

Abstract
Levoglucosan is the 1,6-anhydrosugar of d-glucose formed by pyrolysis of glucans and is found in the environment and industrial waste. Two types of microbial levoglucosan metabolic pathways are known. Although the eukaryotic pathway involving levoglucosan kinase has been well-studied, the bacterial pathway involving levoglucosan dehydrogenase (LGDH) has not been well-investigated. Here, we identified and cloned the lgdh gene from the bacterium Pseudarthrobacter phenanthrenivorans and characterized the recombinant protein. The enzyme exhibited high substrate specificity toward levoglucosan and NAD+ for the oxidative reaction and was confirmed to be LGDH. LGDH also showed weak activities (∼4%) toward l-sorbose and 1,5-anhydro-d-glucitol. The reverse (reductive) reaction using 3-keto-levoglucosan and NADH exhibited significantly lower Km and higher kcat values than those of the forward reaction. The crystal structures of LGDH in the apo and complex forms with NADH, NADH + levoglucosan, and NADH + l-sorbose revealed that LGDH has a typical fold of Gfo/Idh/MocA family proteins, similar to those of scyllo-inositol dehydrogenase, aldose–aldose oxidoreductase, 1,5-anhydro-d-fructose reductase, and glucose–fructose oxidoreductase. The crystal structures also disclosed that the active site of LGDH is distinct from those of these enzymes. The LGDH active site extensively recognized the levoglucosan molecule with six hydrogen bonds, and the C3 atom of levoglucosan was closely located to the C4 atom of NADH nicotinamide. Our study is the first molecular characterization of LGDH, providing evidence for C3-specific oxidation and representing a starting point for future biotechnological use of LGDH and levoglucosan-metabolizing bacteria.
Keywords: dehydrogenase, nicotinamide adenine dinucleotide (NADH), X-ray crystallography, bacterial metabolism, enzyme kinetics, Gfo/Idh/MocA family, l- sorbose, levoglucosan, scyllo-inositol dehydrogenase
Introduction
Levoglucosan (1,6-anhydro-β-d-glucose, LG)3 is formed by pyrolysis of glucans, such as cellulose and starch (1), and frequently appears in the scientific literature as a tracer of biomass burning in atmospheric aerosol (2, 3) and in snow and ice (4). LG and other anhydrosugars are also present in soil, thermochemical processing products of biomass, and municipal waste in considerably large amounts (5–7). Therefore, microbial metabolic pathways of anhydrosugars are recently attracting significant research attention in the context of bioconversion into chemicals and biofuels (8, 9). In the early 1990s, Yasui and co-workers (10, 11) identified two types of microbial metabolic pathways that can introduce LG into the glycolytic pathway using ATP-dependent levoglucosan kinase (EC 2.7.1.-, LGK) or NAD+-dependent levoglucosan dehydrogenase (EC 1.1.1.-, LGDH) (12) as an initial enzyme acting on LG. LGK was found in yeast and fungi, although LGDH was found in bacteria. Studies on naturally LG-fermenting yeasts and fungi, which exhibit LGK activity (13), and heterologous expression of the fungal LGK gene in Escherichia coli (14) demonstrated that the microbial enzymes are promising tools for LG utilization for producing ethanol or lipids (9). Moreover, a recent biochemical and crystallographic study of LGK from Lipomyces starkeyi provided detailed insights into the function and mechanism of the fungal LGK (15). However, the bacterial LGDH pathway is still obscure because the genes for the metabolic pathway have not yet been identified. In 1994, Nakahara et al. (12) purified and characterized LGDH from Arthrobacter sp. I-552 (designated AspLGDH in this work) and proposed a possible metabolic pathway of the bacteria that consist of three steps (Fig. 1A): NAD+-dependent dehydrogenation by LGDH producing 3-keto-levoglucosan (1,6-anhydro-β-d-ribo-hexopyranos-3-ulose, 3-keto-LG); intramolecular hydrolysis producing 3-keto-d-glucose; and NADH-dependent reduction producing d-glucose. The latter two reactions were proposed to be catalyzed by two enzymes other than LGDH (16). Although LGDH was suggested to catalyze the synthesis of 3-keto-LG with the conversion of NAD+ into NADH and H+, it has not been determined which hydroxyl of LG (C2, C3, or C4) is oxidized. Nakahara et al. (12) suggested that the oxidization occurs at the C3 position based on characterization of the crude product by the UV-visible absorption spectrum under alkaline conditions. However, strong bias of the equilibrium of NAD+-dependent dehydrogenase reaction toward the reduced hydroxyl form (reverse reaction) (17) probably hampered successful isolation and characterization of the reaction product in the previous study. Recently, one of us developed a method to synthesize 3-keto-LG, which can be used for the characterization of LGDH, by exploiting the C3-specific LG oxidation of pyranose oxidase (18). 3-Keto-LG was found to be unstable in aqueous solution, even under neutral conditions, due to spontaneous decomposition by β-elimination (18).
Figure 1.

Possible reaction scheme of microbial LG conversion into d-glucose (A), and noncanonical substrates and related molecules for PpLGDH (B). A, steps suggested for Arthrobacter sp. I-552 (12) and B. smithii S-2701M (40).
In this study, we identified and cloned the lgdh gene from the phenanthrene-degrading soil bacteria, Pseudarthrobacter phenanthrenivorans Sphe3, which was formerly designated Arthrobacter phenanthrenivorans (19, 20). From functional and structural characterizations of the heterologously expressed LGDH enzyme, we provided evidence for the C3 oxidation. The crystal structures complexed with NADH and substrates revealed the structural basis for the LG-specific dehydrogenation mechanism.
Results
Enzyme characterization
Through a BLAST search on the bacterial genome database, using the N-terminal amino acid sequences of the 28-kDa subunit-1 (TGNLNVGLIGGGFMGKAHSL) and 21-kDa subunit-2 (TRGGEGPKGPVDVDDEVMTM) of AspLGDH (12), we found several hypothetical proteins: for example, Asphe3_10730 (annotated as putative dehydrogenase) from P. phenanthrenivorans Sphe3 (21); Arth_1911 (oxidoreductase domain protein) from Arthrobacter sp. FB24 (22); and G205_19216 (dehydrogenase) from Arthrobacter nitrophenolicus strain SJCon (23). The three hypothetical proteins show high sequence homology (identity >68%). The two N-terminal sequences of the two subunits of AspLGDH correspond to residues 3–20 and 230–251 of Asphe3_10730 (390 amino acids).
The asphe3_10730 gene cloned from the genomic DNA of P. phenanthrenivorans Sphe3 was expressed as a His6-tag fusion protein in E. coli. The purified Asphe3_10730 protein migrated in SDS-PAGE as a single protein band with an estimated molecular mass of ∼42 kDa, in agreement with the theoretical molecular mass of 44,280 Da. The molecular mass of nondenatured Asphe3_10730 protein estimated by calibrated gel-filtration chromatography was 135 kDa, suggesting that it is tetrameric in solution. The result was not consistent with that of AspLGDH (290 kDa by gel filtration) (12), but crystallographic analysis provided further evidence for tetramer formation, as described below. Because the purified single-polypeptide protein exhibited LGDH activity, as described below, the Asphe3_10730 protein is designated PpLGDH in this paper.
When PpLGDH was incubated with 20 mm LG and 2 mm NAD+ in 50 mm Tris-HCl (pH 8.5) containing 1 mg/ml bovine serum albumin (BSA) as a stabilizer at 40 °C, the absorbance at 340 nm (for NADH) increased with specific activity of 17.8 units/mg (Table 1). The specific activity of PpLGDH was comparable with that of AspLGDH (13.8 units/mg) with 100 mm LG and 2 mm NAD+ at pH 9.0 and 40 °C (12). Table 1 shows specific activities toward various compounds measured by the NAD+ reduction, indicating that this enzyme is highly specific for LG. PpLGDH exhibited weak activities (∼4% compared with that of LG) toward l-sorbose and 1,5-anhydro-d-glucitol, and activity toward d-xylose was also detectable (1.7%, Fig. 1B). The activities toward methyl-α-d-glucopyranoside and d-glucose were barely detectable (∼0.5%), and those toward other substrates (listed in Table 1) were less than 0.2% that of LG. The substrate specificity of PpLGDH was basically similar to that of AspLGDH (30% for sorbose, 18% for xylose, and 8–1% for ribose, glucose, galactose, fructose, and arabinose with 100 mm substrate) (12). PpLGDH was highly specific for the NAD+ cosubstrate (<0.05 units/mg with 2 mm NADP+ and 20 mm LG under the standard assay condition), in agreement with the previous study using AspLGDH (12). PpLGDH was stable up to 60 °C during a 30-min incubation and was stable in a pH range between 5.0 and 8.5 at 40 °C for 30 min, and the apparent optimal temperature was 60 °C (Fig. 2, A–C). The optimal temperature of AspLGDH was reported to be 50 °C (12).
