

Abstract

Majority of current circulating influenza A viruses carry the S31N mutation in their M2 genes, rendering AM2-S31N as a high profile antiviral drug target. With our continuous interest in developing AM2-S31N channel blockers as novel antivirals targeting both oseltamivir-sensitive and -resistant influenza A viruses, we report herein the structure–property relationship studies of AM2-S31N inhibitors. The goal was to identify lead compounds with improved microsomal stability and membrane permeability. Two lead compounds, 10d and 10e, were found to have high mouse and human liver microsomal stability (T1/2 > 145 min) and membrane permeability (>200 nm/s). Both compounds also inhibit both currently circulating oseltamivir-sensitive and -resistant human influenza A viruses (H1N1 and H3N2) with EC50 values ranging from 0.4 to 2.8 μM and a selectivity index of >100. We also showed for the first time that AM2-S31N channel blockers such as 10e inhibited influenza virus replication at both low and high multiply of infection (102–106 pfu/mL) and the inhibition was not cell-type dependent. Overall, these studies have identified two promising lead candidates for further development as antiviral drugs against drug-resistant influenza A viruses.
Keywords: Influenza A virus, AM2 proton channel, AM2-S31N inhibitor, microsomal stability, antiviral
Influenza viruses are negative sense, segmented RNA viruses that are the causative agents for annual influenza epidemic and sporadic influenza pandemics.1 Despite the availability of small molecule antivirals and influenza vaccines, there is an influenza season every year. More concerning is the emerging of influenza pandemic outbreaks that normally occur every 10 to 20 years.2 Part of explanation for the reoccurring influenza virus infection might be because influenza virus not only infects human but also many animals such as swine, migrating birds, chicken, horse, sea lions, etc. As such, there are multiple sources where human can contract the virus. When healthy immunocompetent adults are infected with seasonal influenza viruses, the symptoms are normally mild, and it is rarely fatal.3 Therefore, it might be somewhat surprising to learn that influenza virus infection is currently listed among the top-ten leading causes of deaths in the United Sates.4 The number of influenza virus-related mortality actually surpasses that of breast cancer. There are several factors that might contribute to the surprising death toll of influenza virus infection: (1) influenza virus is transmissible through the airways and can be quickly spread among humans. In each seasonal influenza epidemic, an estimate of 10–20% of the population are infected; (2) mortality rate of influenza virus infection among people in high-risk groups is high.5 They include seniors 65 years or older, people with chronic diseases such as cardiovascular diseases, diabetes, and high blood pressure, and people with compromised immune system. In such cases, influenza virus infection normally serves as a trigger of these pre-existing conditions. Overall influenza virus infection is a persistent public health concern that cannot possibly be ignored.
Currently there are two classes of FDA-approved small molecule influenza antivirals:6 (1) adamantanes such as amantadine and rimantadine. They are channel blockers of the influenza virus AM2 proton channel and inhibit the early stage of viral replication by blocking the virus uncoating. (2) Neuraminidase (NA) inhibitors such as oseltamivir, zanamivir, and peramivir. They are mimics of sialic acid and inhibit the late stage of viral replication by blocking the virus egress. Resistance to both classes of drugs now necessitates the development of newer influenza antivirals.7 Majority of influenza A viruses (>95%) are now resistant to adamantanes due to the AM2-S31N mutation in their M2 genes, and CDC no longer recommends the use of adamantanes in the prophylaxis and treatment of influenza virus infection. Resistance to oseltamivir has been continuously reported, and