned endotoxin-tolerant macrophages with the M2 phenotype, whereas others have described endotoxin-tolerized cells to have a more complex phenotype than typical M1 or M2 macrophages (40, 82). Most recently, endotoxin-tolerized macrophages have been suggested to be an intermediate cell type, shifting between the M1 and M2 phenotypes (83). Understanding the key differences between M2-polarized macrophages and endotoxin-tolerized cells is essential in distinguishing the activation state of macrophages.
There is a plasticity between M1- and M2-polarized macrophages (84). M1 macrophages produce proinflammatory TNF-α (85), which can subsequently shift cells to a more M2-like phenotype, inducing TNF tolerance (72). In addition, IFN-β produced in response to LPS is involved in promoting macrophages to the M1 phenotype (86). There are conflicting reports on the role of IFN-β in endotoxin tolerance; endotoxin tolerance occurs independent of IFN-β production in IFNAR knock-out mice (87), whereas IFN-β production has also been attributed to the development of sepsis in murine models, suggesting an association between a refractory state of monocytes and low IFN-β production (88). Furthermore, IL-27–treated M2 macrophages exhibit suppressed IL-12/23p40 production (32). Macrophages differentiated in the presence of neutralizing IL-27 for 7 days showed a reduced production of M2 cytokine IL-10 in response to LPS stimulation (89). In our study, PMA–THP-1 cells stimulated with LPS produced both TNF-α and IFN-β, and co-stimulation of LPS + IL-27 enhanced production of both cytokines. Therefore, LPS + IL-27 stimulation may shift endotoxin-tolerized PMA–THP-1 cells further toward an M2 phenotype than activated THP-1 cells.
Soluble CD14 promotes LPS responses on CD14low-expressing cells such as THP-1 cells (42, 43, 90). In our study, IL-27 amplified sCD14 production in THP-1 cells but not in PMA–THP-1 cells, primary monocytes, or primary macrophages. Surprisingly, IL-27 added prior to recombinant CD14 induced TNF-α production in the absence of LPS, which may suggest that IL-27 amplified an alternate CD14 signaling mechanism, similar to one discovered recently (91). Specifically, tissue injury releases danger-associated molecular patterns (DAMPs), which induce CD14 internalization to inhibit LPS-TLR4 responses (91). In turn, IL-27 stimulation may affect an unknown cellular mechanism to modulate CD14-mediated processes such as sCD14 production, resulting in continued LPS-mediated responses on the cell surface and restored TNF-α production in tolerized THP-1 cells. Indeed, CD14 is required for TLR4 endocytosis for TRIF-mediated signaling in the endosome (17, 41, 92). However, it is possible that THP-1 cells do not have sufficient CD14 expression to induce endocytosis of TLR4 for induction of IFN-β production via endosomal signaling. In support of this idea, THP-1 cells did not produce detectable levels of IFN-β in all conditions tested. On the other hand, PMA–THP-1 cells, primary monocytes, and primary macrophages exhibited IFN-β production, which was also enhanced by IL-27 pretreatment, indicating that CD14high-expressing cells have sufficient CD14 expression to promote endocytosis for subsequent LPS/TLR4-mediated endosomal signaling. In addition, excessive sCD14 can inhibit LPS-TLR4 responses (42), which may be responsible for the lesser inhibition of tolerance in response to IL-27 in CD14high-expressing cells, as demonstrated in PMA–THP-1 cells, primary monocytes, and primary macrophages. The effects of differential expression of membrane-bound and soluble CD14 in the presence or absence of IL-27 are modeled in Fig. 9.
Figure 9.

A balance between membrane and soluble CD14 modulates the effects of IL-27 on endotoxin tolerance. In untolerized CD14low-expressing cells, LPS-TLR4-CD14 ligation induces NF-κB nuclear translocation and TNF-α production. However, these cells do not have sufficient CD14 to induce TLR4 endocytosis required for IFN-β production. In untolerized CD14high-expressing cells, LPS stimulation results in NF-κB nuclear localization, TNF-α production, and CD14-mediated TLR4 endocytosis, resulting in IFN-β production via endosomal TLR4 signaling. In CD14low-expressing cells, IL-27 enhances sCD14 and TLR4 expression to enhance LPS-TLR4 ligation, resulting in greater NF-κB nuclear localization and TNF-α production. IL-27–treated CD14high-expressing cells also exhibit enhanced TLR4 expression for more TNF-α production. High levels of surface CD14 allow for TLR4 endocytosis and IFN-β production in response to LPS, even though IL-27 does not affect sCD14 in these cells. Endotoxin-tolerized CD14low-expressing cells exhibit reduced NF-κB nuclear translocation, correlating with less TNF-α production compared with untolerized cells. In tolerized CD14high-expressing cells, TLR4 internalization and less sCD14 reduces subsequent responsiveness to LPS, resulting in less TNF-α and IFN-β production. In CD14low-expressing cells, LPS + IL-27 treatment enhances TLR4 expression, resulting in greater LPS signaling and NF-κB nuclear translocation for even greater TNF-α production than untolerized cells. CD14high-expressing cells exposed to LPS + IL-27 internalize TLR4, and because of the excess sCD14, the 2° LPS ligation is inhibited. Together, this results in decreased TNF-α and IFN-β production relative to untolerized cells. However, IL-27–mediated TLR4 up-regulation accounts for increased cytokine production relative to tolerized cells.