Table 1.
Substrate specificity of PpLGDH
Activities for 20 mm substrate were measured in 2 mm NAD+, 1 mg/ml BSA, and 50 mm Tris-HCl (pH 8.5) at 40 °C. Numbers in parentheses indicate relative activity compared with that of LG (100%). The enzyme showed no significant activity (<0.2% compared with that of LG) toward scyllo-inositol, l-glucose, d-galactosan (1,6-anhydro-β-d-galactopyranose), sedoheptulosan (2,7-anhydro-β-d-altro-heptulopyranose), 1,5-anhydro-d-fructose, d-ribose, d-allose, d-fructose, d-galactose, d-mannose, l-arabinose, l-rhamnose, 1,5-anhydro-d-mannitol, sorbitol (d-glucitol), xylitol, dulcitol (galactitol), erythritol, α-d-glucose 1-phosphate, cellobiose, sucrose, lactose, maltose, and trehalose. NADH-dependent reductase activities (reverse reaction) toward 1,5-anhydro-d-fructose and d-glucono-1,5-lactone were also not detected.
| Substrate | Specific activity |
|---|---|
| units/mg | |
| LG | 17.8 (100) |
| l-Sorbose | 0.73 (4.1) |
| 1,5-Anhydro-d-glucitol | 0.65 (3.8) |
| d-Xylose | 0.31 (1.7) |
| Methyl-α-d-glucopyranoside | 0.10 (0.56) |
| d-Glucose | 0.090 (0.50) |
Figure 2.

Enzymatic properties of PpLGDH. A, thermal stability after incubation of the enzyme for 30 min at pH 7.5. B, pH stability after incubation of the enzyme for 30 min at 40 °C. C, effect of temperature on the activity (forward reaction). D, s–v plot of the forward reaction with 2 mm NAD+ at pH 8.5. E, s–v plot of the reverse reaction with 0.2 mm NADH at pH 7.0. F, effect of pH on the forward reaction with 20 mm LG and 2 mm NAD+. G, effect of pH on of the reverse reaction with 2 mm 3-keto-LG and 0.2 mm NADH. See “Experimental procedures” for detailed conditions.
When the LGDH activity was measured under the standard condition (20 mm LG and 2 mm NAD+, Table 1), the reaction quickly reached equilibrium after 0.37% of the substrate (LG) was oxidized. Therefore, we employed pseudo-first–order curve-fitting to accurately measure the velocity as it slowed down at an early stage of the reaction (see “Experimental procedures”). When we measured equilibrium of the LGDH reaction with 4, 10, 20, and 40 mm LG and 2 mm NAD+ at pH 8.5 and 40 °C, the equilibrium constant (Keq = ([product][NADH][H+])/([LG][NAD+])) was (1.21 ± 0.02) × 10−13 m, showing that the equilibrium is highly biased toward the reverse (reducing) reaction. The Keq value of the LGDH reaction was 23-fold smaller than that of the reaction of NADH-dependent l-lactate dehydrogenase, which is one of the most studied dehydrogenases, at 25 °C (Keq = 2.76 × 10−12 m) (24).
Fig. 2D shows the s–v plot of PpLGDH for LG (forward reaction). The Km and kcat values were 7.5 ± 0.6 mm and 4.3 ± 0.1 s−1, respectively, at pH 8.5 and 40 °C. The Km value was smaller than that reported for AspLGDH (14 mm) (12). It is noteworthy that the Km values of LGK determined in previous studies were significantly higher (69–119 mm) (15). The apparent Km value of PpLGDH for NAD+ was 0.34 ± 0.08 mm with 50 mm LG at pH 9.0 and 40 °C (data not shown) and was also comparable with that of AspLGDH (Km for NAD+ = 0.47 mm at pH 9.0 and 40 °C) (12).
We also tried to identify the oxidized reaction product from LG. In a 1H NMR spectrum of the deionized reaction mixture, several signals corresponding to 3-keto-LG were observed (data not shown). However, overlapping signals of the abundant LG hampered complete identification of the reaction product. Instead, we found that PpLGDH efficiently catalyzed NADH-dependent reduction (reverse reaction) of 3-keto-LG, which was previously synthesized (18). A kinetic analysis showed that the reverse reaction activity was significantly higher than that of the forward reaction, providing strong evidence for the C3 position-specific reaction by LGDH. The s–v plot exhibited typical uncompetitive substrate inhibition, which is often observed for NAD(P)H-dependent dehydrogenases (Fig. 2E) (17). The Km, kcat, and Ki values for the reverse reaction were 0.48 ± 0.07 mm, 26 ± 2 s−1, and 9.1 ± 1.7 mm, respectively, at pH 7.0 and 40 °C. Under all conditions we tested, no sigmoidal kinetics was observed. The pH profiles of the forward (Fig. 2F) and reverse (Fig. 2G) reactions indicated that PpLGDH prefers the alkaline and slightly acidic pH conditions for those reactions, respectively. Such pH profiles are also typical for NAD(P)H-dependent dehydrogenases, which use the oxidoreductase reaction between NAD(P)+ and NAD(P)H + H+ and have an acid/base catalyst residue with neutral pKa (typically His) (17, 25). The pKa values estimated from the pH profiles (7.6 ± 0.1 for the forward reaction and 7.1 ± 0.2 for the reverse reaction) suggested that the acid/base catalysis of PpLGDH occurs at His. Additionally, significant reduction in the reverse reaction activity at acidic pH (apparent pKa = 5.7 ± 0.2 by the curve fitting in Fig. 2G) suggested involvement of a second ionizable group in the catalysis.
Crystallography and overall structure
The crystal structures of PpLGDH in apo form, binary complex form with NADH, and ternary complex forms with NADH + LG and NADH + l-sorbose were determined at 1.8–2.4 Å resolutions (Table 2). The tetragonal crystal (space group P43212) of the apo form and the monoclinic crystals (space group P21) of the complex forms contain two and four subunits in the asymmetric unit, respectively. The four subunits in the complex forms and that formed by a crystallographic 2-fold axis in the apo crystal constitute a tetramer related by three 2-fold axes (222 point group symmetry, Fig. 3A). The four chains in the asymmetric unit of the complex structures showed no significant differences; the root mean square deviations (r.m.s.d.) for the Cα atoms were less than 0.92 Å for all of the chain pairs by applying no distance cutoff. The two chains in the apo form crystal also had virtually the same structures (Cα r.m.s.d. = 0.15 Å with 2.0 Å distant cutoff), and a significant deviation was only observed at a functionally irrelevant disordered loop region (residues 223–238, described below). A molecular interface analysis using the PISA server (26) indicated that the tetramer is the most likely state in solution, in accordance with the gel filtration result described above. The interface areas between A–B (or C–D), A–C (or B–D), and A–D (or B–C) chains are ∼1,780, 1,560, and 670 Å2, respectively, and the total estimated ΔGint value of the tetramer formation (26) is approximately −100 kcal/mol. The virtually symmetric quaternary structure of PpLGDH and the simple Michaelis-Menten–type kinetics (Fig. 2, D and E) indicate that it is not an allosteric enzyme.
Table 2.