more alarmingly, the 2007–2009 seasonal influenza virus circulating in North American and Japan was completely resistant to oseltamivir due to the H275Y mutation in its NA gene.7,8 To tackle these drug-resistant viruses, several drug candidates are currently in development,6,9 which include both direct-acting antivirals such as the PA (polymerase acidic protein) endonuclease inhibitor baloxavir marboxil (approved in Japan and in late stage clinical trial in U.S.), polymerase inhibitor T-705 and PB2 inhibitor JNJ-63623872, as well as host-targeting antivirals such as nitazoxanide and DAS181. In addition, a large number of other drug targets are also actively pursued in the early stage of development.10
To design novel antivirals that are active against both oseltamivir-sensitive and -resistant influenza A viruses, we focus on targeting the influenza AM2-S31N proton channel.14−16 AM2-S31N is a high profile antiviral drug target, and more than 95% of current circulating influenza A viruses carry this mutation.17 Therefore, AM2-S31N channel blockers are expected to inhibit both oseltamivir-sensitive and -resistant influenza A viruses. As a proof-of-concept, we have shown that our rationally designed AM2-S31N inhibitors not only have potent channel blockage but also effective antiviral activity against multiple human influenza A viruses that are in circulation in recent years, including both H1N1 and H3N2 viruses that are resistant to either amantadine, oseltamivir, or both.14−16 Importantly, the newly developed AM2-S31N inhibitors showed a higher genetic barrier to drug resistance than amantadine, and drug resistance only emerged under high drug selection pressure after several passages.18,19 To further advance these promising lead compounds to in vivo mice model studies and prove their in vivo antiviral efficacy, we report herein our progress in profiling the metabolism stability of previously reported AM2-S31N inhibitors. One candidate, compound 4, was found to have good metabolic stability. Subsequent structure–activity and −property studies led to the discovery of two lead compounds, 10d and 10e, that have a long half-life in mouse and human liver microsomes (T1/2 > 145 min) as well as a high membrane permeability (Pe > 200 nm/s). Both compounds 10d and 10e showed improved selectivity index compared to that of compound 4. Importantly, compounds 10d and 10e retain potent antiviral activity against both oseltamivir-sensitive and -resistant human H1N1 and H3N2 influenza A viruses (EC50 = 0.4–2.8 μM), rendering them as promising candidates for the next step in vivo metabolic and efficacy studies in mice.
We have discovered several classes of adamantly containing AM2-S31N inhibitors based on structure-guided design and medicinal chemistry optimization.11,12,14−16,20 Representative examples of the most potent AM2-S31N inhibitors were shown in Table 1. They had single to submicromolar efficacy in inhibiting AM2-S31N-containing human influenza A viruses, including current circulating H1N1 and H3N2 strains. As a first step to profile the in vitro absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties, we first tested their microsomal stability in mouse liver microsomes. Surprisingly, all compounds except compound 4 had low microsomal stability (T1/2 < 30 min) (Table 1), precluding them from further progression. Encouragingly, compound 4 showed good microsomal stability with a T1/2 > 145 min, and the predicted liver clearance is less than 38.0 mL/min/kg. Therefore, compound 4 was selected as a lead compound for further optimization.
Table 1. Microsomal Stability Profiling of Previously Reported AM2-S31N Inhibitors.

Given the promising results of compound 4, a library of thiophene containing AM2-S31N inhibitors were synthesized and tested for channel blockage, antiviral activity, cellular cytotoxicity, and microsomal stability. Specifically, two series of new adamantane analogs, the monoaryl adamantanes (10a–10i) and the bis-aryl adamantanes (12a–12i), were designed and synthesized (Figure 1).
Figure 1.