In this study, we suggest that the differential effects of IL-27 on endotoxin tolerance in THP-1 cells compared with PMA–THP-1 cells may also depend on surface expression of CD14. THP-1 cells, which have low CD14 expression, had complete IL-27–mediated restoration of cytokine production in tolerized cells, whereas PMA–THP-1 cells, primary monocytes, and primary macrophages have high CD14 expression and exhibited partially restored cytokine production in tolerized cells treated with IL-27. Elevated CD14 expression on PMA–THP-1 cells may also be responsible for the more prominent LPS-mediated TLR4 internalization compared with THP-1 cells. Indeed, macrophages internalize TLR4 immediately and have reduced CD14 on the surface after 24 h (93). Herein, TLR4 expression remained unchanged in response to LPS treatment of THP-1 cells, similar to our previous observations in CD14-THP-1 cells (28). In contrast, LPS treatment of PMA-THP-1 cells decreased in TLR4 expression. Thus, in THP-1 cells and CD14-THP-1 cells, IL-27–induced up-regulation of TLR4 could be responsible for the IL-27–mediated inhibition of endotoxin tolerance and complete restoration of TNF-α production. Taken together, our data demonstrate a potential role for CD14, IL-27–enhanced TLR4, and TNF-α expression in mediating LPS responsiveness in human monocytes and macrophages.
Experimental procedures
Cell lines, reagents, and stimulations
The THP-1 cell pro-monocytic cell line was purchased from American Type Culture Collection (ATCC, Manassas, VA). Cells were cultured in RPMI 1640 medium (Gibco Life Technologies) supplemented with heat-inactivated 10% fetal bovine serum (FBS; Hyclone Laboratories Inc., Logan, UT). THP-1 cells and THP-1 XBlue cells were differentiated into macrophage-like cells using PMA (10 ng/ml; BioShop Canada Inc., Burlington, Ontario, Canada) for 48 h. THP-1 cells stably transfected with a NF-κB/AP-1–inducible reporter (SEAP) plasmid (THP-1 XBlue cells) and Zeocin selection antibiotic were purchased from InvivoGen (San Diego, CA). THP-1 cells transfected with a CD14-expressing cDNA plasmid (CD14-THP-1 cells) were a kind gift from Dr. Richard Ulevitch (The Scripps Research Institute, La Jolla, CA). G418 sulfate (BioShop Canada) was used for the selection of neomycin-resistant CD14-THP-1 cells. Cells were stimulated with LPS Escherichia coli O111:B4 (10 ng/ml; Sigma-Aldrich) and/or recombinant human IL-27 (100 ng/ml; R&D Systems, Minneapolis, MN) for 24 h at 37 °C with 5% CO2 to induce endotoxin tolerance. Cells were subsequently washed and challenged with LPS (100 ng/ml) for 2–24 h, as indicated.
Primary monocytes and macrophages
Whole blood drawn from healthy volunteers was obtained in accordance with the recommendations of the Canadian Tri-Council Policy Statement: Ethical Conduct for Research Involving Humans and the Queen's University Health Sciences and Affiliated Teaching Hospitals Research Ethics Board (HSREB). All research on human samples was performed in accordance with relevant guidelines/regulations and was approved by the Queen's University HSREB. All subjects gave written informed consent in accordance with the Declaration of Helsinki and as per the protocol approved by the HSREB. For the isolation of primary human monocytes, whole blood was enriched with RosetteSep human monocyte enrichment mixture (STEMCELL Technologies, Vancouver, British Columbia, Canada) as per the manufacturer's instructions. Briefly, enriched blood was diluted 1:1 with PBS with 1 mm EDTA (Bioshop Canada) and 2% FBS (Hyclone). Whole blood was layered on a density gradient medium (Lympholyte-H separation medium; Cedarlane, Burlington, Ontario, Canada) in 50 ml of SepMate conical tubes (STEMCELL Technologies) and centrifuged to separate mononuclear cells from red blood cells. Cells were washed twice with PBS + 1 mm EDTA + 2% FBS and resuspended in RPMI + 10% FBS for immediate stimulation. To obtain primary human macrophages, whole blood was layered on Lympholyte-H as described above. Peripheral blood mononuclear cells were plated in 6-well culture dishes in RPMI supplemented with 25% autologous serum. After 2 h, nonadherent cells were removed, the remaining adherent cells were washed three times with warmed PBS, and RPMI + 25% autologous serum was added. Cells were washed again on days 2 and 4, and fresh RPMI + 25% autologous serum was added. On day 6, cells were washed, and RPMI + 10% FBS was added; cells were stimulated as indicated on day 7.
Flow cytometry
Cells were stained with anti-human CD14-APC/Cy7 and anti-human CD16–Alexa Fluor 647 (BioLegend, San Diego, CA) and by anti-human TLR4—Alexa Fluor 488 (Thermo Fisher Scientific Affymetrix-eBioscience, Waltham, MA). Cells were incubated with LPS—Alexa Fluor 488 (100 ng/ml; Molecular Probes, Eugene, OR) for 1 h at 37 °C. Cells were washed and resuspended in PBS-azide (PBS + 0.01% sodium azide + 2% FBS). For intracellular staining, cells were fixed with 4% paraformaldehyde (Thermo Fisher Scientific) and permeabilized with 0.2% saponin (Honeywell Riedel-de Haën, Morris Plains, NJ) followed by antibody staining. Data were acquired using a CytoFLEX flow cytometer (Beckman Coulter) and analyzed using FlowJo (Ashland, OR), version 10.2.