Data collection and refinement statistics
| Data set | Apo | + NADH | + LG, NADH | + Sorbose, NADH |
|---|---|---|---|---|
| Data collection | ||||
| Beamline | AR-NE3A (KEK-PF) | AR-NW12A (KEK-PF) | BL1A (KEK-PF) | BL26B1 (SPring-8) |
| Space group | P43212 | P21 | P21 | P21 |
| Unit cell | a = b = 95.6 c = 120.8 | a = 80.7 | a = 76.7 | a = 80.2 |
| Dimensions (Å) and angles (°) | b = 93.8 | b = 92.4 | b = 93.5 | |
| c = 101.0 | c = 100.6 | c = 100.9 | ||
| β = 101.0 | β = 104.5 | β = 101.4 | ||
| Mole/ASUa | 2 | 4 | 4 | 4 |
| Resolution (Å)b | 50.0–1.80 (1.83–1.80) | 50.0–1.90 (1.93–1.90) | 50.0–2.40 (2.44–2.40) | 50.0–1.90 (1.93–1.90) |
| Total reflections | 1,050,924 | 407,615 | 170,900 | 431,229 |
| Unique reflectionsb | 74,076 (3,610) | 114,689 (5,628) | 51,934 (2590) | 114,792 (5696) |
| Completeness (%)b | 99.9 (100.0) | 98.7 (97.2) | 99.5 (99.7) | 100.0 (99.9) |
| Redundancyb | 14.2 (13.2) | 3.6 (3.3) | 3.3 (3.4) | 3.8 (3.7) |
| Mean I/σ(I) b | 35.4 (3.4) | 11.4 (1.4) | 16.1 (2.4) | 13.8 (1.7) |
| Rmerge (%)b | 9.3 (69.4) | 10.4 (77.7) | 8.3 (52.0) | 10.3 (72.0) |
| CC1/2a | (0.974) | (0.903) | (0.959) | (0.936) |
| Wilson B-factor (Å2) | 27.84 | 23.47 | 63.79 | 28.60 |
| Refinement | ||||
| Resolution (Å) | 38.28–1.80 | 36.53–1.90 | 34.84–2.41 | 49.49–1.90 |
| No. of reflections | 70,139 | 109,019 | 49,298 | 108,882 |
| Rwork/Rfree (%) | 16.7/20.2 | 16.9/20.4 | 21.5/29.7 | 16.6/20.7 |
| No. of atoms (protein/heterogen/solvent) | 6,350 | 12,439 | 11,518 | 12,478 |
| r.m.s.d. from ideal values | ||||
| Bond lengths (Å) | 0.013 | 0.010 | 0.008 | 0.010 |
| Bond angles (°) | 1.555 | 1.423 | 1.248 | 1.433 |
| No. of solvents and ligands | ||||
| Waters | 484 | 749 | 61 | 767 |
| SO4 | 3 | |||
| MES | 2 | |||
| NADHc | 4 (A, B, C, D) | 3 (A*, B**, C**) | 4 (A*, B*, C*, D*) | |
| Substratec | LG × 2 (A, D) | l-Sorbose × 4 (A, B, C, D) | ||
| Average B-factors (Å2) (chain A/B/C/D) | ||||
| Protein | 28.6/28.4 | 22.6/24.3/25.6/23.5 | 53.1/62.1/72.1/77.8 | 27.1/29.2/30.1/30.8 |
| NADH | 23.0/25.3/28.3/24.8 | 48.3/58.5/65.4/– | 25.0/29.2/30.0/31.1 | |
| Substrate | 62.5/−/−/65.0 | 25.4/26.7/25.7/26.1 | ||
| Solvents | 35.1 | 31.3 | 49.7 | 36.5 |
| Ramachandran plot (%) | ||||
| Favored | 98.1 | 98.2 | 93.7 | 98.0 |
| Allowed | 1.9 | 1.8 | 5.5 | 2.0 |
| Outlier | 0.0 | 0.0 | 0.8 | 0.1 |
| PDB code | 6A3F | 6A3G | 6A3I | 6A3J |
a Number of molecules in the asymmetric unit is shown.
b Values in parentheses are for the highest resolution shell.
c Modeled chains are indicated in parentheses. For NADH, a single asterisk indicates that the nicotinamide moiety was not modeled. Double asterisks indicate that both the nicotinamide and adenine moieties were not modeled.
Figure 3.
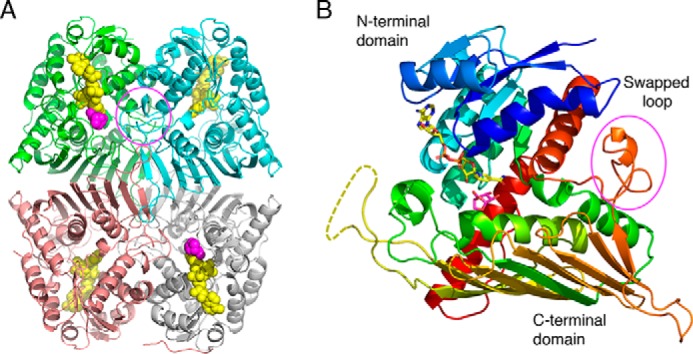
Overall structure of PpLGDH. A composite complex structure with NADH (yellow) and LG (magenta) is shown. The complete NADH molecules observed in the NADH complex structure are superimposed on the complex structure with LG + NADH, whose NADH molecules were only partially observed. A, biological tetramer structure. NADH and LG are shown as spheres. The biological tetramer contained in the asymmetric unit of the LG + NADH complex structure (space group P21) is shown. Chains A–D are shown in green, pink, cyan, and gray, respectively. B, monomer structure in rainbow colors. NADH and LG are shown as sticks. Disordered region (residues 223–238) is shown as a dotted line. The “swapped loop” is indicated in magenta circles.
The monomer has a typical two-domain structure of the glucose–fructose oxidoreductase/inositol dehydrogenase/rhizopine catabolism protein (Gfo/Idh/MocA) family (Fig. 3B) (27). The N-terminal nucleotide-binding domain (before Ala-130) adopts a Rossmann fold and consists of a β-sheet with six strands, flanking five α-helices, and one 310-helix. The C-terminal substrate-binding α/β domain (after Phe-131) consists of a nine-stranded β-sheet, six α-helices, and four 310-helices. The long β-sheet in the C-terminal domain caps the other structural elements in the monomer and mainly contributes to the tetramer interface between A–C (and B–D) chains (Fig. 3A). Residues 223–238 in the C-terminal domain (shown as a yellow dotted line in Fig. 3B) were disordered in all of the four structures and not included in the crystallographic model. This loop is located away from the active site and appears to be functionally irrelevant. A “swapped” loop, which partly forms the active site of the neighboring subunit (residues 316–334, marked in magenta circles in Fig. 3), is present (described below). The N-terminal sequence of the 21-kDa fragment of AspLGDH corresponds to the C-terminal half of the disordered region and subsequent region (residues 230–251), suggesting that AspLGDH was purified as a nicked protein that had been cleaved by proteolysis at the flexible region.
Table 3 shows the result of a structural similarity search using the Dali server (28). Five out of the top six hits were entries of bacterial hypothetical proteins (putative oxidoreductases), which were determined by structural genomics projects. The third hit was scyllo-inositol dehydrogenase (sIDH) from Paracoccus laeviglucosivorans (PlsIDH), whose crystal structure was determined very recently (29). PlsIDH was originally isolated as a catabolic enzyme of l-glucose utilization by P. laeviglucosivorans (30, 31). PlsIDH exhibits both sIDH (Km = 3.7 mm and kcat = 11.8 s−1) and l-glucose dehydrogenase (Km = 59.7 mm and kcat = 17.3 s−1) activities (31). Subsequent hits were 1,5-anhydro-d-fructose (1,5-AF) reductase (AFR) from Sinorhizobium morelense (SmoAFR) (32), aldose–aldose oxidoreductase (AAOR) from Caulobacter crescentus (CcAAOR) (33), glucose–fructose oxidoreductase (GFOR) from Zymomonas mobilis (ZmGFOR) (34), and AFR from Sinorhizobium meliloti (35). However, PpLGDH did not exhibit the activities of sIDH (oxidation of scyllo-inositol and l-glucose), AFR (reduction of 1,5-AF to 1,5-anhydro-d-mannitol), AAOR (both of oxidation and reduction of aldose monosaccharides), and GFOR (coupled reactions of oxidation of d-glucose to d-glucono-1,5-lactone and reduction of d-fructose to d-sorbitol) (Table 1). PlsIDH is an NADH-specific dehydrogenase, whereas the other structural homologs with known functions (AFRs, AAOR, and GFOR) are all NADPH-specific oxidoreductases. The tetramer assembly of LGDH is very similar to those of PlsIDH (29) and ZmGFOR (34), whereas AFRs, CcAAOR, and other Gfo/Idh/MocA family proteins commonly have a dimer structure (27).