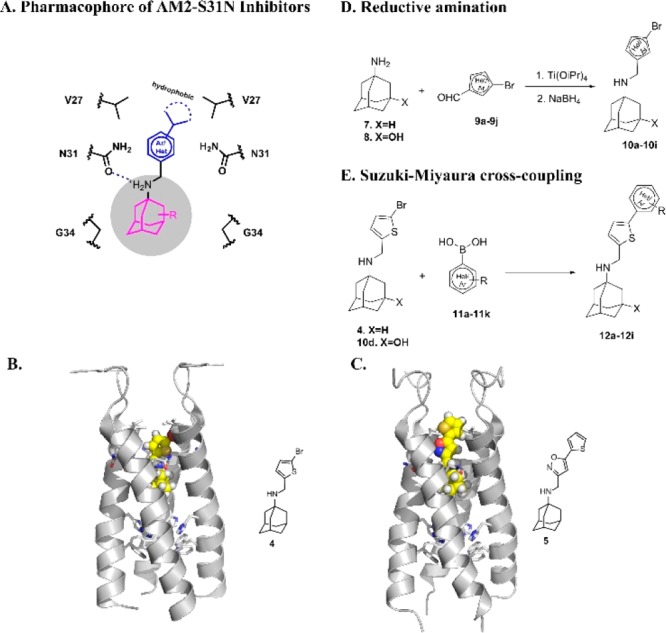
Design and synthesis of AM2-S31N inhibitors. (A) Pharmacophore of AM2-S31N inhibitors. Adamantane cage fits in the hydrophobic cavity formed by Gly34, the ammonium linker forms a hydrogen bond with the Asn31 side chain carbonyl, and the substitution on the aryl group forms hydrophobic interactions with the Val27 side chain. (B) Solution NMR structure of monoaryl AM2-S31N inhibitor 4 in complex with AM2-S31N (19–49) (PDB: 2MUV).12 (C) Solution NMR structure of bis-aryl AM2-S31N inhibitor 5 in complex with AM2-S31N (19–49) (PDB: 2LY0).13 (D) Synthesis of monoaryl adamantanes by reductive amination. (E) Synthesis of biaryl adamantanes by Suzuki–Miyaura cross-coupling reaction.
The design was based on the AM2-S31N inhibitor pharmacophore (Figure 1A), which was derived from previous structure–activity relationship studies.16 Both series of adamantane analogs (10a–10i and 12a–12i) meet the requirements of the AM2-S31N pharmacophore. Therefore, it is likely that they will have potent channel blockage and antiviral activity. Representative examples of monoaryl and bis-aryl AM2-S31N inhibitors are compounds 4 and 5, respectively, and their binding modes to the AM2-S31N channel were determined by solution NMR as shown in Figure 1B,C, respectively.12,13 The monoaryl adamantanes were synthesized using our previously optimized reduction amination condition (Figure 1D),21 and the yields range from 62% to 85%. The bis-aryl adamantanes were synthesized using the expeditious microwave-mediated Suzuki–Miyaura cross-coupling reaction (Figure 1E), and the yields range from 75% to 92%.
The challenge of structure–property relationship studies is that the ADMET properties need to be optimized without negatively affecting the channel blockage and antiviral efficacy. In the first step, we focus on optimizing the mouse liver microsomal stability because mouse is a commonly used animal model for the evaluation of the in vivo antiviral efficacy of influenza antivirals.22,23 Changing the bromide in compound 4 with iodide resulted in compound 10a, which had improved antiviral activity (EC50 = 1.0 ± 0.1 μM vs 2.9 ± 0.8 μM) but drastically reduced microsomal stability (T1/2 = 24.5 min vs >145 min) (Table 2). The selenium analog 10b had similar antiviral activity as the thiophene 4, but it was less metabolically stable (T1/2 = 32.1 min vs >145 min). Replacing the adamantane in 4 with ring-expanded adamantane gave compound 10c, which had increased antiviral activity (EC50 = 0.6 ± 0.1 μM vs 2.9 ± 0.8 μM) but drastically reduced microsomal stability (T1/2 = 9.1 min vs >145 min). In contrast, replacing adamantane in compound 4 with hydroxyl-adamantane yielded compound 10d that not only had improved antiviral activity (EC50 = 1.1 ± 0.2 μM vs 2.9 ± 0.8 μM) but also high microsomal stability (T1/2 > 145 min). Encouraged by this result, we then synthesized the iodide analog 10e. It was found that the iodide analog 10e retained the potent antiviral activity and high microsomal stability (EC50 = 0.9 ± 0.1 μM, T1/2 > 145 min). Both compounds 10d and 10e also had an improved selectivity index than compound 4. Two additional bromothiophene analogs 10f and 10g had reduced antiviral activity compared to compound 10d, and compound 10f was also less metabolically stable than 10d (T1/2 = 100.8 min vs >145 min). The bromothiazole analogs 10h and 10i had drastically reduced channel blockage and antiviral activity, showing less than 40% channel inhibition at 100 μM. For the bis-aryl adamantane analogs 12a–12i, only compounds 12b and 12f had comparable antiviral activity to that of compound 10d. The antiviral EC50 values of 12b and 12f were 2.1 ± 0.2 and 2.5 ± 0.8 μM, respectively. However, both 12b and 12f had reduced selectivity index compared to that of compound 10d; therefore, they were not further pursued. All other bis-aryl compounds (12a, 12c, 12d, 12e, 12g, 12h, and 12i) had significantly reduced antiviral activity as shown by the values of percentage plaque formation at 10 μM. All compounds (10a–10i and 12a–12i) were also tested against the wild-type (WT) AM2 channel, and compounds 4, 10a, and 10c were found to have potent channel blockage against AM2-WT (>77% inhibition at 100 μM). In summary, through the structure–property relationship studies, we identified two lead compounds 10d and 10e that had potent channel blockage, antiviral activity, a high selectivity index, and optimal microsomal stability.
