

Abstract
Recent large‐scale exome sequencing studies have identified mutations in several members of the CHD (Chromodomain Helicase DNA‐binding protein) gene family in neurodevelopmental disorders. Mutations in the CHD2 gene have been linked to developmental delay, intellectual disability, autism and seizures, CHD8 mutations to autism and intellectual disability, whereas haploinsufficiency of CHD7 is associated with executive dysfunction and intellectual disability. In addition to these neurodevelopmental features, a wide range of other developmental defects are associated with mutants of these genes, especially with regards to CHD7 haploinsufficiency, which is the primary cause of CHARGE syndrome. Whilst the developmental expression of CHD7 has been reported previously, limited information on the expression of CHD2 and CHD8 during development is available. Here, we compare the expression patterns of all three genes during mouse development directly. We find high, widespread expression of these genes at early stages of development that gradually becomes restricted during later developmental stages. Chd2 and Chd8 are widely expressed in the developing central nervous system (CNS) at all stages of development, with moderate expression remaining in the neocortex, hippocampus, olfactory bulb and cerebellum of the postnatal brain. Similarly, Chd7 expression is seen throughout the CNS during late embryogenesis and early postnatal development, with strong enrichment in the cerebellum, but displays low expression in the cortex and neurogenic niches in early life. In addition to expression in the brain, novel sites of Chd2 and Chd8 expression are reported. These findings suggest additional roles for these genes in organogenesis and predict that mutation of these genes may predispose individuals to a range of other, non‐neurological developmental defects.
Keywords: autism, CHD2, CHD7, CHD8, development, embryo, expression, mouse, organogenesis
Introduction
Chromatin remodelling factors have emerged as key regulators of gene expression and are often mutated in human disease (Hendrich & Bickmore, 2001; Ronan et al. 2013; Iwase & Martin, 2018). Mammalian chromatin remodelling factors can be subdivided into four families: SWI/SNF (mating type Switching/Sucrose Non‐Fermenting); ISWI (Imitation Switch); INO80 (Inositol requiring 80); and CHD (Chromodomain Helicase DNA‐binding protein; Ho & Crabtree, 2010).
The CHD gene family consists of nine genes (CHD1–CHD9). The encoded proteins utilise the energy from ATP hydrolysis to alter nucleosome positioning, thereby causing local changes in the structure of the chromatin (Marfella & Imbalzano, 2007). CHD1 and CHD2, which belong to the CHD1–2 subfamily, are characterised by the presence of tandem chromodomains and a Snf2 helicase domain – both motifs common to all CHD proteins – in addition to DNA‐binding domains at the C‐terminus (Marfella & Imbalzano, 2007; Liu et al. 2015). CHD3 and CHD4 are structurally similar, but each contains a PHD (Plant Homeo Domain) Zn‐finger‐like domain rather than a DNA‐binding region, forming the second subfamily (Marfella & Imbalzano, 2007). Alongside signature sequence motifs of the CHD family, members of the CHD5–9 subfamily contain a DNA‐binding region alongside various other C‐terminal sequences that alter their function (Marfella & Imbalzano, 2007). The present study focuses on the spatiotemporal pattern of expression of CHD2, CHD7 and CHD8.
The ATP‐dependent activity of CHD2 leads to assembly of chromatin into periodic nucleosome arrays by deposition of various histone proteins, thereby modifying the expression and structure of target sites (Liu et al. 2015; Luijsterburg et al. 2016). Functionally, CHD2 has been reported to maintain pluripotency of stem cells, influence cell fate during myogenesis and interneuron development, and facilitate DNA repair through interaction with histone variant H3.3 (Harada et al. 2012; Rajagopalan et al. 2012; Luijsterburg et al. 2016; Meganathan et al. 2017; Semba et al. 2017).
De novo loss‐of‐function mutations in CHD2 have been reported in patients with autism spectrum disorder (ASD) alongside developmental delay, intellectual disability, increased risk of epileptic seizures and additional behavioural problems (Epi et al. 2013; Chenier et al. 2014, 2017; O'Roak et al. 2014; Pinto et al. 2016; Lebrun et al. 2017). The association between CHD2 haploinsufficiency and epileptic encephalopathy, or Lennox–Gastaut or Dravet syndrome, is also well‐established, and variants of CHD2 are recognised risk factors for photosensitivity in epilepsy (Carvill et al. 2013; Suls et al. 2013; Lund et al. 2014; Galizia et al. 2015). CHD2 mutations are commonly identified in patients with chronic lymphocytic leukaemia, frequently in conjuncture with alterations in functional pathways associated with brain development (Rodriguez et al. 2015).
Homozygous Chd2 mutant mice die around birth due to unknown causes (Marfella et al. 2006). Heterozygous mice exhibit reduced growth and viability and range of phenotypic abnormalities, which include extramedullary haematopoiesis, susceptibility to lymphomas, cardiomyopathy, liver inflammation, glomerulopathy and various other renal defects (Marfella et al. 2006, 2008; Nagarajan et al. 2009; Rajagopalan et al. 2012). More recently, Chd2 knockdown has been demonstrated to decrease Pax6+ radial glial cell numbers, a cell type in which it is highly expressed, and to promote neuronal and intermediate progenitor production, implying an important balancing role for CHD2 in progenitor renewal and cortical development (Shen et al. 2015). At present, limited expression data for Chd2 are available. Quantitative analyses of Chd2 in the adult mouse demonstrate that Chd2 is widely expressed in a number of tissues, including the heart, brain, lungs, thymus, lymphoid tissue and skeletal muscle (Marfella et al. 2006; Nagarajan et al. 2009). Macroscopic analysis of whole embryos stained for Chd2 showed expression in the developing heart, forebrain, eye, dorsal facial region and limbs between E10.5 and E15.5 (Kulkarni et al. 2008). These data show that Chd2 expression is apparent in many tissues during development and in the adult mouse, although a true spatiotemporal pattern of expression is yet to be defined.
CHD7 is thought to maintain an open chromatin conformation at putative regulatory elements (Feng et al. 2017; Whittaker et al. 2017b). CHD7 facilitates neural stem cell (NSC) multipotency in the developing brain and quiescence in the adult as both differentiation potential and stem cell depletion rates are correlated with the levels of CHD7 (Jones et al. 2015; Fujita et al. 2016; Feng et al. 2017; Yamamoto et al. 2018). As well as maintaining multipotency, CHD7 has also been shown to directly control lineage identity in NSCs through coordination of transcription factors in the neural crest (Chai et al. 2018). In a similar vein, CHD7 is required for the formation of migratory neural crest cells and, accordingly, induced pluripotent stem cells derived from patients with CHD7 mutations exhibit defective delamination, migration and motility (Bajpai et al. 2010; Prasad et al. 2012; Okuno et al. 2017). Finally, CHD7 has been shown to have multiple roles in cerebellar development, consistent with the observation that individuals harbouring CHD7 mutations exhibit vermis hypoplasia (Yu et al. 2013; Donovan et al. 2017; Whittaker et al. 2017,b).
Haploinsufficiency of the CHD7 gene is the major cause of CHARGE syndrome (Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and/or development, Genitalia and/or urinary abnormalities, and Ear abnormalities and deafness), and mutations have also been reported in patients with Kallmann syndrome (Kim et al. 2008; Jongmans et al. 2009). Some of the CHD7 mutations in patients with CHARGE syndrome have been shown to result in defective nucleosome remodelling activity in vitro, directly linking chromatin remodelling defects with disease (Bouazoune & Kingston, 2012). Chd7 −/− embryos do not survive beyond E11, indicating early requirements for this gene during embryonic development, whereas heterozygotes exhibit features similar to those associated with CHARGE syndrome (Bosman et al. 2005; Hurd et al. 2007). Akin to Chd2, Chd7 expression during development is not limited to one tissue type. Chd7 has been shown to be expressed in the developing eye, inner ear, olfactory epithelium, dorsal root ganglia, lung, kidneys, gut and throughout the neural ectoderm, including the neural crest (Bosman et al. 2005; Aramaki et al. 2007; Hurd et al. 2007; Engelen et al. 2011; Fujita et al. 2014, 2016; Gage et al. 2015). More recently, preserved expression of Chd7 has been seen in the adult cerebellum (Whittaker et al. 2017a).
In vitro evidence has suggested a central role of CHD8 in transcription and transcriptional elongation (Yuan et al. 2007; Nishiyama et al. 2009; Rodriguez‐Paredes et al. 2009; Yates et al. 2010). CHD8S, a partial N‐terminal fragment of CHD8, also referred to as Duplin, acts as a regulator of β‐catenin‐mediated transcription – largely causing transcriptional repression (Sakamoto et al. 2000; Kobayashi et al. 2002; Nishiyama et al. 2004, 2012; Thompson et al. 2008, 2017).