Table 3.
Result of structural similarity search using Dali server
| Protein | Source organism | PDB (chain) | Z score | r.m.s.d. | Naligna | %seqb |
|---|---|---|---|---|---|---|
| Å | ||||||
| Putative oxidoreductasec | K. flavida | 4H3V (A) | 54.2 | 1.4 | 373 | 42 |
| Putative oxidoreductasec | K. pneumoniae | 4GQA (C) | 52.3 | 1.5 | 373 | 39 |
| scyllo-Inositol dehydrogenase (PlsIDH) | P. laeviglucosivorans | 5YAB (C) | 50.5 | 1.5 | 368 | 38 |
| Putative oxidoreductasec | R. etli | 4FB5 (A) | 49.0 | 1.6 | 367 | 35 |
| Putative oxidoreductasec | S. meliloti | 3V5N (A) | 40.9 | 1.8 | 330 | 25 |
| Putative oxidoreductasec | P. syringae | 3DTY (A) | 40.0 | 2.0 | 337 | 21 |
| 1,5-Anhydro-d-fructose reductase (SmoAFR) | S. morelense | 2GLX (A) | 38.4 | 2.3 | 322 | 26 |
| Aldose–aldose oxidoreductase (CcAAOR) | C. crescentus | 5A03 (C) | 37.8 | 2.0 | 324 | 24 |
| Glucose–fructose oxidoreductase (ZmGFOR) | Z. mobilis | 1H6D (A) | 37.1 | 2.1 | 316 | 23 |
| 1,5-Anhydro-d-fructose reductase (SmeAFR) | S. eliloti | 4KOA (A) | 37.0 | 2.1 | 316 | 23 |
a The number of aligned residues is shown.
b Sequence identity is shown.
c The uncharacterized protein whose structure was determined by structural genomics projects is shown.
Active site
In the NADH-only complex structure of PpLGDH, the electron density of NADH was unambiguously observed for the entire molecule (Fig. 4A). The nicotinamide ribose and adenine ribose moieties are both in an anti-C2′-endo conformation. The NADH molecule is recognized by the main-chain atoms of Phe-13, Met-14, Thr-81, Lys-104, and Ala-130 and the side chain atoms of Glu-43, Asn-83, Glu-103, Lys-104, Asn-132, Trp-175, and Tyr-335. The conformation and protein interactions of the nicotinamide group, its ribose, and the pyrophosphate group are similar to those found in PlsIDH, AAOR, AFRs, and GFOR (29, 32, 33, 36). The conformation of the adenosine moiety (anti-C2′-endo) is similar to the NADH-dependent PlsIDH, although that is clearly different from the NADPH-dependent oxidoreductases (AAOR, AFR, and GFOR), in which the ribose moiety was in a syn-C3′-endo conformation. These NADPH-dependent oxidoreductases have a positively charged binding pocket for the 2′-phosphate group formed by the side chains of Ser, Thr, and Lys (or Arg). In PpLGDH, the side chain of Glu-43 forms bifurcated hydrogen bonds with the hydroxyls of ribose, in accordance with the sharp cosubstrate specificity toward NAD+ over NADP+, as described above.
Figure 4.
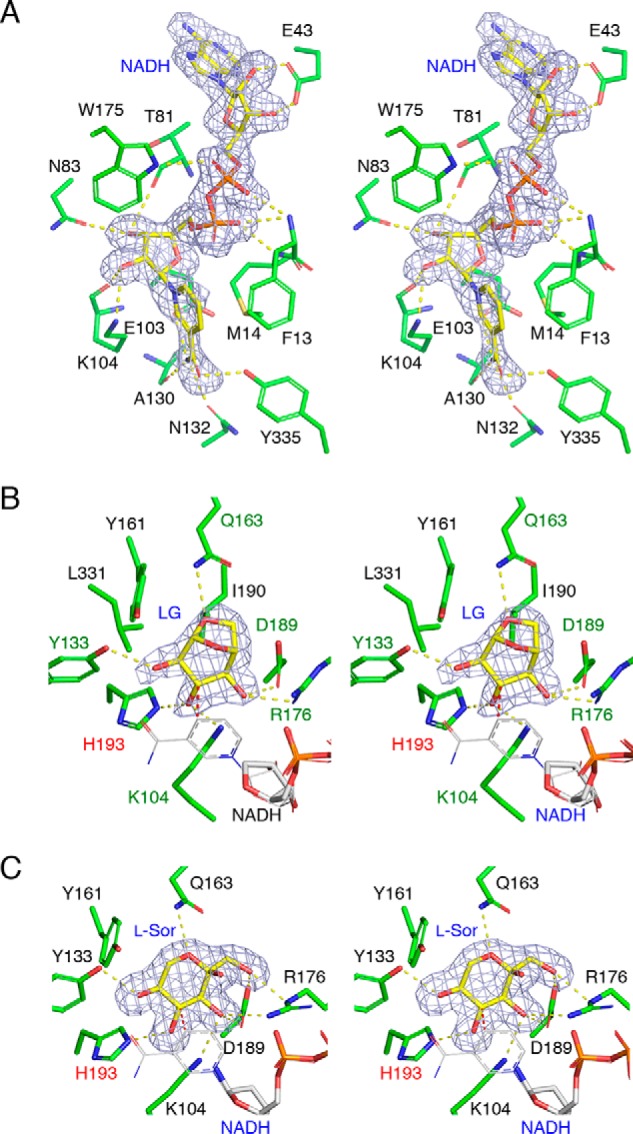
Stereoviews of the substrate-binding sites. mFo − Fc omit electron density maps (3.0σ, gray mesh) are shown for each ligand. Protein residues and ligands are shown as green and yellow sticks, respectively. Hydrogen bonds are shown as yellow dotted lines. A, NADH-binding site of the NADH complex structure. B, active site of NADH + LG complex structure. Residues forming direct hydrogen bonds are designated by green labels. C, active site of NADH + l-sorbose complex structure. B and C, partially disordered NADH molecule and superimposed complete NADH molecule (from the NADH complex) are shown as thick sticks and thin lines in gray color. Interatomic distance between the C4 atom of NADH nicotinamide and the target carbon atom (C3 of LG and C4 of l-sorbose) are 2.2–2.3 Å and shown as red dotted lines. His-193 (designated by red label) is a possible proton donor residue.
In the NADH + LG complex structure, the electron density of NADH was relatively ambiguous, especially for the nicotinamide moiety, and LG was observed in chains A and D (Table 2). As shown in Fig. 4B, electron density of LG in chain A was unambiguously observed. The three hydroxyl groups (C2, C3, and C4) and the bicyclic group were clearly seen. The LG molecule adopts a conformation between B3, O and 1S3, and the three hydroxyl groups are all equatorial. The Cremer-Pople parameters (37) of the LG molecules in the A/D chains are ϕ = 194.3/190.2°, ψ = 94.7/93.3°, and Q = 0.85/0.85. The C2, C3, and C4 hydroxyl groups and the O6 atom form hydrogen bonds with the side chains of Tyr-133, Lys-10, and His-193, Arg-176 and Asp-189, and Gln-163, respectively. When the nicotinamide group of the NADH complex structure was superimposed (thin gray lines in Fig. 4B), the C4 atom is located near the hydrogen atom side of C3 (C4–C3 atom distance = 2.3 Å). This structure gives a strong support for a C3-specific dehydrogenase reaction.