Confocal microscopy
Cells were washed with warmed PBS, fixed with 4% paraformaldehyde, and permeabilized using 0.1% Triton X-100. Cells were probed with mouse anti-human NF-κB p65 mAb (1:100 dilution; Abcam, Cambridge, UK) and rabbit anti-human NF-κB p50/p105 polyclonal antibody (Cell Signaling Technology, Danvers, MA). Subsequently, cells were stained with donkey anti-rabbit IgG (H+L) Alexa Fluor 488 (1:500; Molecular Probes) and donkey anti-mouse IgG (H+L) Alexa Fluor 568 (Life Technologies) secondary antibodies. Nuclei were visualized using NucRed® Live 647 ReadyProbes® (Molecular Probes). PMA–THP-1 cells were cultured on 35-mm glass-bottom dishes (MatTek Corp., Ashland, MA). THP-1 cells were stained and then visualized using ibiTreat μ-Slide VI0.4 slides (ibidi, Planegg, Germany). Images were captured using a FV1000 confocal laser-scanning microscope (Olympus, Center Valley, PA) equipped with FV10 ASW 4.01 software through a 60X/1.42A oil immersion objective at a digital zoom factor of 4 to ensure that pixel intensities were in the linear range within each channel. Blinded and using differential interference contrast (DIC) and NucRed images as guides, we measured the relative pixel intensities of NF-κB p65 and p50 in identically sized regions (1.275 μm2) in the nucleus and cytoplasm of the same cell for two cells/per image with three images captured for each specimen (n = 18 cells/condition). The nuclear/cytoplasmic ratios were calculated for each condition.
NF-κB/AP-1 activation assay
THP-1 XBlue and PMA–THP-1 XBlue cells were stimulated as described above to produce NF-κB/AP-1-inducible SEAP, the production of which was quantified by incubating 20 μl of cell-free supernatant with 180 μl of QUANTI-BlueTM buffer (InvivoGen) at 37 °C for 4 h according to the manufacturer's instructions. Optical density was measured at 650 nm on the Varioskan spectrophotometer (Thermo Fisher Scientific).
ELISA
Cytokine production in cell-free supernatants was quantified according to the manufacturer's instructions for human TNF-α (Ready-SET-Go!, Thermo Fisher Scientific Affymetrix-eBioscience), human soluble CD14, human LBP, and human IFN-β (DuoSet, R&D Systems). Absorbance was measured at 450 nm on the ELx800 microplate reader (BioTek, Winooski, VT).
Statistical analyses
Analyses were performed in GraphPad Prism 6. Individual sets of data were combined showing mean ± S.E. of replicate sets of experiments. One-way analysis of variance with Holm-Sidak multiple comparisons test was performed between all relevant conditions and confirmed using the Wilcoxon matched pairs signed-rank test between individual pairs. In Figs. 1–8: ns, not significant; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.001.
Author contributions
C. P. and K. G. conceptualization; C. P., V. M., and K. G. data curation; C. P., V. M., R. L. F., B. W. B., and K. G. formal analysis; C. P. and K. G. writing-original draft; C. P., V. M., R. L. F., B. W. B., and K. G. writing-review and editing; R. L. F. and B. W. B. resources; R. L. F. and B. W. B. software; C. P., R. L. F., B. W. B., and K. G. methodology; K. G. supervision; K. G. and B. W. B. funding acquisition; K. G. investigation; K. G. project administration.
Supplementary Material
Acknowledgments
We thank our generous blood donors for their contributions to this study. We thank Andrew Day (Senior Biostatistician at Kingston General Hospital-Clinical Research Centre and the Critical Care Clinical Evaluation Research Unit) for assistance with statistical analyses.
This work was supported by Grants 342168 (to K. G.) and 418719 (to B. W. B.) from the Natural Sciences and Engineering Research Council of Canada (NSERC). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5.
1° and 2° are used to distinguish the first and second doses, respectively, of medium control, LPS, IL-27, or LPS + IL-27.
- LPS
- lipopolysaccharide
- TNF
- tumor necrosis factor
- LBP
- LPS-binding protein
- TLR4
- Toll-like receptor 4
- TRIF
- Toll/IL-1R domain–containing adaptor-inducing IFN-β
- IFN
- interferon
- IL
- interleukin
- PMA
- phorbol 12-myristate 13-acetate
- med
- medium control
- SEAP
- secreted embryonic alkaline phosphatase
- sCD14
- soluble CD14
- FBS
- fetal bovine serum
- DIC
- differential interference contrast.