Recurrent de novo mutations in CHD8 have been linked to ASD. A significant body of literature, including case reports and large exome sequencing studies, have identified CHD8 mutations in individuals with ASD (Zahir et al. 2007; Neale et al. 2012; O'Roak et al. 2012; Sanders et al. 2012; Talkowski et al. 2012; Bernier et al. 2014; Wilkinson et al. 2015; Merner et al. 2016; Stolerman et al. 2016; Wang et al. 2016). It is one of the highest confidence risk genes for autism identified to date. ASD is highly heterogeneous, but can be identified by a repertoire of behavioural features in patients: social impairment, communication impairment, repetitive behaviours, and sometimes accompanied by an array of other conditions such as epilepsy, dyslexia, dyspraxia and attention deficit hyperactivity disorder (Leyfer et al. 2006; Brieber et al. 2007; Canitano, 2007; Dziuk et al. 2007; Helbig et al. 2009; Taurines et al. 2012). The effects of CHD8 mutation may also manifest as characteristic physical features, including macrocephaly, facial dysmorphia and gastrointestinal disturbance, perhaps defining CHD8‐related ASD as a distinct subtype (Bernier et al. 2014).
CHD8 is recruited to promoters of highly expressed genes in NSCs, and reduced expression of CHD8 in mouse and human cells have been shown to precipitate dysregulation of ASD‐related genes and alter cortical neurogenesis (Sugathan et al. 2014; Cotney et al. 2015; Wang et al. 2015; Wilkinson et al. 2015; Durak et al. 2016). In Chd8 +/− mice behavioural changes have been documented alongside characteristic neurodevelopmental changes pertaining to altered neurogenesis and long‐range connectivity, brain overgrowth and craniofacial anomalies (Katayama et al. 2016; Gompers et al. 2017; Platt et al. 2017; Suetterlin et al. 2018). Chd8 −/− embryos die by E7.5 of development. The early embryonic lethality associated with CHD8 loss has been proposed to be caused by aberrant p53‐mediated apoptosis as a consequence of loss of CHD8‐mediated repression of p53 target genes (Nishiyama et al. 2004). As the mutants do not survive, the developmental roles after E7.5 are not known. The expression pattern of Chd8 has been described between E7.5 and E10.5 in the mouse using a CHD8s/Duplin antisense riboprobe (Nishiyama et al. 2004). Whole embryo analysis showed expression predominantly in the brain, face and limb buds. Microarray data have been used to quantify the level of Chd8 expression in developing mouse, macaque and human brains. A regional expression heatmap showed widespread expression, highest in the early pre‐natal period (Bernier et al. 2014). Platt et al. (2017) demonstrated a similar temporal pattern of quantitative expression in the mouse brain, and further showed that Chd8 is expressed in almost all neuronal populations. Despite these insights, no study to date has characterised the expression pattern of full‐length Chd8 in all tissues of the developing mouse from mid‐gestation and through early life. Given the strong association of CHD8 mutations with ASD and other physical abnormalities, determining a comprehensive spatiotemporal expression pattern of CHD8 during development is of great interest.
In the present study, we investigated the expression patterns of three CHD genes with strong evidence for important functions in brain development and neurodevelopmental disorders. The expression pattern of Chd8 was compared with Chd7 and Chd2. As these genes tend to be widely expressed during early development, we focused on later embryonic stages. We report novel expression sites for all three genes during development, with examples of overlapping, complementary and distinct expression patterns.
Materials and methods
Animals
Timed‐mated CD1 embryos and pups were produced in our in‐house facility. Noon on the day a vaginal plug was detected was designated as embryonic day 0.5 (E0.5). The day of birth was designated as postnatal day (P)0. All experimental procedures were approved by the institutional Local Ethical Review Panel and the UK Home Office.
Primer design and probe synthesis
Primers were designed to amplify a 455‐bp fragment of exon 37 of Chd8 from mouse genomic DNA: forward 5’‐TCTCTGCCTTTTATGCCGTTTG‐3’; reverse 5’‐CACCTCCTGAAGTCTTGGGTTTC‐3’ with T7 recognition sequence added to the reverse primers in a polymerase chain reaction reaction. The resulting DNA template was used for the synthesis of digoxigenin‐labelled antisense or sense mRNA probes. A Chd7 probe template was made with primer pairs that amplify a 222‐bp fragment of Chd7 exon 3 from mouse genomic DNA: forward 5’‐TTGGTAAAGATGACTTCCCTGGTG‐3’; reverse 5’‐GTTTTGGCGTGACAGTTTTTGC‐3’. A Chd2 625‐bp probe template was amplified from mouse brain cDNA using primer pairs: forward 5’‐AGAAGAGCGTCCTCACAAAGACTG‐3’; reverse 5’‐ TTTTTCCTCAGGGTCCACAGG‐3’.
Sample preparation
Embryos and brains were dissected in ice‐cold diethylpyrocarbonate‐treated phosphate‐buffered saline (DEPC PBS) and fixed in 4% paraformaldehyde (PFA) overnight. After several washes in DEPC PBS, embryos or brains were placed in cassettes immersed in 70% ethanol. The samples were processed in a Leica ASP300 tissue processor following a standard protocol. The processed samples were embedded in wax, sectioned sagittally at 10 μm using a Leica RM2145 microtome, placed on Superfrost Plus slides and left to dry at 42 °C for 48 h.
In situ hybridisation
E12.5, E14.5, P0, P7 and P20 sagittal sections on slides were deparaffinised in xylene and rehydrated in decreasing series of ethanol concentrations. This was followed by DEPC PBS washes. Proteinase K (50 μg mL−1 in DEPC PBS) was added and sections were incubated for 10 min at 37 °C.
The slides were then washed in DEPC PBS, refixed in 4% PFA for 10 min and washed again in DEPC PBS. Sections were acetylated (0.25% acetic anhydride, 0.1 m triethanolamine, DEPC water at pH 7.5) for 10 min, after which they were again washed in DEPC PBS three times. Sections were dehydrated in 70% ethanol (5 min) and 95% ethanol (a few seconds), and left to air‐dry for a few minutes. Probe‐hybridisation mix [300 μL; 2 μL of probe per mL hybridisation solution; 50% dextran sulphate, 50% formamide, 1% Denhardt's solution, 0.3 m NaCl (sodium chloride), 20 mm Tris‐HCl (pH 8), 10 mm NaPO4 (sodium phosphate), 5 mm EDTA (ethylenediaminetetraacetic acid), 250 μg mL−1 yeast tRNA, 1% sarcosyl, sterile water] pre‐heated to 80 °C was added to each slide and covered with parafilm. The slides were then arranged in a humid chamber (50% formamide/water) and incubated overnight at 65 °C.
The following day the slides were washed in high‐stringency [HIS; formamide, 0.1% SSC (saline‐sodium citrate), sterile distilled water] wash for 30 min at 65 °C followed by RNase buffer (0.5 m NaCl, 10 mm Tris‐HCl pH 7.5, 5 mm EDTA, distilled water) at 37 °C for 10 min (3 ×). Slides were treated with RNase buffer with 20 μg mL−1 RNase A at 37 °C for 30 min followed by a single wash in RNase buffer at 37 °C for 15 min. The slides were again washed twice in HIS at 65 °C for 20 min each. Then, 2 × SSC and 0.1 × SSC washes for 15 min were performed twice followed by PBT (PBS, 0.1% Tween 20) washes at room temperature. Sections were blocked with 10% heat‐inactivated goat serum in PBT for 1 h at room temperature, before a 3‐h incubation in alkaline phosphatase coupled with anti‐dioxygenin antibody (1 : 500 dilution; Roche) and 1% heat‐inactivated goat serum in PBT. At the end of incubation, slides were washed four times with PBT for 15 min each at room temperature followed by freshly prepared NTMT buffer (100 mM NaCl, 100 mM Tris‐HCl at pH 9.5, 50 mM MgCl2, 0.1% Tween‐20, sterile distilled water and 0.5 mg mL−1 levamisole) twice at room temperature. Finally, the slides were incubated in darkness in BM purple (Roche) and 0.5 mg mL−1 levamisole at room temperature overnight.
When signal appeared on sections, the reaction was stopped by washing in PBS at room temperature for 5 min. Slides were dehydrated with an increasing series of ethanol washes followed by xylene before being mounted with di‐N‐butyl phthalate in xylene (DPX) and left to air‐dry.