The l-sorbose molecule bound to PpLGDH adopts the α-l-sorbopyranose form (Figs. 1B and 4C). The side chains of Arg-176 and Asp-139 form hydrogen bonds with the C1 and C3 hydroxyls, and the side chain of Tyr-133 forms a hydrogen bond with the C5 hydroxyl. The C4 hydroxyl forms hydrogen bonds with the side chain of His-193. The distance between the C4 atom of l-sorbose and the C4 atom of superimposed NADH molecule is 2.2 Å, suggesting that the dehydrogenase reaction toward l-sorbose occurs at the C4 hydroxyl, and the product is likely 4-keto-l-sorbose.
Discussion
Structural comparison with other oxidoreductases
Gfo/Idh/MocA family enzymes share a similar overall three-dimensional fold, although their sequence identity between most of the members is very low, and the extent of different function is large (27). Here, we also found that several monosaccharide oxidoreductases exhibit significant structural similarity with LGDH, but they show low sequence homology (23–38% identity). Fig. 5A shows a phylogenetic tree of PpLGDH and its homologs. PpLGDH and its close uncharacterized homologs (G205_19216 and Arth_1911), AFRs, and two monosaccharide-specific oxidoreductases (CcAAOR and ZmGFOR) form separate clades, whereas PlsIDH and other uncharacterized structural homologs do not form clear clades.
Figure 5.

Phylogenetic tree (A) and partial amino acid sequence alignment (B) with uncharacterized close homologs (Arth_1911 and G205_19216) and structural homologs in Gfo/Idh/MocA family. Putative oxidoreductases are designated by PDB codes. B, key amino acid residues in the active site of PpLGDH are indicated above the sequences. The swapped loop region of PpLGDH and PlsIDH is boxed by magenta lines.
To investigate characteristic features for substrate recognition, the active sites of these homologs were compared (Fig. 6). Fig. 6, A and B, shows superimposition of the PpLGDH structure with the structures of PlsIDH (scyllo-inosose complex and l-glucono-1,5-lactone complex), and Fig. 5B shows partial amino acid sequence alignment of PpLGDH and its homologs. A long “swapped” loop (residues 303–321 indicated by the magenta box in Fig. 5B), which extends to the neighboring subunit, is a characteristic feature of PlsIDH (29). The loop is directly involved in the substrate recognition (Fig. 6A). His-318 in the swapped loop of PlsIDH forms a hydrogen bond with O6 hydroxyl of scyllo-inosose (oxidized product of scyllo-inositol) and O3 hydroxyl of l-glucono-1,5-lactone (oxidized product of l-glucose, Fig. 6B). In the l-glucono-1,5-lactone complex, the side chains of Arg-178 and Asp-191 displace to avoid the protruding C6 hydroxyl (shown as blue arrows in Fig. 6B). In PpLGDH, there is a corresponding loop, which partly forms the active site of a neighboring subunit (shown as magenta in Fig. 6A). However, Leu-331 is present in PpLGDH at the corresponding position of His-318 in PlsIDH and appears to prevent binding of the sIDH substrates (Fig. 6B). In PpLGDH, the side chains of Arg-176 and Asp-189 are slightly displaced to accommodate the protruding C1 hydroxyl of l-sorbose (Fig. 4C) compared with the LG complex (Fig. 4B). Fig. 6, C and D, shows superimpositions of the PpLGDH structure (LG complex) with the structures of SmoAFR (acetate complex) and CcAAOR (d-sorbitol complex and d-xylose complex). Lys-104, Arg-176, and Asp-189 of PpLGDH are conserved in these enzymes, whereas other residues are not. His-193 is conserved in PlsIDH and AFRs, and the corresponding residue is suggested to be involved in the acid/base catalysis (29, 32). The corresponding residue in CcAAOR is Tyr-189 (Fig. 6D). The Lys–Tyr dyad in CcAAOR and ZmGFOR was suggested to be essential for the catalysis, but the mutual role of these residues is still unclear (33). The presence of Tyr-133 and Gln-163, which play critical roles in binding of the C2 hydroxyl and O6 atom of LG, appears to be a characteristic of LGDH. In PlsIDH, Glu-165 is present at the corresponding position of Gln-163 in PpLGDH (Fig. 6B).
Figure 6.

Structural comparison with homologous enzymes. A, superimposition at subunit interface of PpLGDH (chain A in green and chain C in pink) and scyllo-inosose complex of PlsIDH (PDB code 5YAQ, chain B in cyan and chain A in dark blue). The swapped loops are shown in magenta (PpLGDH) and light blue (PlsIDH). B–D, active sites of PpLGDH (green) and structural homologs (cyan and blue) are superimposed. B, scyllo-inosose complex (PDB code 5YAQ, cyan) and d-glucono-1,5-lactone complex (PDB code 5YAP, blue) of PlsIDH. The side chains from the neighboring subunit (His-318 and Leu-311) are underlined. The side chain displacements of Arg-178 and Asp-191 in PlsIDH are shown by blue arrows. C, acetate complex of SmoAFR (PDB code 2GLX, ligand in yellow). D, d-sorbitol complex of CcAAOR (PDB code 5A06, ligand in yellow). d-Xylose (PDB code 5A03, gray sticks) is also shown.
From the multiple sequence alignment (Fig. 5B), the two close homologs from Pseudarthrobacter and Arthrobacter species (Arth_1911 and G205_19216) and two putative proteins from Kribbella flavida (PDB code 4H3V) and Klebsiella pneumoniae (PDB code 4GQA) were suggested to have LGDH activity because both residues corresponding to Tyr-133 and Gln-163 are conserved. From the His residue conservation in the swapped loop, putative proteins from Rhizobium etli (PDB code 4FB5), Sinorhizobium meliloti (PDB code 3V5N), and Pseudomonas syringae (PDB code 3DTY) probably have sIDH activity.
Reaction mechanism and substrate recognition of LGDH
From the biochemical and structural analyses, we concluded that the reversible dehydrogenase reactions of LGDH are C3-specific. In the forward (oxidative) reaction, hydride transfer must occur from the C3 hydrogen of LG to the pro-R side of the C4 atom of NAD+ (Fig. 4B). His-193 is the most likely candidate of the proton donor for the O3 atom by considering the apparent pKa values estimated by the pH profiles of the forward and reverse reactions. As to candidates of the second ionizable group of the reverse reaction (pKa = 5.7), Asp-189 and Glu-103 were found in the active site (Fig. 4, A and B). The presence of the nearby Arg-176 residue would reduce the pKa of Asp-189; thus, we suggest that Glu-103 is the most likely candidate for the second ionizable group involved in the reverse reaction. The protonation of Glu-103 probably weakens the fixation of the amide group of NADH and reduces the activity.
We also determined the complex structure with the second-best substrate, l-sorbose (4.1% activity compared with that of LG). The three hydroxyl groups (C3, C4, and C5) of α-l-sorbopyranose are recognized by LGDH in a similar manner to the C2, C3, and C4 hydroxyl groups of LG. The third and fourth preferred substrates (1,5-anhydro-d-glucitol and d-xylose) also have a similar structural feature; three hydroxyl groups on a pyranose ring have a down-up-down (all equatorial) configuration. In addition, the absence of a hydroxyl or a methoxyl group at the C1 position is a key feature for the substrates of LGDH (Table 1). As shown in Figs. 4C and 5B, the hydrophobic interaction and steric hindrance by the side chains of Leu-331 and Tyr-161 are responsible for limiting the specificity at the C1 (LG) and C6 (l-sorbose) atoms.
It has been shown that LG predominantly adopts the inverted chair (1C4) ring conformations, in which the three hydroxyl groups are axial and form an intramolecular hydrogen-bonding interaction in the gas phase (38) and in crystals (39). The all-axial 1C4 conformation was also observed in the LG molecule bound to LGK (15). Therefore, the all-equatorial B3, O–1S3 conformation observed in LGDH is likely a high-energy conformation. However, LGDH exhibited a significantly lower Km value than that of LGK. This result is probably because the LG molecule is stabilized by more extensive interactions in the active site of LGDH, such as hydrogen bonds, hydrophobic interactions, and shape complementarity.