Table 2. Channel Blockage, Antiviral Activity, Cytotoxicity, and Mouse Liver Microsomal Stability of AM2-S31N Inhibitors.

Values represent the mean of three independent measurements ± standard deviation. Compounds were tested at 100 μM concentration.
Antiviral activity was tested with the A/California/07/2009 (H1N1) virus, which contains the AM2-S31N mutant, in plaque assay. Values represent the mean of two independent measurements ± standard deviation.
Cytotoxicity was tested by incubating MDCK cells with compounds for 48 h, and the cells were stained with neutral red.24 N.T. = not tested.
The membrane permeability of the two lead compounds 10d and 10e was further profiled (Table 3). Compounds 10d and 10e were predicted by the Schrödinger Glide QikProp program to have high membrane permeability and oral absorption (Table 3). Next, to experimentally determine their membrane permeability, compounds 10d and 10e were tested in the parallel artificial membrane permeability assay (PAMPA), which is commonly used as an in vitro model of passive, transcellular permeation. Both compounds 10d and 10e showed high membrane permeability with Pe greater than 200 nm/s in the Egg-PAMPA assay, indicating they can passively diffuse through the transcellular membrane. Both compounds 10d and 10e also did not violate the Lipinski rule of five. The microsomal stability of compounds 10d and 10e was further confirmed in human liver microsomal stability test, and both compounds 10d and 10e were found to have a long half-life with T1/2 > 145 min. Taken together, compounds 10d and 10e appear to be promising leads for further development.
Table 3. PK Predictions, Membrane Permeability, Physiochemical Properties, and Mouse Microsomal Stability of Lead Compounds 10d and 10e.

Membrane permeability and cLogP were calculated by the Schrödinger Glide QikProp program.
One of the major therapeutic advantages of AM2-S31N inhibitors is that they have no cross-resistance with neuraminidase inhibitors such as oseltamivir.18 Therefore, they are expected to have potent antiviral activity against both oseltamivir-sensitive and -resistant influenza A viruses. To prove this hypothesis, we tested the antiviral activity of compounds 10d and 10e against several human influenza A viruses, including both H1N1 and H3N2 strains (Table 4). All viruses contain the AM2-S31N mutation in their M2 genes and are resistant to amantadine. In addition, the four oseltamivir-resistant strains encode the H275Y mutant in their neuraminidase gene, which confer to their resistance to oseltamivir. It was found that compounds 10d and 10e inhibited all seven influenza A viruses with EC50 values ranging from 0.4 to 2.8 μM.
Table 4. Antiviral Activity of Compounds 10d and 10e in Inhibiting Oseltamivir-Sensitive and -Resistant Influenza A Viruses.