Results
Chd2, Chd7 and Chd8 gene expression in mouse embryos at E12.5 and E14.5
At E12.5, Chd7 and Chd8 expression was apparent throughout the neuroepithelium of the developing central nervous system (CNS; Fig. 1A,B). Chd8 transcript signals were observed throughout the ventricular and subventricular regions of the neocortex and in the hindbrain (Fig. 1Aa,Ab), including the cerebellum where expression was evident in the ventricular zone (VZ), rhombic lip (RL) and the isthmus (Fig. 1Aa). Both the VZ and RL are germinal centres where progenitor cells are born that later migrate and populate the cerebellum (White & Sillitoe, 2013). Notably, Chd8 expression could also be observed at the lower RL and floor plate region of the hindbrain, extending to the spinal cord and dorsal root ganglia (Figs 1A, S1A). Chd8 expression can be observed throughout the neural tube with no evident mediolateral or dorsoventral gradient (Fig. S1A–D). Other regions of interest showing high Chd8 expression included the diencephalon and areas adjacent to the hypothalamus and pituitary gland (Fig. 1A). Chd8 expression was observed throughout the craniofacial region, including the tongue and olfactory epithelium (Fig. 1A). Elsewhere, other organs of the embryo also showed substantial Chd8 expression, with signals present in the intersomitic regions, lungs, gut, genital tubercle and tail (Fig. 1A).
Figure 1.
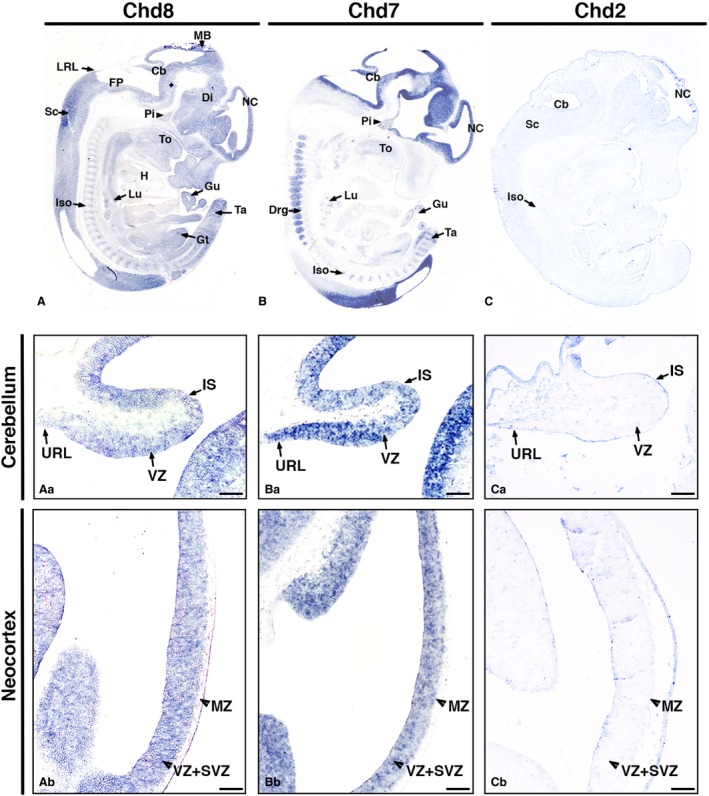
Distinct Chd8, Chd7 and Chd2 expression patterns at E12.5. In situ hybridisation on sagittal sections of mouse embryos at developmental stage E12.5 using antisense riboprobes to detect Chd8, Chd7 and Chd2 mRNA, anterior to the right (A–C). Gene expression is indicated by purple/blue staining. Note the widespread Chd8 expression in most embryonic tissues (A), the high, localised expression of Chd7, specifically in the developing nervous system (B), and very low, widespread expression of Chd2 (C). High‐magnification images of the developing cerebellum (Aa–Ca) demonstrate the presence of Chd8 (Aa) and Chd7 (Ba) transcripts throughout the neuroepithelium, with little Chd2 expression evident (Ca). High‐magnification images through the neocortex show widespread Chd8 expression (Ab), note that Chd7 expression tends to be higher on the ventricular side (Bb) and that there is little discernible Chd2 expression (Cb). Other regions with relatively strong signals were the nasal epithelium, tail, genital tubercle, intersomitic mesoderm, spinal cord, mid brain, diencephalon, tongue and pituitary. Scale bars: 100 μm. Cb, cerebellum; Di, diencephalon; Drg, dorsal root ganglia; FP, floor plate; Gt, genital tubercle; Gu, gut; H, heart; Is, isthmus; Iso, intersomitic mesoderm; Lu, lungs; LRL, lower rhombic lip; MB, midbrain; MZ, molecular zone; NC, neocortex; Pi, pituitary; Sc, spinal cord; SVZ, subventricular zone; Ta, tail; To, tongue; URL, upper rhombic lip; VZ, ventricular zone.
As with Chd8, Chd7 mRNA transcripts were observed throughout the embryo (Fig. 1B). Expression was found in the ventricular region of the developing brain and spinal cord. Chd7 mRNA transcripts were present in both the ventricular and subventricular zones of the neocortex (Fig. 1Bb). In the hindbrain, Chd7 was expressed in all regions, including the upper RL of the cerebellum, the lower RL and floor plate (Fig. 1Ba). Chd7 expression was also observed in the diencephalon and the pituitary (Fig. 1B). Within the neural tube, expression was present in both cranial and caudal poles (Fig. S1E,F). Additionally, in transverse sections, Chd7 was noted to be enriched in the VZ and displayed a ventral to dorsal gradient within the developing spinal cord (Fig. S1G,H). Extensive expression was also observed outside the CNS. In the head region, diffuse Chd7 expression was present in the tongue. Other organs with expression included the dorsal root ganglia, intersomitic regions, gut, lungs and the tail (Fig. 1B).
At E12.5, low Chd2 mRNA transcript signals were observed in many tissues in the developing mouse (Fig. 1C), differentiated from background by use of a sense control (Fig. S2). Diffuse Chd2 expression was observed within the brain (Fig. 1Ca,Cb), intersomitic regions and the spinal cord. Despite this increased signal in brain tissue, the level of expression was low, suggesting that, at this stage, Chd2 is expressed ubiquitously at low levels throughout the embryo.
As several tissues outside of the CNS expressed both Chd7 and Chd8 strongly, these were compared directly at higher power. Sites of expression included the cochlea, lungs, eyes and kidneys (Fig. 2A–D,F–I). For both, distinct expression levels were observed at the vestibulocochlear ganglion and cochlear epithelium in the ear (Fig. 2A,F). In the kidney, expression levels were high in the mesenchyme and metanephric tubule epithelium (Fig. 2B,G), whereas in the lung, expression was observed in the pulmonary epithelium (Fig. 2C,H). Both transcripts were also observed in the neural retina/optic cup and retinal pigmented epithelium of the eyes, with Chd8 transcripts present widely throughout the surrounding mesenchyme and craniofacial tissues (Fig. 2D,I). Interestingly, Chd8 also showed high expression in the incisor primordium, where no Chd7 expression was seen (Fig. 2E).
Figure 2.
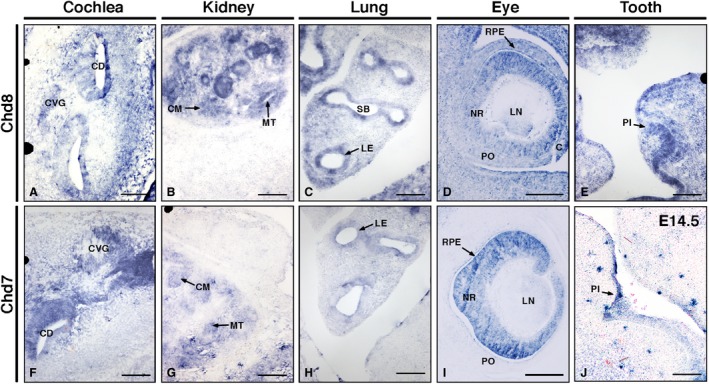
Chd7 and Chd8 are expressed in multiple organs at E12.5. In situ hybridisation images of Chd8 (A–E) and Chd7 (F–I) transcripts around the cochlea of the inner ear (A,F), kidney (B,G), lung (C,H), eye (D,I) and tooth (E,J). Scale bars: 100 μm. C, cornea; CD, cochlear duct, CVG, cochlea‐vestibular ganglia; CM, condensing mesenchyme; LE, lung epithelium; LN, lens; MT, metanephric tubule; NR, neuroretina; PI, primordium of incisor; PO, pre‐optic cup; RPE, retinal pigmented epithelium; SB, segmental bronchus.