References
- 1. López-Collazo E., and del Fresno C. (2013) Pathophysiology of endotoxin tolerance: Mechanisms and clinical consequences. Crit. Care 17, 242 10.1186/cc13110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. West M. A., and Koons A. (2008) Endotoxin tolerance in sepsis: Concentration-dependent augmentation or inhibition of LPS-stimulated macrophage TNF secretion by LPS pretreatment. J. Trauma 65, 893–900 10.1097/TA.0b013e3181877fde [DOI] [PubMed] [Google Scholar]
- 3. El Gazzar M., Yoza B. K., Hu J. Y., Cousart S. L., and McCall C. E. (2007) Epigenetic silencing of tumor necrosis factor α during endotoxin tolerance. J. Biol. Chem. 282, 26857–26864 10.1074/jbc.M704584200 [DOI] [PubMed] [Google Scholar]
- 4. Deng J. C., Cheng G., Newstead M. W., Zeng X., Kobayashi K., Flavell R. A., and Standiford T. J. (2006) Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J. Clin. Invest. 116, 2532–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kox M., de Kleijn S., Pompe J. C., Ramakers B. P., Netea M. G., van der Hoeven J. G., Hoedemaekers C. W., and Pickkers P. (2011) Differential ex vivo and in vivo endotoxin tolerance kinetics following human endotoxemia. Crit. Care Med. 39, 1866–1870 10.1097/CCM.0b013e3182190d5d [DOI] [PubMed] [Google Scholar]
- 6. Draisma A., Pickkers P., Bouw M. P., and van der Hoeven J. G. (2009) Development of endotoxin tolerance in humans in vivo. Crit. Care Med. 37, 1261–1267 10.1097/CCM.0b013e31819c3c67 [DOI] [PubMed] [Google Scholar]
- 7. Xiong Y., and Medvedev A. E. (2011) Induction of endotoxin tolerance in vivo inhibits activation of IRAK4 and increases negative regulators IRAK-M, SHIP-1, and A20. J. Leukoc. Biol. 90, 1141–1148 10.1189/jlb.0611273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Finberg R. W., Re F., Popova L., Golenbock D. T., and Kurt-Jones E. A. (2004) Cell activation by Toll-like receptors: Role of LBP and CD14. J. Endotoxin Res. 10, 413–418 10.1177/09680519040100060601,10.1179/096805104225006273 [DOI] [PubMed] [Google Scholar]
- 9. Fitzgerald K. A., Rowe D. C., and Golenbock D. T. (2004) Endotoxin recognition and signal transduction by the TLR4/MD2 complex. Microbes Infect. 6, 1361–1367 10.1016/j.micinf.2004.08.015 [DOI] [PubMed] [Google Scholar]
- 10. Shin H. J., Lee H., Park J. D., Hyun H. C., Sohn H. O., Lee D. W., and Kim Y. S. (2007) Kinetics of binding of LPS to recombinant CD14, TLR4, and MD-2 proteins. Mol. Cells 24, 119–124 [PubMed] [Google Scholar]
- 11. Yamamoto M., Sato S., Hemmi H., Hoshino K., Kaisho T., Sanjo H., Takeuchi O., Sugiyama M., Okabe M., Takeda K., and Akira S. (2003) Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301, 640–643 10.1126/science.1087262 [DOI] [PubMed] [Google Scholar]
- 12. Lu Y. C., Yeh W. C., and Ohashi P. S. (2008) LPS/TLR4 signal transduction pathway. Cytokine 42, 145–151 10.1016/j.cyto.2008.01.006 [DOI] [PubMed] [Google Scholar]
- 13. Shen H., Tesar B. M., Walker W. E., and Goldstein D. R. (2008) Dual signaling of MyD88 and TRIF is critical for maximal TLR4-induced dendritic cell maturation. J. Immunol. 181, 1849–1858 10.4049/jimmunol.181.3.1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Husebye H., Halaas Ø., Stenmark H, Tunheim G., Sandanger Ø., Bogen B., Brech A., Latz E., and Espevik T. (2006) Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 25, 683–692 10.1038/sj.emboj.7600991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tanimura N., Saitoh S., Matsumoto F., Akashi-Takamura S., and Miyake K. (2008) Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem. Biophys. Res. Commun. 368, 94–99 10.1016/j.bbrc.2008.01.061 [DOI] [PubMed] [Google Scholar]
- 16. Shuto T., Kato K., Mori Y., Viriyakosol S., Oba M., Furuta T., Okiyoneda T., Arima H., Suico M. A., and Kai H. (2005) Membrane-anchored CD14 is required for LPS-induced TLR4 endocytosis in TLR4/MD-2/CD14 overexpressing CHO cells. Biochem. Biophys. Res. Commun. 338, 1402–1409 10.1016/j.bbrc.2005.10.102 [DOI] [PubMed] [Google Scholar]
- 17. Zanoni I., Ostuni R., Marek L. R., Barresi S., Barbalat R., Barton G. M., Granucci F., and Kagan J. C. (2011) CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 147, 868–880 10.1016/j.cell.2011.09.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kamakura M., Morisawa K., Komi H., Tomatani A., Saito F., Konishi Y., Jin Y., Manabe T., Kuroda M., Imai S., Mizuguchi H., and Taniguchi T. (2006) Regulation of IL-27p28 gene by lipopolysaccharide in dendritic DC2.4 cells. Biochem. Biophys. Res. Commun. 349, 1372–1377 10.1016/j.bbrc.2006.09.004 [DOI] [PubMed] [Google Scholar]
- 19. Molle C., Nguyen M., Flamand V., Renneson J., Trottein F., De Wit D., Willems F., Goldman M., and Goriely S. (2007) IL-27 synthesis induced by TLR ligation critically depends on IFN regulatory factor 3. J. Immunol. 178, 7607–7615 10.4049/jimmunol.178.12.7607 [DOI] [PubMed] [Google Scholar]