Possible bacterial degradation pathway of LG
In this study, we demonstrated that LGDH produces 3-keto-LG. Nakahara et al. (12) suggested that the degradation steps by Arthrobacter sp. I-552 subsequently proceed through two enzymatic reactions: intramolecular hydrolysis of 3-keto-LG to 3-keto-glucose and NADH-dependent reduction to d-glucose. Involvement of the additional enzymes in the course of glucose formation from LG was demonstrated by anion-exchange chromatogram of the crude extract of the bacteria (16). In addition to the LGDH-containing fraction, at least two separate fractions were required to reconstruct the full glucose-forming activity. Recently, Iwazaki et al. (40) reported isolation of LG-utilizing thermophilic bacteria (Bacillus smithii and Parageobacillus thermoglucosidasius strains). B. smithii S-2701M showed the best growth on LG, and anion-exchange chromatography of its crude extract exhibited a similar separation pattern of fractions containing the LGDH activity and the LGDH-dependent glucose-forming activity. In addition to Pseudarthrobacter and Arthrobacter species (Actinobacteria), the amino acid sequence alignment with the putative enzymes (Fig. 5B) suggested that LGDH is also present in K. flavida (Actinobacteria) and K. pneumoniae (Gammaproteobacteria). Further studies are required to identify the other enzymes for the glucose-forming activity and to explore distribution of possible LG-utilizing metabolic pathways among wide arrays of bacteria.
Concluding remarks
Here, we identified and cloned the LGDH gene from bacteria. LGDH was highly specific to LG and could be efficiently produced by the heterologous expression system of E. coli. Therefore, the enzyme has a potential for developing a novel fluorometric system (41) to detect LG in the environment. Moreover, understanding the molecular basis of the bacterial assimilation pathway will contribute to future developments in microbial bioconversion of LG that is present in industrial waste.
Experimental procedures
Materials
LG and l-glucose were purchased from Sigma. 1,5-Anhydro-d-fructose and 1,5-anhydro-d-mannitol were purchased from Carbosynth (Compton, Berkshire, UK). l-Sorbose was purchased from Nacalai Tesque (Kyoto, Japan). scyllo-Inositol was purchased from Tokyo Chemical Industry (Tokyo, Japan). 3-Keto-LG was enzymatically synthesized, as described previously (18). Other chemicals were purchased from Wako Pure Chemical Industries (Osaka, Japan). P. phenanthrenivorans Sphe3 (16027T) was purchased from RIKEN BRC (Tsukuba, Japan).
Gene cloning and protein preparation
The genomic DNA of P. phenanthrenivorans Sphe3 was prepared using Wizard Genomic DNA purification kit (Promega, WI). The gene encoding Asphe3_10730 (GenBankTM accession number ADX72256) was amplified from the genomic DNA via a PCR performed using Eazy-A Taq polymerase (Agilent Technologies, Santa Clara, CA). The oligonucleotide primers 5′-CATATGCAGAACCTCAACGTCG-3′ and 5′-AAGCTTTCACGCGCTGATCTGCG-3′ were used. The amplified product was ligated into the pGEM-T easy vector (Promega), transformed into E. coli JM109, and detected using blue/white screening. Sequence of the insert was confirmed by DNA sequencing. The plasmid was extracted and then digested with NdeI and HindIII. The insert fragment was purified by agarose gel extraction after electrophoresis, ligated into the expression vector pET28a (Novagen, Madison, WI) using ligation high (Toyobo, Osaka, Japan), and introduced into E. coli DH5α. E. coli C43 (DE3) harboring the expression plasmid was used to produce recombinant protein with an N-terminal His6 tag. The cells were grown at 37 °C in 1.5 liter of Luria-Bertani medium (1% tryptone, 0.5% yeast extract, and 0.5% NaCl) containing 50 μg/ml kanamycin until the absorbance reached 0.6 at 600 nm. The expression was then induced using 0.1 mm isopropyl β-d-thiogalactopyranoside and continued at 30 °C for 12 h. The cells were harvested via centrifugation at 7,300 × g for 10 min and suspended in 25 mm Tris-HCl buffer (pH 7.5). The cells were disrupted via sonication (Branson Sonifier250D; Branson Ultrasonics Division of Emerson Japan, Kanagawa, Japan), and the supernatant was collected via centrifugation at 12,000 × g for 45 min. The protein was purified to homogeneity using nickel-affinity chromatography (cOmplete His tag purification resin, Roche Diagnostics, Basel, Switzerland) and gel-filtration chromatography (HiLoad 16/60 Superdex 200 pg column, GE Healthcare, Buckinghamshire, UK). Solutions of 25 and 500 mm imidazole in 25 mm Tris-HCl (pH 7.5) were used for the wash and elution buffers for the nickel-affinity chromatography, respectively. A solution of 500 mm NaCl in 25 mm Tris-HCl (pH 7.5) was used for gel-filtration chromatography, and the relative molecular mass of the protein was determined using molecular standards of thyroglobulin (669 kDa), γ-globulin (158 kDa), ovalbumin (44 kDa), and myoglobin (17 kDa). The protein solution was desalted and concentrated using Vivaspin Turbo 15, MWCO 10,000 (Sartorius, Göttingen, Germany). The protein concentrations were determined using the BCA protein assay kit (ThermoFisher Scientific, Waltham, MA) with BSA as a standard.
Enzyme assay and kinetic parameter determination
The increase (forward reaction) or decrease (reverse reaction) in NADH was monitored by the absorbance at 340 nm using a temperature-controlled microplate reader (Synergy H1, BioTek, Winooski, VT, or Multiscan Go, ThermoFisher Scientific). The standard assay conditions for the forward reaction were 20 mm (or 50–100 mm) substrate (e.g. LG), 2 mm NAD+, and 1 mg/ml BSA in 50 mm Tris-HCl buffer (pH 8.5) at 40 °C. We normally used the assay condition with 20 mm substrate because high substrate concentration (100 mm), which was used by Nakahara et al. (12), caused various effects on the solution depending on substrates. An appropriate concentration of enzyme was added to start the reaction. For Synergy H1 and Multiscan Go microplate readers, 96- and 384-well plates were used, and volumes of the assay solution were 200 and 100 μl per well, respectively. Because of the strong equilibrium bias of the NAD+-dependent dehydrogenase reaction toward the reduced hydroxyl form, an increase of A340 for NADH slowed down immediately after starting the reaction when the activity was high. In such cases, the initial velocity (= A∞k) was calculated by fitting the data to the following equation (pseudo-first-order expression): At = A∞(1 − e−kt) + offset. One unit was defined as the amount of enzyme that reduces 1 μmol of NAD+ per min. The standard assay conditions for the reverse reaction were 2 mm substrate (e.g. 3-keto-LG), 0.2 mm NADH, and 1 mg/ml BSA in 50 mm HEPES-NaOH buffer (pH 7.0) at 40 °C.
The thermal and pH stabilities were evaluated by measuring the residual activity of the forward reaction under the standard assay condition (100 mm LG) after incubation of the enzyme at 30–70 °C for 30 min in 25 mm potassium-phosphate buffer (pH 7.5) and at various pH values at 40 °C for 30 min, respectively. For pH stability, the following 50 mm buffers were used: sodium acetate (pH 3.0–5.0), MES-NaOH (pH 6.0), MOPS-NaOH (pH 7.0), Tris-HCl (pH 8.0–9.0), CHES-NaOH (pH 9.0–10.0), and CAPS-NaOH (pH 11.0). The effect of temperature on the activity was evaluated by measuring the forward reaction activity under the standard assay condition (50 mm LG) by changing the temperature between 30 and 70 °C. The effect of pH on the forward (50 mm substrate) and reverse reaction activities was determined under standard conditions by substituting the buffer with the following 50 mm buffers: MES-NaOH (pH 5.5–6.5), HEPES-NaOH (pH 6.5–8.0), and Tris-HCl (pH 8.0–9.0). Of note, we adopted the assay condition with 50–100 mm LG for the purpose of comparing the pH and thermal features of PpLGDH with those of AspLGDH (12).