In E14.5 embryos, several sites of prominent Chd8 expression could be seen (Fig. 3A). In the head, abundant Chd8 transcripts were observed in the forebrain, midbrain, RL and VZ of the cerebellum (Fig. 3Aa). In the neocortex, significant expression was revealed in the ventricular, subventricular and mantle zones (Fig. 3Ab). Prominent expression was also seen in the basal forebrain, including the ganglionic eminences, suggesting a role for Chd8 in the generation of γ‐aminobutyric acid (GABA)‐ergic interneurons (Fig. 3A). Diffuse or low Chd8 expression was observed in the diencephalon and midbrain region. Extending from the hindbrain region, the spinal cord also showed low expression. Elsewhere in the head, expression was seen in the olfactory epithelium, the tongue and the ventral incisor. Other organs continued to show Chd8 expression as at E12.5, including the lungs, gut and kidneys. In addition, at E14.5, Chd8 transcripts were detected within the heart, thyroid, thymus, liver, gastric epithelium, trigeminal ganglion and digits of the hind limb (Fig. 3A).
Figure 3.
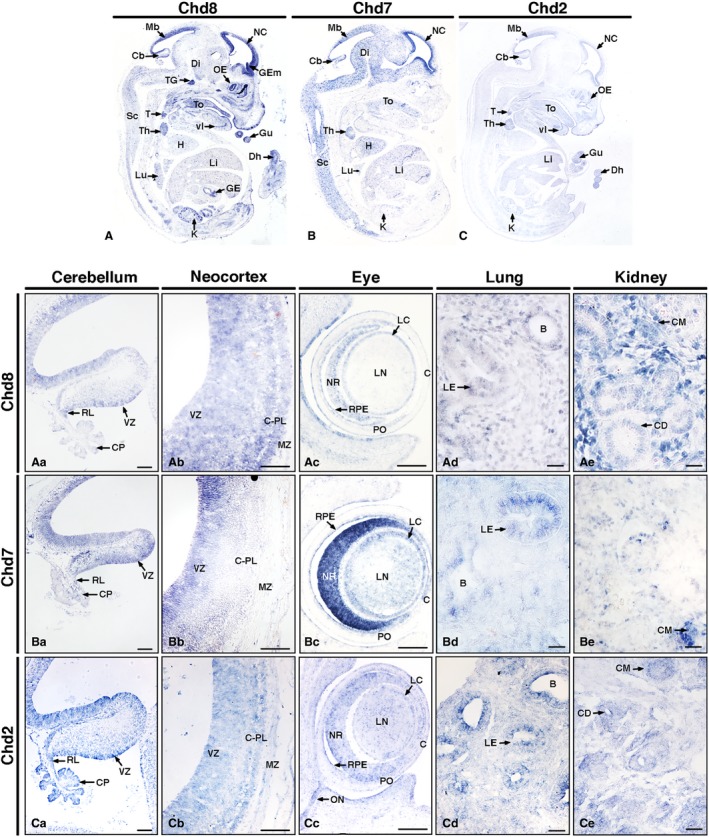
Distinct nervous system and organ‐specific expression patterns of Chd8, Chd7 and Chd2 in E14.5 mouse embryos. In situ hybridisation on sagittal sections of E14.5 mouse embryos (A–C), anterior to the right. Note distinct Chd8, Chd7 and Chd2 expression patterns throughout the embryos with notably higher levels in the developing nervous system. Beyond the nervous system, other notable regions of expression included various organs and glands, for example the thymus and thyroid, heart and kidneys. High‐magnification images (Aa–Ce) revealed specific expression patterns in the cerebellum (Aa–Ca), neocortex (Ab–Cb), eye (Ac–Cc), lung (Ad–Cd) and kidney (Ae–Ce). Scale bars: 100 μm. B, bronchus; C, cornea; Cb, cerebellum; CD, collecting duct; CM, condensing mesenchyme; CP, choroid plexus; C‐PL, cortical plate; Di, diencephalon; Dh, digit of hindlimb; GE, gastric epithelium; GEm, ganglionic eminence; Gu, gut; H, heart; K, kidney; LC, lens capsule; LE, lung epithelium; Li, liver; LN, lens; Lu, lung; Mb, mid brain roof plate; MZ, marginal zone; NC, neocortex; NR, neural retina; OE, olfactory epithelium; ON, optic nerve and surrounding structures; PO, pre‐optic cup; RL, rhombic lip; RPE, retinal pigmented epithelium; Sc, spinal cord; T, thyroid; To, tongue; Ta, tail; Th, thymus; TG, trigeminal ganglion; vI, ventral incisor; VZ, ventricular zone.
Comparable to its expression at E12.5, high levels of Chd7 mRNA transcripts were present most prominently in the ventricular region of the neocortex (Fig. 3Bb) in accordance with previous expression analyses (Engelen et al. 2011). Signals were also observed in the midbrain region extending to the hindbrain (Fig. 3B). Within the cerebellum, significant Chd7 signals were observed at the RL and VZ of the fourth ventricle (Fig. 3Ba). Widespread Chd7 expression was also present in the diencephalon (Fig. 3B). Extending from the hindbrain, the spinal cord showed widespread Chd7 signal. In the oral region, the tongue and incisor primordium showed Chd7 expression. Note that its expression in the tooth appears to occur later in development, at E14.5, than its family member Chd8 (Fig. 2J). Other organs such as the lungs, thymus, heart, kidneys and liver, which showed significant Chd8 expression, also displayed Chd7 expression (Fig. 3B).
Chd2 expression at E14.5 was still low and widespread, but was markedly elevated in certain regions compared with E12.5 (Fig. 3C). Prominent signals were detected in the neocortex (Fig. 3Cb), RL and the VZ of the cerebellum (Fig. 3Ca). In the craniofacial region, the tongue, incisor primordium and olfactory epithelium all stained for Chd2. Specific expression signals outside of the head were noted in the kidney, liver, thymus, lung, thyroid, gut, digits of the hindlimb and myogenic tissue (Fig. 3C).
Much like at E12.5, both Chd8 and Chd7 transcripts could be detected in the kidneys, lungs and eyes (Fig. 3Ac–Ae,Bc–Be). Additionally, Chd2 expression could also be detected in these tissues at this stage (Fig. 3Cc–Ce). mRNA transcripts of all three genes were detected in the condensing mesenchyme of the kidney (Fig. 3Ae,Be,Ce), epithelium of the lung (Fig. 3Ad,Bd,Cd) and neural retina, optic cup and lens of the eyes, with particularly strong expression of Chd7 seen in the retina (Fig. 3Ac,Bc,Cc). Notably, a Chd2 signal was also detected in the anatomical space containing the optic nerve and its surrounding structures (Fig. 3Cc).
Distinct Chd2, Chd7 and Chd8 expression patterns in the postnatal mouse brain
In order to define the domains of Chd8 expression in the postnatal brain, in situ hybridisation on brain sections at P0 were carried out. At this stage, widespread expression of Chd8 was observed (Fig. 4A,A’). Closer examination revealed expression throughout the cerebellum (Fig. 4Aa), and a slight enrichment of Chd8 expression towards the outer neocortex (Fig. 4Ab). Other Chd8‐expressing regions of interest include the hippocampus, hypothalamus and olfactory bulb (Fig. 4A,A’).
Figure 4.