- 20. Trinchieri G., Pflanz S., and Kastelein R. A. (2003) The IL-12 family of heterodimeric cytokines: New players in the regulation of T cell responses. Immunity 19, 641–644 10.1016/S1074-7613(03)00296-6 [DOI] [PubMed] [Google Scholar]
- 21. Gee K., Guzzo C., Mat C., Nor F., Ma W., and Kumar A. (2009) The IL-12 family of cytokines in infection, inflammation and autoimmune disorders. Inflamm. Allergy Drug Targets 8, 40–52 [DOI] [PubMed] [Google Scholar]
- 22. Vignali D. A., and Kuchroo V. K. (2012) IL-12 family cytokines: Immunological playmakers. Nat. Immunol. 13, 722–728 10.1038/ni.2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takeda A., Hamano S., Yamanaka A., Hanada T., Ishibashi T., Mak T. W., Yoshimura A., and Yoshida H. (2003) Cutting edge: Role of IL-27/WSX-1 signaling for induction of T-bet through activation of STAT1 during initial Th1 commitment. J. Immunol. 170, 4886–4890 10.4049/jimmunol.170.10.4886 [DOI] [PubMed] [Google Scholar]
- 24. Hibbert L., Pflanz S., de Waal Malefyt R., and Kastelein R. A. (2003) IL-27 and IFN-α signal via Stat1 and Stat3 and induce T-Bet and IL-12Rβ2 in naive T cells. J. Interferon Cytokine Res. 23, 513–522 10.1089/10799900360708632 [DOI] [PubMed] [Google Scholar]
- 25. Pflanz S., Hibbert L., Mattson J., Rosales R., Vaisberg E., Bazan J. F., Phillips J. H., McClanahan T. K., de Waal Malefyt R., and Kastelein R. A. (2004) WSX-1 and glycoprotein 130 constitute a signal-transducing receptor for IL-27. J. Immunol. 172, 2225–2231 10.4049/jimmunol.172.4.2225 [DOI] [PubMed] [Google Scholar]
- 26. Guzzo C., Mat N. F., and Gee K. (2010) Interleukin-27 induces a STAT1/3-and NF-κB-dependent proinflammatory cytokine profile in human monocytes. J. Biol. Chem. 285, 24404–24411 10.1074/jbc.M110.112599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guzzo C., Ayer A., Basta S., Banfield B. W., and Gee K. (2012) IL-27 enhances LPS-induced proinflammatory cytokine production via upregulation of TLR4 expression and signaling in human monocytes. J. Immunol. 188, 864–873 10.4049/jimmunol.1101912 [DOI] [PubMed] [Google Scholar]
- 28. Petes C., Wynick C., Guzzo C., Mehta D., Logan S., Banfield B. W., Basta S., Cooper A., and Gee K. (2017) IL-27 enhances LPS-induced IL-1β in human monocytes and murine macrophages. J. Leukoc. Biol. 102, 83–94 10.1189/jlb.3A0316-098R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petes C., Mariani M. K., Yang Y., Grandvaux N., and Gee K. (2018) IL-6 inhibits IL-27- and IL-30-mediated inflammatory responses in human monocytes. Front. Immunol. 9, 256 10.3389/fimmu.2018.00256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kalliolias G. D., and Ivashkiv L. B. (2008) IL-27 activates human monocytes via STAT1 and suppresses IL-10 production but the inflammatory functions of IL-27 are abrogated by TLRs and p38. J. Immunol. 180, 6325–6333 10.4049/jimmunol.180.9.6325 [DOI] [PubMed] [Google Scholar]
- 31. Hölscher C., Hölscher A., Rückerl D., Yoshimoto T., Yoshida H., Mak T., Saris C., and Ehlers S. (2005) The IL-27 receptor chain WSX-1 differentially regulates antibacterial immunity and survival during experimental tuberculosis. J. Immunol. 174, 3534–3544 10.4049/jimmunol.174.6.3534 [DOI] [PubMed] [Google Scholar]
- 32. Rückerl D., Hessmann M., Yoshimoto T., Ehlers S., and Hölscher C. (2006) Alternatively activated macrophages express the IL-27 receptor α chain WSX-1. Immunobiology 211, 427–436 10.1016/j.imbio.2006.05.008 [DOI] [PubMed] [Google Scholar]
- 33. Iyer S. S., Ghaffari A. A., and Cheng G. (2010) Lipopolysaccharide-mediated IL-10 transcriptional regulation requires sequential induction of type I IFNs and IL-27 in macrophages. J. Immunol. 185, 6599–6607 10.4049/jimmunol.1002041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Auwerx J. (1991) The human leukemia cell line, THP-1: A multifacetted model for the study of monocyte-macrophage differentiation. Experientia 47, 22–31 10.1007/BF02041244 [DOI] [PubMed] [Google Scholar]
- 35. Chen R. F., Wang L., Cheng J. T., and Yang K. D. (2012) Induction of IFNα or IL-12 depends on differentiation of THP-1 cells in dengue infections without and with antibody enhancement. BMC Infectious Diseases 12, 340 10.1186/1471-2334-12-340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang J., Zhang L., Yu C., Yang X. F., and Wang H. (2014) Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark. Res. 2, 1 10.1186/2050-7771-2-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ziegler-Heitbrock L., Ancuta P., Crowe S., Dalod M., Grau V., Hart D. N., Leenen P. J., Liu Y. J., MacPherson G., Randolph G. J., Scherberich J., Schmitz J., Shortman K., Sozzani S., Strobl H., et al. (2010) Nomenclature of monocytes and dendritic cells in blood. Blood 116, e74–80 10.1182/blood-2010-02-258558 [DOI] [PubMed] [Google Scholar]