The antiviral activity of compound 10e was further validated when MDCK cells were infected with the A/California/07/2009 (H1N1) or the A/Wisconsin/67/2005 (H3N2) virus at high MOIs, a condition that mimics the late stage of treatment when the virus was already amplified in the host. It was found that compound 10e significantly suppressed the viral replication when MOI was as high as 106 pfu/mL for both influenza strains (Figure 2). In contrast, oseltamivir carboxylate was only effective at low MOIs (101 to 103 pfu/mL) and was not effective at high MOIs (104–106 pfu/mL). Overall, compound 10e showed potent inhibition against human H1N1 and H3N2 influenza A strains at both low and high MOIs.
Figure 2.
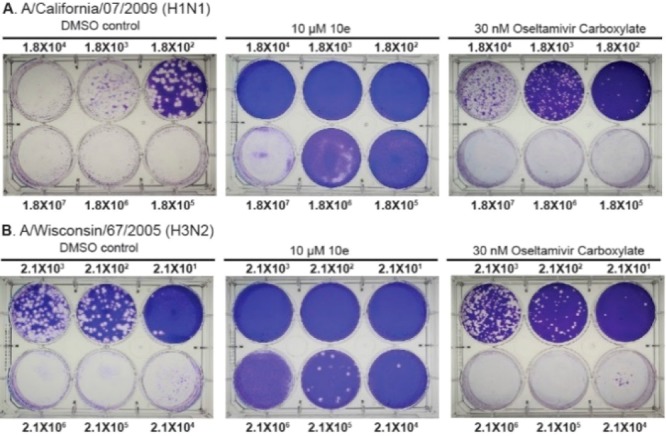
Antiviral activity of compound 10e in inhibiting A/California/ 07/2009 (H1N1) (A) and A/Wisconsin/67/2005 (H3N2) (B) viruses at different MOIs (101 −106 pfu/mL) using plaque assay. The numbers shown are viral titers at initial infection. The images shown were representative results from two repeats.
To test whether the antiviral efficacy of compound 10e is cell-type dependent, we performed viral titer reduction assay using the A/California/07/2009 (H1N1) virus and the A549 cells. In this assay, confluent A549 cells were infected with the A/California/07/2009 (H1N1) virus at MOI of 0.001, the cell culture supernatant was collected at 24, 48, and 72 h postinfection, and the viral titer was quantified by plaque assay. Compared with the DMSO control, treatment with 10 μM of compound 10e significantly reduced the viral titers of 1.8, 2.1, and 2.9 log10 units at 24, 48, and 72 h, respectively (Figure 3). In comparison, oseltamivir carboxylate only delayed viral replication and showed no effect at 72 hpi under similar drug selection pressure (10-fold of EC50 value).
Figure 3.
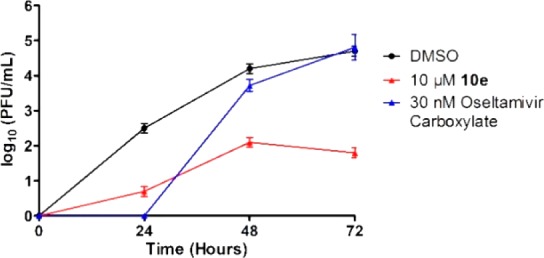
Antiviral activity of compound 10e in inhibiting A/California/07/2009 (H1N1) virus replication in A549 cells. The results shown were from two repeats.
The binding of compound 10e in the AM2-S31N channel was modeled by the Schrödinger Glide standard precision docking program. In the energy-minimized docking pose (Figure 4), compound 10e fitted inside the channel with the thiophene group facing toward the N-terminal of the channel. The hydroxyl group from 10e forms a hydrogen bond with the backbone amide carbonyl from one of the helixes, and the ammonium from 10e forms another hydrogen bond with the side chain amide carbonyl from the neighboring helix. The iodothiophene substitution forms hydrophobic interactions with the V27 side chain methyls. Overall, the docking pose of compound 10e is similar to that of compound 4 (PDB: 2MUV) (Figure 1b).
Figure 4.