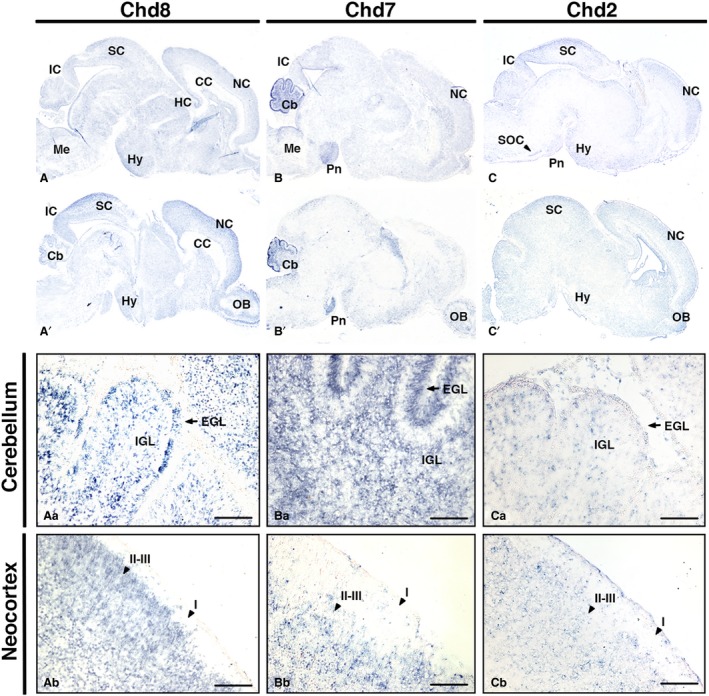
Comparative Chd8, Chd7 and Chd2 expression patterns in the newborn mouse brain. Sagittal sections through newborn mouse brain (anterior to the right), hybridised with antisense RNA probes to detect Chd8 (A,A’), Chd7 (B,B’) and Chd2 (C,C’) transcripts in blue. Representative medial (top) and lateral (bottom) sections are shown for each. Note widespread expression of Chd8, highly localised Chd7 expression in the cerebellum and pons, and Chd2 in the neocortex, midbrain and cerebellum. High‐magnification views of the cerebellum (Aa–Ca) and neocortex (Ab–Cb) are shown. Scale bars: 100 μm. Cb, cerebellum; EGL, external granule cell layer; HC, hippocampus; Hy, hypothalamus; I, cortical layer I; II–III, cortical layers II–III; IC, inferior colliculus; IGL, internal granule cell layer; Me, medulla; NC, neocortex; OB, olfactory bulb; Pn, pons; SC, superior colliculus; SOC, superior olivary complex.
At this stage, a comparable widespread pattern of expression was seen for Chd7 (Fig. 4B,B’,C,C’), with Chd7 exhibiting particularly strong expression in the cerebellum and pons (Fig. 4Ba,Ca). Chd7 was highly expressed within the cerebellum in contrast to Chd2 and Chd8 for which moderately strong and more diffuse expression was seen (Fig. 4Aa–Ca). Chd2 transcripts were enriched in the outer neocortex, hypothalamic area, superior olivary complex and basal pons (Fig. 4C).
The expression patterns of these genes in the P7 brain were similarly widespread with continued expression in the cerebellum, neocortex and hippocampus (Fig. 5A–C,A’–C’). Interestingly, all three genes appear to be expressed within the rostral migratory stream (RMS), suggesting a role for the CHD family in coordinating the formation of the infant olfactory system. High‐power images demonstrated prominent expression of all three genes in the cerebellum (Fig. 5Aa–Ca), the dentate gyrus (DG) and cornu ammonis 1–3 (CA1–3) of the hippocampus (Fig. 5Ab–Cb) and neocortex, enriched in layers II–III of the neocortex (Fig. 5Ac‐Cc). This cortical distribution is particularly marked for both Chd7 and Chd8, where a distinct band of high signal density can be appreciated. Much like in the P0 brain, Chd7 was most strongly expressed in the cerebellum.
Figure 5.
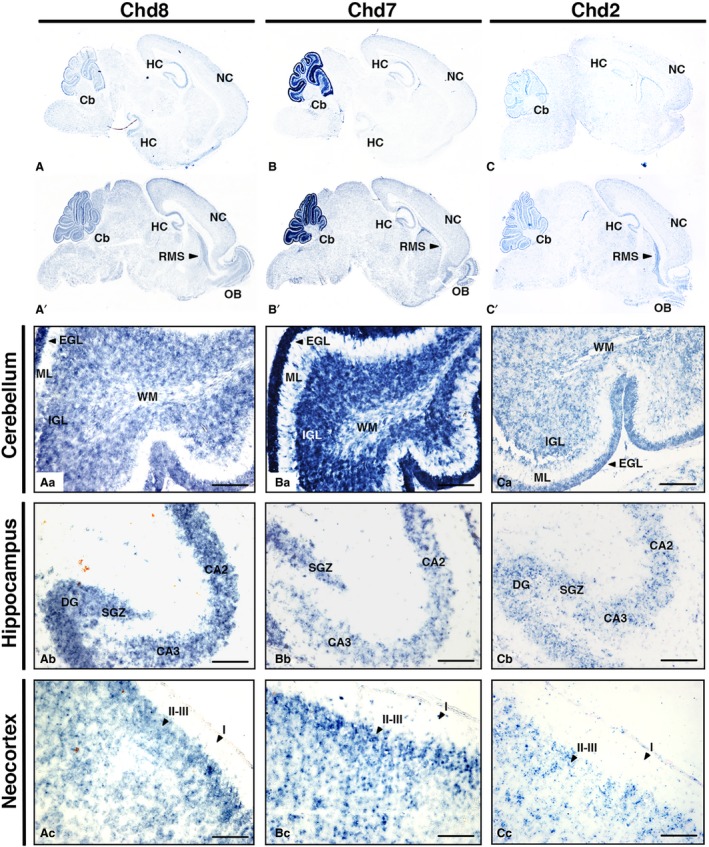
Chd8, Chd7 and Chd2 are expressed in the early postnatal cerebellum, hippocampus, neocortex and RMS. Sagittal sections through postnatal day 7 (P7) mouse brain (anterior to the right), hybridised with antisense RNA probes to detect Chd8 (A,A’), Chd7 (B,B’) and Chd2 (C,C’) transcripts, visualised in blue. Representative lateral (top) and medial (bottom) sections are shown for each. Higher magnification images to visualise specific expression domains in the cerebellum (Aa–Ac), hippocampus (Ba–Bc) and neocortex (Ca–Cc) are shown. Scale bars: 100 μm. CA1–3, cornu ammonis 1–3; Cb, cerebellum; DG, dentate gyrus; EGL, external granule cell layer; GL, glomerular layer; HC, hippocampus; I, cortical layer I; II–III, cortical layers II–III; IGL, internal granule cell layer; IPL, internal plexiform layer; ML, molecular layer; NC, neocortex; OB, olfactory bulb; RMS, rostral migratory stream; SGZ, subgranular zone; WM, white matter.
At P20, Chd8 and Chd2 expression was prominent in the cerebellum, neocortex, hippocampus, RMS and olfactory bulb (Fig. 6A,A’,C,C’). Chd7 was most prominent in the cerebellum, with low expression in the hippocampus, RMS and olfactory bulb (Fig. 6B,B’,Bc). All three genes were expressed in the maturing granule cell layer of the cerebellum (Fig. 6Aa–Ca), and the DG and CA1–3 of the hippocampus (Fig. 6Ab–Cb). Chd2 and Chd7 expression in the hippocampus was much lower and more diffuse compared with the prominent expression of Chd8 (Fig. 6Ab–Cb). Clear expression of Chd2 and Chd8 was noted in the neocortex, whilst Chd7 expression was very low in comparison (Fig. 6Ac–Cc).
Figure 6.

Comparison of Chd8, Chd7 and Chd2 expression patterns in the P20 mouse brain. Sagittal sections through P20 mouse brain (anterior to the right), hybridised with antisense RNA probes to detect Chd8 (A,A’), Chd7 (B,B’) and Chd2 (C,C’) transcripts, visualised in blue. Representative lateral (top) and medial (bottom) sections are shown for each. Note high expression of all three genes in the cerebellum, with widespread Chd8 and Chd2 expression remaining in the neocortex. High‐magnification images of the cerebellum (Aa–Ca), hippocampus (Ab–Cb) and neocortex (Ac–Cc) are shown. Scale bars: 100 μm. CA1–3, cornu ammonis 1–3; Cb, cerebellum; DG, dentate gyrus; GCL, granule cell layer; HC, hippocampus; I, cortical layer I; II–III, cortical layers II–III; ML, molecular layer; NC, neocortex; OB, olfactory bulb; RMS, rostral migratory stream; SGZ, subgranular zone; WM, white matter.
Discussion
The results of the current study demonstrate that all three genes are widely expressed, and show little evidence of restricted temporal and spatial expression patterns during embryonic development. Although expression seemingly occurs in many different tissues in utero, it can be noted that neurological tissue in particular expresses these members of the CHD family at a high level; an observation that is not wholly unsurprising considering the phenotypic manifestations of mutations of these genes.
CHD gene expression in the embryo
Chd8 is widely expressed in embryonic stages E12.5 and E14.5, consistent with a continued role for CHD8 during early stages of development, after E7.5 when Chd8 −/− mouse embryos were demonstrated to die due to apoptosis (Nishiyama et al. 2004). Recent work also implicated a role for CHD8 in suppressing p53 and the transactivation of genes under p53 control by preventing the process of apoptosis (Nishiyama et al. 2009). This could explain the early embryonic lethality observed. Moreover, the suggested role of CHD8 in transcription and elongation together with its role in controlling the expression of CCNE2 and TYMS, which are involved in the G1/S phase of the cell cycle, reinforce its possible role in normal gene regulation and cell proliferation, respectively (Rodriguez‐Paredes et al. 2009), hence normal development.