- 38. Wong K. L., Yeap W. H., Tai J. J., Ong S. M., Dang T. M., and Wong S. C. (2012) The three human monocyte subsets: implications for health and disease. Immunologic Research 53, 41–57 10.1007/s12026-012-8297-3 [DOI] [PubMed] [Google Scholar]
- 39. Zhang G., and Ghosh S. (2000) Molecular mechanisms of NF-κB activation induced by bacterial lipopolysaccharide through Toll-like receptors. J. Endotoxin Res. 6, 453–457 10.1179/096805100101532414,10.1177/09680519000060060701 [DOI] [PubMed] [Google Scholar]
- 40. Biswas S. K., and Lopez-Collazo E. (2009) Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 30, 475–487 10.1016/j.it.2009.07.009 [DOI] [PubMed] [Google Scholar]
- 41. Kim D., and Kim J. Y. (2014) Anti-CD14 antibody reduces LPS responsiveness via TLR4 internalization in human monocytes. Mol. Immunol. 57, 210–215 10.1016/j.molimm.2013.09.009 [DOI] [PubMed] [Google Scholar]
- 42. Kitchens R. L., Thompson P. A., Viriyakosol S., O'Keefe G. E., and Munford R. S. (2001) Plasma CD14 decreases monocyte responses to LPS by transferring cell-bound LPS to plasma lipoproteins. J. Clin. Invest. 108, 485–493 10.1172/JCI200113139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shive C. L., Jiang W., Anthony D. D., and Lederman M. M. (2015) Soluble CD14 is a nonspecific marker of monocyte activation. AIDS 29, 1263–1265 10.1097/QAD.0000000000000735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Santucci M. B., Ciaramella A., Mattei M., Sumerska T., and Fraziano M. (2003) Batimastat reduces Mycobacterium tuberculosis-induced apoptosis in macrophages. Int. Immunopharmacol. 3, 1657–1665 10.1016/S1567-5769(03)00202-9 [DOI] [PubMed] [Google Scholar]
- 45. Deschner J., Singhal A., Long P., Liu C. C., Piesco N., and Agarwal S. (2003) Cleavage of CD14 and LBP by a protease from Prevotella intermedia. Arch. Microbiol. 179, 430–436 10.1007/s00203-003-0548-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pedron T., Girard R., and Chaby R. (1995) Variation of LPS-binding capacity, epitope expression, and shedding of membrane-bound CD14 during differentiation of human monocytes. J. Immunol. 155, 1460–1471 [PubMed] [Google Scholar]
- 47. Bazil V., and Strominger J. (1991) Shedding as a mechanism of down-modulation of CD14 on stimulated human monocytes. J. Immunol. 147, 1567–1574 [PubMed] [Google Scholar]
- 48. Bufler P., Stiegler G., Schuchmann M., Hess S., Krüger C., Stelter F., Eckerskorn C., Schütt C., and Engelmann H. (1995) Soluble lipopolysaccharide receptor (CD14) is released via two different mechanisms from human monocytes and CD14 transfectants. Eur. J. Immunol. 25, 604–610 10.1002/eji.1830250244 [DOI] [PubMed] [Google Scholar]
- 49. Kagan J. C., Su T., Horng T., Chow A., Akira S., and Medzhitov R. (2008) TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 9, 361–368 10.1038/ni1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wynick C., Petes C., Tigert A., and Gee K. (2016) Lipopolysaccharide-mediated induction of concurrent IL-1β and IL-23 expression in THP-1 cells exhibits differential requirements for caspase-1 and cathepsin B activity. J. Interferon Cytokine Res. 36, 477–487 10.1089/jir.2015.0134 [DOI] [PubMed] [Google Scholar]
- 51. Cavaillon J. M., Adrie C., Fitting C., and Adib-Conquy M. (2003) Endotoxin tolerance: Is there a clinical relevance? J. Endotoxin Res. 9, 101–107 10.1179/096805103125001487,10.1177/09680519030090020501 [DOI] [PubMed] [Google Scholar]
- 52. Cavaillon J. M., and Adib-Conquy M. (2006) Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Critical Care 10, 233 10.1186/cc5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cohen J. (2002) The immunopathogenesis of sepsis. Nature 420, 885–891 10.1038/nature01326 [DOI] [PubMed] [Google Scholar]
- 54. Liew F. Y., Xu D., Brint E. K., and O'Neill L. A. (2005) Negative regulation of toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 5, 446–458 10.1038/nri1630 [DOI] [PubMed] [Google Scholar]
- 55. Wheeler D. S., Lahni P. M., Denenberg A. G., Poynter S. E., Wong H. R., Cook J. A., and Zingarelli B. (2008) Induction of endotoxin tolerance enhances bacterial clearance and survival in murine polymicrobial sepsis. Shock 30, 267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Biswas S. K., and Shalova I. N. (2012) Endotoxin tolerance as a key mechanism for immunosuppression, in Immunosuppression: Role in Health and Diseases (Kapur S. and Portela M., ed) Chap. 2, pp. 21–40, InTech Open Ltd., London [Google Scholar]
- 57. Rinchai D., Khaenam P., Kewcharoenwong C., Buddhisa S., Pankla R., Chaussabel D., Bancroft G. J., and Lertmemongkolchai G. (2012) Production of interleukin-27 by human neutrophils regulates their function during bacterial infection. Eur. J. Immunol. 42, 3280–3290 10.1002/eji.201242526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wong H. R., Lindsell C. J., Lahni P., Hart K. W., and Gibot S. (2013) Interleukin-27 as a sepsis diagnostic biomarker in critically ill adults. Shock 40, 382–386 10.1097/SHK.0b013e3182a67632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cao J., Xu F., Lin S., Song Z., Zhang L., Luo P., Xu H., Li D., Zheng K., Ren G., and Yin Y. (2014) IL-27 controls sepsis-induced impairment of lung antibacterial host defence. Thorax 69, 926–937 10.1136/thoraxjnl-2014-205777 [DOI] [PubMed] [Google Scholar]