Crystallography
For crystallization, buffer of the protein solution was changed to 25 mm Tris-HCl (pH 7.5) using Amicon Ultra-4 centrifugal filter devices, 30,000 MWCO (Millipore, Billerica, MA). Crystals of PpLGDH were grown at 20 °C using the hanging drop vapor diffusion method by mixing 0.5 μl of a 10 mg/ml protein solution with an equal volume of a reservoir solution. Apo crystals were grown using a reservoir solution containing 0.1 m sodium cacodylate (pH 6.5) and 1.25 m ammonium sulfate and cryoprotected by a reservoir solution supplemented with 20% glycerol. Cocrystals were grown using a basal reservoir solution containing 0.1 m MES-NaOH (pH 6.0), 5% PEG3000, and 30% PEG200. The basal reservoir solution was supplemented with 20 mm NADH (NADH complex), 20 mm NADH, and 100 mm LG (NADH + LG), or 20 mm NADH and 100 mm l-sorbose (NADH + l-sorbose) for growth of cocrystals. The crystals were flash-cooled by dipping into liquid nitrogen. X-ray diffraction data were collected at 100 K on beamlines at the Photon Factory of the High Energy Accelerator Research Organization KEK, Tsukuba, Japan) and SPring-8 (Hyogo, Japan). The data set was processed using HKL2000 (42). The initial phase was determined by the molecular replacement method using Molrep (43), and the structure of a putative oxidoreductase protein from K. flavida (PDB code 4H3V) was used as a search model. Manual model building and refinement were carried out using Coot (44) and Refmac5 (45). Molecular graphic images were prepared using PyMOL (Schrödinger, LLC, New York).
Author contributions
M. S., M. N., C. Y., T. A., M. K., and S. F. formal analysis; M. S., M. N., C. Y., and M. K. investigation; M. S., M. N., C. Y., T. A., M. K., and S. F. writing-review and editing; C. Y., T. A., and M. K. methodology; C. Y., T. A., and S. F. project administration; M. K. and S. F. conceptualization; M. K. resources; S. F. supervision; S. F. funding acquisition; S. F. validation; S. F. visualization; S. F. writing-original draft.
Acknowledgments
We thank the staff of the Photon Factory and SPring-8 for the X-ray data collection and Prof. Emeritus Hirofumi Shoun whose research inspired this work.
This work was supported in part by the Platform Project for Support in Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from the Japan Agency for Medical Research and Development (AMED) and JSPS-KAKENHI (15H02443 to S. F.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 6A3F, 6A3G, 6A3I, and 6A3J) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- LG
- levoglucosan
- AAOR
- aldose–aldose oxidoreductase
- 1,5-AF
- 1,5-anhydro-d-fructose
- AFR
- 1,5-anhydro-d-fructose reductase
- AspLGDH
- levoglucosan dehydrogenase from Arthrobacter sp. I-552
- CcAAOR
- aldose–aldose oxidoreductase from Caulobacter crescentus
- GFOR
- glucose–fructose oxidoreductase
- LGDH
- levoglucosan dehydrogenase
- PlsIDH
- scyllo-inositol dehydrogenase from Paracoccus laeviglucosivorans
- PpLGDH
- levoglucosan dehydrogenase from Pseudarthrobacter phenanthrenivorans Sphe3
- sIDH
- scyllo-inositol dehydrogenase
- SmoAFR
- 1,5-anhydro-d-fructose reductase from Sinorhizobium morelense
- ZmGFOR
- glucose–fructose oxidoreductase from Zymomonas mobilis
- LGK
- levoglucosan kinase
- PDB
- Protein Data Bank
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid
- r.m.s.d.
- root mean square deviation.
References
- 1. Lakshmanan C. M., Galor B., and Hoelscher H. E. (1969) Production of levoglucosan by pyrolysis of carbohydrates. Ind. Eng. Chem. Prod. Res. Dev. 8, 261–267 10.1021/i360031a010 [DOI] [Google Scholar]
- 2. Schkolnik G., and Rudich Y. (2006) Detection and quantification of levoglucosan in atmospheric aerosols: a review. Anal. Bioanal. Chem. 385, 26–33 10.1007/s00216-005-0168-5 [DOI] [PubMed] [Google Scholar]
- 3. Vicente A., Alves C., Calvo A. I., Fernandes A. R., Nunes T., Monteiro C., Almeida S. M., and Pio C. (2013) Emission factors and detailed chemical composition of smoke particles from the 2010 wildfire season. Atmos. Environ. 71, 295–303 10.1016/j.atmosenv.2013.01.062 [DOI] [Google Scholar]
- 4. You C., and Xu C. (2018) Review of levoglucosan in glacier snow and ice studies: recent progress and future perspectives. Sci. Total Environ. 616, 1533–1539 [DOI] [PubMed] [Google Scholar]
- 5. Vancampenhout K., De Vos B., Wouters K., Swennen R., Buurman P., and Deckers J. (2012) Organic matter of subsoil horizons under broadleaved forest: highly processed or labile and plant-derived? Soil Biol. Biochem. 50, 40–46 10.1016/j.soilbio.2012.03.005 [DOI] [Google Scholar]
- 6. Wallner P., Kundi M., Moshammer H., Scharf S., Schmutzer M., Weiss S., Hohenblum P., and Hutter H. P. (2013) Urinary levoglucosan levels in Austrian communities differing in agrarian quota. Int. J. Hyg. Environ. Heal. 216, 280–283 10.1016/j.ijheh.2012.05.001 [DOI] [PubMed] [Google Scholar]
- 7. Hamaguchi N., Hirai H., Aizawa K., and Takada M. (2015) Production of water-soluble indigestible polysaccharides using activated carbon. J. Appl. Glycosci. 62, 7–13 10.5458/jag.jag.JAG-2014_009 [DOI] [Google Scholar]
- 8. Islam Z. U., Zhisheng Y., Hassan el B, Dongdong C., and Hongxun Z. (2015) Microbial conversion of pyrolytic products to biofuels: a novel and sustainable approach toward second-generation biofuels. J. Ind. Microbiol. Biotechnol. 42, 1557–1579 10.1007/s10295-015-1687-5 [DOI] [PubMed] [Google Scholar]
- 9. Bacik J. P., and Jarboe L. R. (2016) Bioconversion of anhydrosugars: emerging concepts and strategies. IUBMB Life 68, 700–708 10.1002/iub.1533 [DOI] [PubMed] [Google Scholar]
- 10. Kitamura Y., Abe Y., and Yasui T. (1991) Metabolism of levoglucosan (1,6-anhydro-β-d-glucopyranose) in microorganisms. Agric. Biol. Chem. 55, 515–521 10.1080/00021369.1991.10870617 [DOI] [Google Scholar]
- 11. Kitamura Y., and Yasui T. (1991) Purification and some properties of levoglucosan (1,6-anhydro-β-d-glucopyranose) kinase from the yeast Sporobolomyces salmonicolor. Agric. Biol. Chem. 55, 523–529 10.1080/00021369.1991.10870618 [DOI] [Google Scholar]
- 12. Nakahara K., Kitamura Y., Yamagishi Y., Shoun H., and Yasui T. (1994) Levoglucosan dehydrogenase involved in the assimilation of levoglucosan in Arthrobacter sp. I-552. Biosci. Biotechnol. Biochem. 58, 2193–2196 10.1271/bbb.58.2193 [DOI] [PubMed] [Google Scholar]
- 13. Lian J., Garcia-Perez M., and Chen S. (2013) Fermentation of levoglucosan with oleaginous yeasts for lipid production. Bioresour. Technol. 133, 183–189 10.1016/j.biortech.2013.01.031 [DOI] [PubMed] [Google Scholar]
- 14. Layton D. S., Ajjarapu A., Choi D. W., and Jarboe L. R. (2011) Engineering ethanologenic Escherichia coli for levoglucosan utilization. Bioresour. Technol. 102, 8318–8322 10.1016/j.biortech.2011.06.011 [DOI] [PubMed] [Google Scholar]
- 15. Bacik J. P., Klesmith J. R., Whitehead T. A., Jarboe L. R., Unkefer C. J., Mark B. L., and Michalczyk R. (2015) Producing glucose 6-phosphate from cellulosic biomass: structural insights into levoglucosan bioconversion. J. Biol. Chem. 290, 26638–26648 10.1074/jbc.M115.674614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yasui T., Kitamura Y., Nakahara K., and Abe Y. (1991) Metabolism of levoglucosan (1,6-anhydro-β-d-glucopyranose) in bacteria. Agric. Biol. Chem. 55, 1927–1929 10.1080/00021369.1991.10870881 [DOI] [Google Scholar]