Similar to Chd8, the widespread Chd2 and Chd7 expression suggests they also have important roles in early developmental processes and organogenesis. These data are consistent with the evidence that neither Chd2 nor Chd7 homozygotes thrive past early development (Bosman et al. 2005; Marfella et al. 2006; Hurd et al. 2007). The CHD7 gene is the dominant cause of CHARGE syndrome, which is characterised by defects in the eye, brain, ear, heart and genitalia; areas in which we observed high levels of Chd7 expression (Vissers et al. 2004; Janssen et al. 2012). There are also reports of scoliosis caused by CHD7 mutations (Gao et al. 2007), which might relate to the expression we observed in the inter‐somitic mesoderm. FAM124B was reported to be a component of a CHD7‐ and CHD8‐containing complex (Batsukh et al. 2010; Batsukh et al. 2012), suggesting that this multi‐protein complex could be functional in cells where Chd7 and Chd8 are co‐expressed. Whereas CHD7 mutations are clearly linked to multi‐organ defects in the context of CHARGE syndrome (Vissers et al. 2004; Gao et al. 2007; Van de Laar et al. 2007; Janssen et al. 2012; Patten et al. 2012), a clear role for CHD8 in organogenesis has not been reported. Here, however, we show that Chd8 is expressed in many developing organs, including the lumen of stomach and midgut, an observation that may explicate the gastrointestinal complications associated with CHD8 mutations in patients with ASD (Bernier et al. 2014).
In the case of Chd2, heterozygous mice most notably display an array of gross kidney abnormalities, which might pertain to the high levels of expression of this gene we observed in the developing kidney (Marfella et al. 2008). Despite this association, the absence of reported renal dysgenesis in humans harbouring CHD2 mutations might indicate divergent functions for this gene in the human kidney, or a degree of functional redundancy with other CHD genes. Notably, at E14.5 the Chd2 expression pattern was markedly similar to Chd8 and, indeed, several regions of the embryo at this stage expressed these two genes in exclusion of Chd7, suggesting the possibility that they may serve similar functions. Some such regions include the dorsal hindlimb, thyroid, gut and olfactory epithelium.
CHD genes in brain development
CHD2 and CHD8 mutations share a well‐established link with ASD, a disorder that is widely regarded to be caused by aberrant neurodevelopment (Neale et al. 2012; O'Roak et al. 2012; Sanders et al. 2012; Lebrun et al. 2017). Our study demonstrates high levels of both Chd2 and Chd8 expression in the developing brain, especially during embryonic development. Additionally, preserved expression of both was revealed in key areas of the perinatal (P0) and postnatal brain (P7 and P20), including the cerebellum, hippocampus and neocortex – regions of the brain that are implicated in ASD (Riedel & Micheau, 2001; Allen et al. 2005; de Anda et al. 2012; Donovan & Basson, 2017). Within the neocortex, expression of Chd2 and Chd8 appears to be particularly prominent within the outer layers, distinctly layers II–III of the postnatal brain, areas in which high numbers of other ASD risk genes are also enriched (Parikshak et al. 2013).
Taken together, and bolstered by evidence of aberrant neurodevelopmental phenotypes associated with mutants of these genes (Epi et al. 2013; Chenier et al. 2014; O'Roak et al. 20142017; Katayama et al. 2016; Pinto et al. 2016; Gompers et al. 2017; Lebrun et al. 2017, 2014, 2016; Platt et al. 2017; Suetterlin et al. 2018), our data suggest that Chd2 and Chd8 are expressed in a spatiotemporally appropriate way such that impairment in their expression might precipitate some of the neurological changes seen in patients with ASD. With both genes expressed in the subgranular zone of the hippocampus, our data further support the notion that CHD2 and CHD8 might regulate neurogenesis (Shen et al. 2015; Durak et al. 2016), akin to the reported role of their counterpart CHD7 (Feng et al. 2013; Jones et al. 2015).
In addition to its role in adult neurogenesis, the diverse, temporally distinct functions of CHD7 during cerebellar development (Yu et al. 2013; Donovan et al. 2017; Whittaker et al. 2017,b) are consistent with its pronounced expression in the postnatal cerebellum. In view of the role of CHD7 in neurodevelopment, our study supports the notion that its mutation might account for the cerebellar hypoplasia associated with CHARGE syndrome. Furthermore, its expression in the olfactory bulb and RMS throughout postnatal development further bolsters the link between Kallmann syndrome, characterised by anosmia and hypogonadism, and CHD7 mutation (Jongmans et al. 2009).
Finally, the reported expression of Chd2 in the postnatal hippocampus invites a potential link between the dysfunction and deficiency of hippocampal interneurons documented in epileptic encephalopathies, temporal lobe epilepsy and seizures associated with ASD and its proposed role in interneuron development (Lado et al. 2013; Liu et al. 2014; Frye et al. 2016; Meganathan et al. 2017). Furthermore, Chd2 expression in the eye and related structures during early development might also pertain to the association of CHD2 mutation with photosensitivity in epilepsy (Carvill et al. 2013; Suls et al. 2013; Lund et al. 2014; Galizia et al. 2015).
In conclusion, in addition to their established roles in early brain development, our expression analyses also implicate Chd2 and Chd8, alongside Chd7, in organogenesis. Our data also implicate all three genes in the process of postnatal neurogenesis due to their expression in the neurogenic niches of the adult brain. Additional studies will be necessary to further define the function of these genes in these developmental processes. The gene expression data reported here will provide invaluable information and reference points to guide these future studies.
Supporting information
Fig. S1. Distinct Chd8 and Chd7 expression patterns within the neural tube at E12.5. In situ hybridisation on sagittal (anterior to the right) and transverse sections of mouse embryos at developmental stage E12.5 using antisense riboprobes to detect Chd8 and Chd7 mRNA (A–H). Gene expression is indicated by purple/blue staining. Note that both Chd8 and Chd7 are expressed throughout the length of the neural tube (A,B,E,F). Whilst Chd8 displays no mediolateral or dorsoventral gradient in transverse sections, Chd7 shows distinct enrichment in the ventricular zone of the developing CNS and a ventral to dorsal gradient within the spinal cord. Drg, dorsal root ganglia; Iso, intersomitic region; LGE, lateral ganglionic eminence; MGE, medial ganglionic eminence; No, notochord; NT, neural tube; SE, surface ectoderm.
Fig. S2. Sense control sections at E12.5. In situ hybridisation on sagittal (anterior to the right) sections of mouse embryos at developmental stage E12.5 using sense riboprobes to Chd8, Chd7 and Chd2 mRNA. Note that for all three genes there is little to no hybridisation or staining using the sense riboprobe in contrast to what is seen when using the anti‐sense probe.
Acknowledgement
This work was supported by an Anatomical Society Undergraduate Summer Vacation Scholarship to CO.