- 60. Scicluna B. P., and van der Poll T. (2012) Interleukin-27: A potential new sepsis biomarker exposed through genome-wide transcriptional profiling. Crit. Care 16, 188 10.1186/cc11893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pearl J. E., Khader S. A., Solache A., Gilmartin L., Ghilardi N., deSauvage F., and Cooper A. M. (2004) IL-27 signaling compromises control of bacterial growth in mycobacteria-infected mice. J. Immunol. 173, 7490–7496 10.4049/jimmunol.173.12.7490 [DOI] [PubMed] [Google Scholar]
- 62. Ziegler-Heitbrock L. (2001) The p50-homodimer mechanism in tolerance to LPS. J. Endotoxin Res. 7, 219–222 10.1179/096805101101532701,10.1177/09680519010070030401 [DOI] [PubMed] [Google Scholar]
- 63. Ziegler-Heitbrock H., Wedel A., Schraut W., Ströbel M., Wendelgass P., Sternsdorf T., Bäuerle P., Haas J. G., and Riethmüller G. (1994) Tolerance to lipopolysaccharide involves mobilization of nuclear factor kappa B with predominance of p50 homodimers. J. Biol. Chem. 269, 17001–17004 [PubMed] [Google Scholar]
- 64. Baer M., Dillner A., Schwartz R. C., Sedon C., Nedospasov S., and Johnson P. F. (1998) Tumor necrosis factor α transcription in macrophages is attenuated by an autocrine factor that preferentially induces NF-κB p50. Mol. Cell. Biol. 18, 5678–5689 10.1128/MCB.18.10.5678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Carmody R. J., Ruan Q., Palmer S., Hilliard B., and Chen Y. H. (2007) Negative regulation of Toll-like receptor signaling by NF-κB p50 ubiquitination blockade. Science 317, 675–678 10.1126/science.1142953 [DOI] [PubMed] [Google Scholar]
- 66. Escoll P., del Fresno C., García L., Vallés G., Lendínez M. J., Arnalich F., and López-Collazo E. (2003) Rapid up-regulation of IRAK-M expression following a second endotoxin challenge in human monocytes and in monocytes isolated from septic patients. Biochem. Biophys. Res. Commun. 311, 465–472 10.1016/j.bbrc.2003.10.019 [DOI] [PubMed] [Google Scholar]
- 67. van't Veer C., van den Pangaart P. S., van Zoelen M. A., de Kruif M., Birjmohun R. S., Stroes E. S., de Vos A. F., and van der Poll T. (2007) Induction of IRAK-M is associated with lipopolysaccharide tolerance in a human endotoxemia model. J. Immunol. 179, 7110–7120 10.4049/jimmunol.179.10.7110 [DOI] [PubMed] [Google Scholar]
- 68. del Fresno C., Soler-Rangel L., Soares-Schanoski A., Gómez-Piña V., González-León M. C., Gómez-García L., Mendoza-Barberá E., Rodríguez-Rojas A., García F., Fuentes-Prior P., Arnalich F., and López-Collazo E. (2007) Inflammatory responses associated with acute coronary syndrome up-regulate IRAK-M and induce endotoxin tolerance in circulating monocytes. J. Endotoxin Res. 13, 39–52 10.1177/0968051907078623 [DOI] [PubMed] [Google Scholar]
- 69. Sly L. M., Rauh M. J., Kalesnikoff J., Song C. H., and Krystal G. (2004) LPS-induced upregulation of SHIP is essential for endotoxin tolerance. Immunity 21, 227–239 10.1016/j.immuni.2004.07.010 [DOI] [PubMed] [Google Scholar]
- 70. Wertz I. E., and Dixit V. M. (2010) Signaling to NF-κB: Regulation by ubiquitination. Cold Spring Harb. Perspect. Biol. 2, a003350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen J., and Ivashkiv L. B. (2010) IFN-γ abrogates endotoxin tolerance by facilitating Toll-like receptor-induced chromatin remodeling. Proc. Natl. Acad. Sci. U.S.A. 107, 19438–19443 10.1073/pnas.1007816107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Park S. H., Park-Min K. H., Chen J., Hu X., and Ivashkiv L. B. (2011) TNF induces endotoxin tolerance mediated by GSK3 in macrophages. Nat. Immunol. 12, 607–615 10.1038/nri3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Huber R., Bikker R., Welz B., Christmann M., and Brand K. (2017) TNF tolerance in monocytes and macrophages: Characteristics and molecular mechanisms. J. Immunol. Res. 2017, 9570129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Geissmann F., Manz M. G., Jung S., Sieweke M. H., Merad M., and Ley K. (2010) Development of monocytes, macrophages, and dendritic cells. Science 327, 656–661 10.1126/science.1178331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Shi C., and Pamer E. G. (2011) Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 11, 762–774 10.1038/nri3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kalliolias G. D., Gordon R. A., and Ivashkiv L. B. (2010) Suppression of TNF-α and IL-1 signaling identifies a mechanism of homeostatic regulation of macrophages by IL-27. J. Immunol. 185, 7047–7056 10.4049/jimmunol.1001290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang N., Liang H., and Zen K. (2014) Molecular mechanisms that influence the macrophage M1–M2 polarization balance. Front. Immunol. 