- 17. Holbrook J. J., Liljas A., Steindel S. J., and Rossmann M. G. (1975) in The Enzymes (Boyer P. D., ed), pp. 191–292, Academic Press, New York [Google Scholar]
- 18. Kitaoka M. (2017) Synthesis of 3-keto-levoglucosan using pyranose oxidase and its spontaneous decomposition via β-elimination. J. Appl. Glycosci. 64, 99–107 10.5458/jag.jag.JAG-2017_013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kallimanis A., Kavakiotis K., Perisynakis A., Spröer C., Pukall R., Drainas C., and Koukkou A. I. (2009) Arthrobacter phenanthrenivorans sp. nov., to accommodate the phenanthrene-degrading bacterium Arthrobacter sp. strain Sphe3. Int. J. Syst. Evol. Microbiol. 59, 275–279 10.1099/ijs.0.000984-0 [DOI] [PubMed] [Google Scholar]
- 20. Busse H. J. (2016) Review of the taxonomy of the genus Arthrobacter, emendation of the genus Arthrobacter sensu lato, proposal to reclassify selected species of the genus Arthrobacter in the novel genera Glutamicibacter gen. nov., Paeniglutamicibacter gen. nov., Pseudoglutamicibacter gen. nov., Paenarthrobacter gen. nov., and Pseudarthrobacter gen. nov., and emended description of Arthrobacter roseus. Int. J. Syst. Evol. Microbiol. 66, 9–37 10.1099/ijsem.0.000702 [DOI] [PubMed] [Google Scholar]
- 21. Kallimanis A., Labutti K. M., Lapidus A., Clum A., Lykidis A., Mavromatis K., Pagani I., Liolios K., Ivanova N., Goodwin L., Pitluck S., Chen A., Palaniappan K., Markowitz V., Bristow J., et al. (2011) Complete genome sequence of Arthrobacter phenanthrenivorans type strain (Sphe3). Stand. Genomic Sci. 4, 123–130 10.4056/sigs.1393494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nakatsu C. H., Barabote R., Thompson S., Bruce D., Detter C., Brettin T., Han C., Beasley F., Chen W., Konopka A., and Xie G. (2013) Complete genome sequence of Arthrobacter sp. strain FB24. Stand. Genomic Sci. 9, 106–116 10.4056/sigs.4438185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vikram S., Kumar S., Vaidya B., Pinnaka A. K., and Raghava G. P. (2013) Draft genome sequence of the 2-chloro-4-nitrophenol-degrading bacterium Arthrobacter sp. strain SJCon. Genome Announc. 1, e0005813 10.1128/genomeA.00058-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hakala M. T., Glaid A. J., and Schwert G. W. (1956) Lactic dehydrogenase. II. Variation of kinetic and equilibrium constants with temperature. J. Biol. Chem. 221, 191–209 [PubMed] [Google Scholar]
- 25. Clarke A. R., Atkinson T., and Holbrook J. J. (1989) From analysis to synthesis: new ligand binding sites on the lactate dehydrogenase framework. Part I. Trends Biochem. Sci. 14, 101–105 10.1016/0968-0004(89)90131-X [DOI] [PubMed] [Google Scholar]
- 26. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 27. Taberman H., Parkkinen T., and Rouvinen J. (2016) Structural and functional features of the NAD(P) dependent Gfo/Idh/MocA protein family oxidoreductases. Protein Sci. 25, 778–786 10.1002/pro.2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Holm L., and Laakso L. M. (2016) Dali server update. Nucleic Acids Res. 44, W351–W355 10.1093/nar/gkw357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shimizu T., Takaya N., and Nakamura A. (2012) An l-glucose catabolic pathway in Paracoccus species 43P. J. Biol. Chem. 287, 40448–40456 10.1074/jbc.M112.403055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fukano K., Ozawa K., Kokubu M., Shimizu T., Ito S., Sasaki Y., Nakamura A., and Yajima S. (2018) Structural basis of l-glucose oxidation by scyllo-inositol dehydrogenase: implications for a novel enzyme subfamily classification. PLoS One 13, e0198010 10.1371/journal.pone.0198010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakamura A. (2015) Paracoccus laeviglucosivorans sp. nov., an l-glucose-utilizing bacterium isolated from soil. Int. J. Syst. Evol. Microbiol. 65, 3878–3884 10.1099/ijsem.0.000508 [DOI] [PubMed] [Google Scholar]
- 32. Dambe T. R., Kühn A. M., Brossette T., Giffhorn F., and Scheidig A. J. (2006) Crystal structure of NADP(H)-dependent 1,5-anhydro-d-fructose reductase from Sinorhizobium morelense at 2.2 A resolution: construction of a NADH-accepting mutant and its application in rare sugar synthesis. Biochemistry 45, 10030–10042 10.1021/bi052589q [DOI] [PubMed] [Google Scholar]
- 33. Taberman H., Andberg M., Koivula A., Hakulinen N., Penttilä M., Rouvinen J., and Parkkinen T. (2015) Structure and function of Caulobacter crescentus aldose–aldose oxidoreductase. Biochem. J. 472, 297–307 10.1042/BJ20150681 [DOI] [PubMed] [Google Scholar]
- 34. Nurizzo D., Halbig D., Sprenger G. A., and Baker E. N. (2001) Crystal structures of the precursor form of glucose–fructose oxidoreductase from Zymomonas mobilis and its complexes with bound ligands. Biochemistry 40, 13857–13867 10.1021/bi011355d [DOI] [PubMed] [Google Scholar]
- 35. Schu M., Faust A., Stosik B., Kohring G. W., Giffhorn F., and Scheidig A. J. (2013) The structure of substrate-free 1,5-anhydro-d-fructose reductase from Sinorhizobium meliloti 1021 reveals an open enzyme conformation. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69, 844–849 10.1107/S1744309113019490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kingston R. L., Scopes R. K., and Baker E. N. (1996) The structure of glucose–fructose oxidoreductase from Zymomonas mobilis: an osmoprotective periplasmic enzyme containing non-dissociable NADP. Structure 4, 1413–1428 10.1016/S0969-2126(96)00149-9 [DOI] [PubMed] [Google Scholar]
- 37. Cremer D., and Pople J. A. (1975) A general definition of ring puckering coordinates. J. Am. Chem. Soc. 97, 1354–1358 10.1021/ja00839a011 [DOI] [Google Scholar]
- 38. Uriarte I., Écija P., Lozada-Garcia R., Çarçabal P., and Cocinero E. J. (2018) Investigating the conformation of the bridged monosaccharide levoglucosan. Chemphyschem 19, 766–773 10.1002/cphc.201701242 [DOI] [PubMed] [Google Scholar]
- 39. Smrcok L., Sládkovicová M., Langer V., Wilson C. C., and Koós M. (2006) On hydrogen bonding in 1,6-anhydro-β-d-glucopyranose (levoglucosan): X-ray and neutron diffraction and DFT study. Acta Crystallogr. B. 62, 912–918 10.1107/S010876810602489X [DOI] [PubMed] [Google Scholar]
- 40. Iwazaki S., Hirai H., Hamaguchi N., and Yoshida N. (2018) Isolation of levoglucosan-utilizing thermophilic bacteria. Sci. Rep. 8, 4066 10.1038/s41598-018-22496-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guilbault G. G., and Kramer D. N. (1965) Fluorometric procedure for measuring the activity of dehydrogenases. Anal. Chem. 37, 1219–1221 10.1021/ac60229a011 [DOI] [PubMed] [Google Scholar]
- 42. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 43. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 10.1107/S0907444909042589 [DOI] [PubMed] [Google Scholar]
- 44. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]