References
- Allen G, McColl R, Barnard H, et al. (2005) Magnetic resonance imaging of cerebellar‐prefrontal and cerebellar‐parietal functional connectivity. NeuroImage 28, 39–48. [DOI] [PubMed] [Google Scholar]
- de Anda FC, Rosario AL, Durak O, et al. (2012) Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat Neurosci 15, 1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramaki M, Kimura T, Udaka T, et al. (2007) Embryonic expression profile of chicken CHD7, the ortholog of the causative gene for CHARGE syndrome. Birth Defects Res A Clin Mol Teratol 79, 50–57. [DOI] [PubMed] [Google Scholar]
- Bajpai R, Chen DA, Rada‐Iglesias A, et al. (2010) CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463, 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batsukh T, Pieper L, Koszucka AM, et al. (2010) CHD8 interacts with CHD7, a protein which is mutated in CHARGE syndrome. Hum Mol Genet 19, 2858–2866. [DOI] [PubMed] [Google Scholar]
- Batsukh T, Schulz Y, Wolf S, et al. (2012) Identification and characterization of FAM124B as a novel component of a CHD7 and CHD8 containing complex. PLoS ONE 7, e52640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier R, Golzio C, Xiong B, et al. (2014) Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosman EA, Penn AC, Ambrose JC, et al. (2005) Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum Mol Genet 14, 3463–3476. [DOI] [PubMed] [Google Scholar]
- Bouazoune K, Kingston RE (2012) Chromatin remodeling by the CHD7 protein is impaired by mutations that cause human developmental disorders. Proc Natl Acad Sci USA 109, 19 238–19 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieber S, Neufang S, Bruning N, et al. (2007) Structural brain abnormalities in adolescents with autism spectrum disorder and patients with attention deficit/hyperactivity disorder. J Child Psychol Psychiatry 48, 1251–1258. [DOI] [PubMed] [Google Scholar]
- Canitano R (2007) Epilepsy in autism spectrum disorders. Eur Child Adolesc Psychiatry 16, 61–66. [DOI] [PubMed] [Google Scholar]
- Carvill GL, Heavin SB, Yendle SC, et al. (2013) Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 45, 825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai M, Sanosaka T, Okuno H, et al. (2018) Chromatin remodeler CHD7 regulates the stem cell identity of human neural progenitors. Genes Dev 32, 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenier S, Yoon G, Argiropoulos B, et al. (2014) CHD2 haploinsufficiency is associated with developmental delay, intellectual disability, epilepsy and neurobehavioural problems. J Neurodev Disord 6, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotney J, Muhle RA, Sanders SJ, et al. (2015) The autism‐associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat Commun 6, 6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan APA, Basson MA (2017) The neuroanatomy of autism – a developmental perspective. J Anat 230, 4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan APA, Yu T, Ellegood J, et al. (2017) Cerebellar vermis and midbrain hypoplasia upon conditional deletion of Chd7 from the embryonic mid‐hindbrain region. Front Neuroanat 11, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durak O, Gao F, Kaeser‐Woo YJ, et al. (2016) Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat Neurosci 19, 1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziuk MA, Gidley Larson JC, Apostu A, et al. (2007) Dyspraxia in autism: association with motor, social, and communicative deficits. Dev Med Child Neurol 49, 734–739. [DOI] [PubMed] [Google Scholar]
- Engelen E, Akinci U, Bryne JC, et al. (2011) Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat Genet 43, 607–611. [DOI] [PubMed] [Google Scholar]
- Epi KC, Epilepsy Phenome/Genome P, Allen AS, et al. (2013) De novo mutations in epileptic encephalopathies. Nature 501, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Khan MA, Bellvis P, et al. (2013) The chromatin remodeler CHD7 regulates adult neurogenesis via activation of SoxC transcription factors. Cell Stem Cell 13, 62‐72. [DOI] [PubMed] [Google Scholar]
- Feng W, Kawauchi D, Korkel‐Qu H, et al. (2017) Chd7 is indispensable for mammalian brain development through activation of a neuronal differentiation programme. Nat Commun 8, 14 758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye RE, Casanova MF, Fatemi SH, et al. (2016) Neuropathological mechanisms of seizures in autism spectrum disorder. Front Neurosci 10, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita K, Ogawa R, Kawasaki S, et al. (2014) Roles of chromatin remodelers in maintenance mechanisms of multipotency of mouse trunk neural crest cells in the formation of neural crest‐derived stem cells. Mech Dev 133, 126–145. [DOI] [PubMed] [Google Scholar]
- Fujita K, Ogawa R, Ito K (2016) CHD7, Oct3/4, Sox2, and Nanog control FoxD3 expression during mouse neural crest‐derived stem cell formation. FEBS J 283, 3791–3806. [DOI] [PubMed] [Google Scholar]
- Gage PJ, Hurd EA, Martin DM (2015) Mouse models for the dissection of CHD7 functions in eye development and the molecular basis for ocular defects in charge syndrome. Invest Ophthalmol Vis Sci 56, 7923–7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galizia EC, Myers CT, Leu C, et al. (2015) CHD2 variants are a risk factor for photosensitivity in epilepsy. Brain 138, 1198–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Gordon D, Zhang D, et al. (2007) CHD7 gene polymorphisms are associated with susceptibility to idiopathic scoliosis. Am J Hum Genet 80, 957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompers AL, Su‐Feher L, Ellegood J, et al. (2017) Germline Chd8 haploinsufficiency alters brain development in mouse. Nat Neurosci 20, 1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A, Okada S, Konno D, et al. (2012) Chd2 interacts with H3.3 to determine myogenic cell fate. EMBO J 31, 2994–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig I, Mefford HC, Sharp AJ, et al. (2009) 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet 41, 160–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich B, Bickmore W (2001) Human diseases with underlying defects in chromatin structure and modification. Hum Mol Genet 10, 2233–2242. [DOI] [PubMed] [Google Scholar]
- Ho L, Crabtree GR (2010) Chromatin remodelling during development. Nature 463, 474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd EA, Capers PL, Blauwkamp MN, et al. (2007) Loss of Chd7 function in gene‐trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mamm Genome 18, 94–104. [DOI] [PubMed] [Google Scholar]
- Iwase S, Martin DM (2018) Chromatin in nervous system development and disease. Mol Cell Neurosci 87, 1‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen N, Bergman JE, Swertz MA, et al. (2012) Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat 33, 1149–1160. [DOI] [PubMed] [Google Scholar]
- Jones KM, Saric N, Russell JP, et al. (2015) CHD7 maintains neural stem cell quiescence and prevents premature stem cell depletion in the adult hippocampus. Stem Cells 33, 196–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongmans MC, van Ravenswaaij‐Arts CM, Pitteloud N, et al. (2009) CHD7 mutations in patients initially diagnosed with Kallmann syndrome – the clinical overlap with CHARGE syndrome. Clin Genet 75, 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama Y, Nishiyama M, Shoji H, et al. (2016) CHD8 haploinsufficiency results in autistic‐like phenotypes in mice. Nature 537, 675–679. [DOI] [PubMed] [Google Scholar]
- Kim HG, Kurth I, Lan F, et al. (2008) Mutations in CHD7, encoding a chromatin‐remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet 83, 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Kishida S, Fukui A, et al. (2002) Nuclear localization of Duplin, a beta‐catenin‐binding protein, is essential for its inhibitory activity on the Wnt signaling pathway. J Biol Chem 277, 5816–5822. [DOI] [PubMed] [Google Scholar]
- Kulkarni S, Nagarajan P, Wall J, et al. (2008) Disruption of chromodomain helicase DNA binding protein 2 (CHD2) causes scoliosis. Am J Med Genet A 146a, 1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lado FA, Rubboli G, Capovilla G, et al. (2013) Pathophysiology of epileptic encephalopathies. Epilepsia 54(Suppl 8), 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun N, Parent P, Gendras J, et al. (2017) Autism spectrum disorder recurrence, resulting of germline mosaicism for a CHD2 gene missense variant. Clin Genet 92, 669–670. [DOI] [PubMed] [Google Scholar]
- Leyfer OT, Folstein SE, Bacalman S, et al. (2006) Comorbid psychiatric disorders in children with autism: interview development and rates of disorders. J Autism Dev Disord 36, 849–861. [DOI] [PubMed] [Google Scholar]
- Liu YQ, Yu F, Liu WH, et al. (2014) Dysfunction of hippocampal interneurons in epilepsy. Neurosci Bull 30, 985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JC, Ferreira CG, Yusufzai T (2015) Human CHD2 is a chromatin assembly ATPase regulated by its chromo‐ and DNA‐binding domains. J Biol Chem 290, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg MS, de Krijger I, Wiegant WW, et al. (2016) PARP1 links CHD2‐mediated chromatin expansion and H3.3 deposition to DNA repair by non‐homologous end‐joining. Mol Cell 61, 547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund C, Brodtkorb E, Oye AM, et al. (2014) CHD2 mutations in Lennox‐Gastaut syndrome. Epilepsy Behav 33, 18–21. [DOI] [PubMed] [Google Scholar]
- Marfella CGA, Imbalzano AN (2007) The Chd family of chromatin remodelers. Mutat Res 618, 30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marfella CG, Ohkawa Y, Coles AH, et al. (2006) Mutation of the SNF2 family member Chd2 affects mouse development and survival. J Cell Physiol 209, 162–171. [DOI] [PubMed] [Google Scholar]