5, 614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Italiani P., and Boraschi D. (2014) From monocytes to M1/M2 macrophages: Phenotypical vs. functional differentiation. Front. Immunol. 5, 514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ivashkiv L. B. (2011) Inflammatory signaling in macrophages: Transitions from acute to tolerant and alternative activation states. Eur. J. Immunol. 41, 2477–2481 10.1002/eji.201141783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pena O. M., Pistolic J., Raj D., Fjell C. D., and Hancock R. E. (2011) Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J. Immunol. 186, 7243–7254 10.4049/jimmunol.1001952 [DOI] [PubMed] [Google Scholar]
- 81. Porta C., Rimoldi M., Raes G., Brys L., Ghezzi P., Di Liberto D., Dieli F., Ghisletti S., Natoli G., and De Baetselier P. (2009) Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor κB. Proc. Natl. Acad. Sci. U.S.A. 106, 14978–14983 10.1073/pnas.0809784106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shalova I. N., Lim J. Y., Chittezhath M., Zinkernagel A. S., Beasley F., Hernández-Jiménez E., Toledano V., Cubillos-Zapata C., Rapisarda A., Chen J., Duan K., Yang H., Poidinger M., Melillo G., Nizet V., et al. (2015) Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1α. Immunity 42, 484–498 10.1016/j.immuni.2015.02.001 [DOI] [PubMed] [Google Scholar]
- 83. Lin Y. W., Lee B., Liu P. S., and Wei L. N. (2016) Receptor-interacting protein 140 orchestrates the dynamics of macrophage M1/M2 polarization. J. Innate Immun. 8, 97–107 10.1159/000433539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Moghaddam A. S., Mohammadian S., Vazini H., Taghadosi M., Esmaeili S. A., Mardani F., Seifi B., Mohammadi A., Afshari J. T., and Sahebkar A. (2018) Macrophage plasticity, polarization and function in health and disease. J. Cell. Physiol. 233, 6425–6440 10.1002/jcp.26461,10.1002/jcp.26429 [DOI] [PubMed] [Google Scholar]
- 85. Wu X., Xu W., Feng X., He Y., Liu X., Gao Y., Yang S., Shao Z., Yang C., and Ye Z. (2015) TNF-a-mediated inflammatory macrophage polarization contributes to the pathogenesis of steroid-induced osteonecrosis in mice. Int. J. Immunopathol. Pharmacol. 28, 351–361 10.1177/0394632015593228 [DOI] [PubMed] [Google Scholar]
- 86. Xie C., Liu C., Wu B., Lin Y., Ma T., Xiong H., Wang Q., Li Z., Ma C., and Tu Z. (2016) Effects of IRF1 and IFN-β interaction on the M1 polarization of macrophages and its antitumor function. Int. J. Mol. Med. 38, 148–160 10.3892/ijmm.2016.2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Karimi Y., Poznanski S. M., Vahedi F., Chen B., Chew M. V., Lee A. J., and Ashkar A. A. (2017) Type I interferon signalling is not required for the induction of endotoxin tolerance. Cytokine 95, 7–11 10.1016/j.cyto.2017.01.017 [DOI] [PubMed] [Google Scholar]
- 88. Rackov G., Shokri R., De Mon M. Á., Martínez-A C., and Balomenos D. (2017) The role of IFN-β during the course of sepsis progression and its therapeutic potential. Front. Immunol. 8, 493 10.3389/fimmu.2017.00493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. d'Almeida S. M., Kauffenstein G., Roy C., Basset L., Papargyris L., Henrion D., Catros V., Ifrah N., Descamps P., Croue A., Jeannin P., Grégoire M., Delneste Y., and Tabiasco J. (2016) The ecto-ATPDase CD39 is involved in the acquisition of the immunoregulatory phenotype by M-CSF-macrophages and ovarian cancer tumor-associated macrophages: Regulatory role of IL-27. Oncoimmunology 5, e1178025 10.1080/2162402X.2016.1178025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kitchens R. L., Thompson P. A., Munford R. S., and O'Keefe G. E. (2003) Acute inflammation and infection maintain circulating phospholipid levels and enhance lipopolysaccharide binding to plasma lipoproteins. J. Lipid Res. 44, 2339–2348 10.1194/jlr.M300228-JLR200 [DOI] [PubMed] [Google Scholar]
- 91. Zanoni I., Tan Y., Di Gioia M., Springstead J. R., and Kagan J. C. (2017) By capturing inflammatory lipids released from dying cells, the receptor CD14 induces inflammasome-dependent phagocyte hyperactivation. Immunity 47, 697–709.e3 10.1016/j.immuni.2017.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Rajaiah R., Perkins D. J., Ireland D. D., and Vogel S. N. (2015) CD14 dependence of TLR4 endocytosis and TRIF signaling displays ligand specificity and is dissociable in endotoxin tolerance. Proc. Natl. Acad. Sci. U.S.A. 112, 8391–8396 10.1073/pnas.1424980112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Nomura F., Akashi S., Sakao Y., Sato S., Kawai T., Matsumoto M., Nakanishi K., Kimoto M., Miyake K., Takeda K., and Akira S. (2000) Cutting edge: Endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J. Immunol. 164, 3476–3479 10.4049/jimmunol.164.7.3476 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.