- Marfella CG, Henninger N, LeBlanc SE, et al. (2008) A mutation in the mouse Chd2 chromatin remodeling enzyme results in a complex renal phenotype. Kidney Blood Press Res 31, 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meganathan K, Lewis EMA, Gontarz P, et al. (2017) Regulatory networks specifying cortical interneurons from human embryonic stem cells reveal roles for CHD2 in interneuron development. Proc Natl Acad Sci USA 114, E11 180–E11 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merner N, Forgeot d'Arc B, Bell SC, et al. (2016) A de novo frameshift mutation in chromodomain helicase DNA‐binding domain 8 (CHD8): a case report and literature review. Am J Med Genet A 170A, 1225–1235. [DOI] [PubMed] [Google Scholar]
- Nagarajan P, Onami TM, Rajagopalan S, et al. (2009) Role of chromodomain helicase DNA‐binding protein 2 in DNA damage response signaling and tumorigenesis. Oncogene 28, 1053–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, et al. (2012) Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama M, Nakayama K, Tsunematsu R, et al. (2004) Early embryonic death in mice lacking the beta‐catenin‐binding protein Duplin. Mol Cell Biol 24, 8386–8394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama M, Oshikawa K, Tsukada Y, et al. (2009) CHD8 suppresses p53‐mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat Cell Biol 11, 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama M, Skoultchi AI, Nakayama KI (2012) Histone H1 recruitment by CHD8 is essential for suppression of the Wnt‐beta‐catenin signaling pathway. Mol Cell Biol 32, 501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno H, Renault Mihara F, Ohta S, et al. (2017) CHARGE syndrome modeling using patient‐iPSCs reveals defective migration of neural crest cells harboring CHD7 mutations. eLife 6, e21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Girirajan S, et al. (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Stessman HA, Boyle EA, et al. (2014) Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun 5, 5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak N, Luo R, Zhang A, et al. (2013) Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten SA, Jacobs‐McDaniels NL, Zaouter C, et al. (2012) Role of Chd7 in zebrafish: a model for CHARGE syndrome. PLoS ONE 7, e31650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto AM, Bianciardi L, Mencarelli MA, et al. (2016) Exome sequencing analysis in a pair of monozygotic twins re‐evaluates the genetics behind their intellectual disability and reveals a CHD2 mutation. Brain Dev 38, 590–596. [DOI] [PubMed] [Google Scholar]
- Platt RJ, Zhou Y, Slaymaker IM, et al. (2017) Chd8 mutation leads to autistic‐like behaviors and impaired striatal circuits. Cell Rep 19, 335–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad MS, Sauka‐Spengler T, LaBonne C (2012) Induction of the neural crest state: control of stem cell attributes by gene regulatory, post‐transcriptional and epigenetic interactions. Dev Biol 366, 10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan S, Nepa J, Venkatachalam S (2012) Chromodomain helicase DNA‐binding protein 2 affects the repair of X‐ray and UV‐induced DNA damage. Environ Mol Mutagen 53, 44–50. [DOI] [PubMed] [Google Scholar]
- Riedel G, Micheau J (2001) Function of the hippocampus in memory formation: desperately seeking resolution. Prog Neuropsychopharmacol Biol Psychiatry 25, 835–853. [DOI] [PubMed] [Google Scholar]
- Rodriguez D, Bretones G, Quesada V, et al. (2015) Mutations in CHD2 cause defective association with active chromatin in chronic lymphocytic leukemia. Blood 126, 195–202. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Paredes M, Ceballos‐Chavez M, Esteller M, et al. (2009) The chromatin remodeling factor CHD8 interacts with elongating RNA polymerase II and controls expression of the cyclin E2 gene. Nucleic Acids Res 37, 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronan JL, Wu W, Crabtree GR (2013) From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet 14, 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto I, Kishida S, Fukui A, et al. (2000) A novel beta‐catenin‐binding protein inhibits beta‐catenin‐dependent Tcf activation and axis formation. J Biol Chem 275, 32 871–32 878. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, et al. (2012) De novo mutations revealed by whole‐exome sequencing are strongly associated with autism. Nature 485, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba Y, Harada A, Maehara K, et al. (2017) Chd2 regulates chromatin for proper gene expression toward differentiation in mouse embryonic stem cells. Nucleic Acids Res 45, 8758–8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T, Ji F, Yuan Z, et al. (2015) CHD2 is required for embryonic neurogenesis in the developing cerebral cortex. Stem Cells 33, 1794–1806. [DOI] [PubMed] [Google Scholar]
- Stolerman ES, Smith B, Chaubey A, et al. (2016) CHD8 intragenic deletion associated with autism spectrum disorder. Eur J Med Genet 59, 189–194. [DOI] [PubMed] [Google Scholar]
- Suetterlin P, Hurley S, Mohan C, et al. (2018) Altered neocortical gene expression, brain overgrowth and functional over‐connectivity in Chd8 haploinsufficient mice. Cereb Cortex 28, 2192–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugathan A, Biagioli M, Golzio C, et al. (2014) CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci USA 111, E4468–E4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suls A, Jaehn JA, Kecskes A, et al. (2013) De novo loss‐of‐function mutations in CHD2 cause a fever‐sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet 93, 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Rosenfeld JA, Blumenthal I, et al. (2012) Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149, 525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taurines R, Schwenck C, Westerwald E, et al. (2012) ADHD and autism: differential diagnosis or overlapping traits? A selective review. Atten Defic Hyperact Disord 4, 115–139. [DOI] [PubMed] [Google Scholar]
- Thompson BA, Tremblay V, Lin G, et al. (2008) CHD8 is an ATP‐dependent chromatin remodeling factor that regulates beta‐catenin target genes. Mol Cell Biol 28, 3894–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Laar I, Dooijes D, Hoefsloot L, et al. (2007) Limb anomalies in patients with CHARGE syndrome: an expansion of the phenotype. Am J Med Genet A 143a, 2712–2715. [DOI] [PubMed] [Google Scholar]
- Vissers LE, van Ravenswaaij CM, Admiraal R, et al. (2004) Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet 36, 955–957. [DOI] [PubMed] [Google Scholar]
- Wang P, Lin M, Pedrosa E, et al. (2015) CRISPR/Cas9‐mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Mol Autism 6, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Guo H, Xiong B, et al. (2016) De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun 7, 13 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JJ, Sillitoe RV (2013) Development of the cerebellum: from gene expression patterns to circuit maps. Wiley Interdiscip Rev Dev Biol 2, 149‐164. [DOI] [PubMed] [Google Scholar]
- Whittaker DE, Kasah S, Donovan APA, et al. (2017) Distinct cerebellar foliation anomalies in a CHD7 haploinsufficient mouse model of CHARGE syndrome. Am J Med Genet C Semin Med Genet 175, 465‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker DE, Riegman KL, Kasah S, et al. (2017b) The chromatin remodeling factor CHD7 controls cerebellar development by regulating reelin expression. J Clin Invest 127, 874–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson B, Grepo N, Thompson BL, et al. (2015) The autism‐associated gene chromodomain helicase DNA‐binding protein 8 (CHD8) regulates noncoding RNAs and autism‐related genes. Transl Psychiatry 5, e568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Takenaka C, Yoda Y, et al. (2018) Differentiation potential of Pluripotent Stem Cells correlates to the level of CHD7. Sci Rep 8, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates JA, Menon T, Thompson BA, et al. (2010) Regulation of HOXA2 gene expression by the ATP‐dependent chromatin remodeling enzyme CHD8. FEBS Lett 584, 689–693. [DOI] [PubMed] [Google Scholar]
- Yu T, Meiners LC, Danielsen K, et al. (2013) Deregulated FGF and homeotic gene expression underlies cerebellar vermis hypoplasia in CHARGE syndrome. Elife 2, e01305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan CC, Zhao X, Florens L, et al. (2007) CHD8 associates with human Staf and contributes to efficient U6 RNA polymerase III transcription. Mol Cell Biol 27, 8729–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahir F, Firth HV, Baross A, et al. (2007) Novel deletions of 14q11.2 associated with developmental delay, cognitive impairment and similar minor anomalies in three children. J Med Genet 44, 556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Distinct Chd8 and Chd7 expression patterns within the neural tube at E12.5. In situ hybridisation on sagittal (anterior to the right) and transverse sections of mouse embryos at developmental stage E12.5 using antisense riboprobes to detect Chd8 and Chd7 mRNA (A–H). Gene expression is indicated by purple/blue staining. Note that both Chd8 and Chd7 are expressed throughout the length of the neural tube (A,B,E,F). Whilst Chd8 displays no mediolateral or dorsoventral gradient in transverse sections, Chd7 shows distinct enrichment in the ventricular zone of the developing CNS and a ventral to dorsal gradient within the spinal cord. Drg, dorsal root ganglia; Iso, intersomitic region; LGE, lateral ganglionic eminence; MGE, medial ganglionic eminence; No, notochord; NT, neural tube; SE, surface ectoderm.
Fig. S2. Sense control sections at E12.5. In situ hybridisation on sagittal (anterior to the right) sections of mouse embryos at developmental stage E12.5 using sense riboprobes to Chd8, Chd7 and Chd2 mRNA. Note that for all three genes there is little to no hybridisation or staining using the sense riboprobe in contrast to what is seen when using the anti‐sense probe.