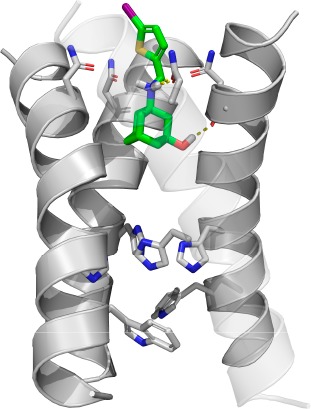
Docking model of compound 10e in the transmembrane domain of AM2-S31N (PDB: 2LY0).13 The transparency of the front helix was set as 0.7 for clarity.
Drug discovery is a lengthy and expansive endeavor,25 and lead compounds can fail in any step during the early preclinical development phase or the later human clinical trials. Therefore, it is essential to provide additional backup compounds with favorable PK properties for the following in vivo animal and human studies. Herein, we report our progress of optimizing the mouse microsomal stability and cell membrane permeability of thiophene-containing AM2-S31N inhibitors. Starting from a promising lead compound 4, we were able to identify two compounds 10d and 10e with improved antiviral efficacy and selectivity index. The optimized lead compounds 10d and 10e retained high mouse liver microsomal stability (T1/2 > 145 min) and had favorable membrane permeability in the PAMPA assay (Pe > 200 nm/s) and a high selectivity index (SI > 100). As a demonstration of the therapeutic value of AM2-S31N inhibitors, compounds 10d and 10e were found to have potent antiviral potency against several oseltamivir-sensitive and -resistant human influenza A viruses. Compound 10e was also able to inhibit A/California/07/2009 (H1N1) and A/Wisconsin/67/2005 (H3N2) at MOIs ranging from 102 to 106 pfu/mL, and the antiviral activity of compound 10e was further confirmed in human A549 cell line. Taken together, the potent antiviral efficacy, a high selectivity index, a long half-life in mouse liver microsomes, and a high membrane permeability of the identified lead compounds 10d and 10e warrant their further development as orally bioavailable influenza antivirals. Indeed, several FDA-approved oral drugs such as rivaroxaban, chlorothen, brotizolam, and lornoxicam similarly contain halogen-deactivated thiophene,26 which reassures continuous development of this series of compounds.
Glossary
ABBREVIATIONS
- WT
wild type
- DMEM
Dulbecco’s modified Eagle’s medium
- MDCK
Madin–Darby Canine Kidney
- TEVC
two-electrode voltage clamp
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00336.
Synthesis procedures; characterization of compounds; antiviral and cytotoxicity assay; electrophysiological assay; mouse microsomal stability assay; membrane permeability assay (PDF)
Author Contributions
# Y.H. and R.K.H. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This research is supported by the startup funding from the University of Arizona and the NIH grant AI119187 to J.W.
The authors declare no competing financial interest.
Supplementary Material
References
- Webster R. G.; Monto A. S.; Braciale T. J.; Lamb R. A.. Textbook of Influenza; Wiley: 2013. [Google Scholar]
- https://www.cdc.gov/flu/pandemic-resources/basics/past-pandemics.html (Accessed on 09/28/2018).
- Clinical Signs and Symptoms of Influenza. https://www.cdc.gov/flu/professionals/acip/clinical.htm (accessed on 09/28/2018).
- Leading Causes of Death. https://www.cdc.gov/nchs/fastats/leading-causes-of-death.htm (accessed on 09/28/2018).
- People at High Risk of Developing Serious Flu–Related Complications, https://www.cdc.gov/flu/about/disease/high_risk.htm (accessed on 09/28/2018).
- Shaw M. L. The Next Wave of Influenza Drugs. ACS Infect. Dis. 2017, 3, 691–694. 10.1021/acsinfecdis.7b00142. [DOI] [PubMed] [Google Scholar]
- Hurt A. C. The epidemiology and spread of drug resistant human influenza viruses. Curr. Opin. Virol. 2014, 8, 22–29. 10.1016/j.coviro.2014.04.009. [DOI] [PubMed] [Google Scholar]
- Matsuzaki Y.; Mizuta K.; Aoki Y.; Suto A.; Abiko C.; Sanjoh K.; Sugawara K.; Takashita E.; Itagaki T.; Katsushima Y.; Ujike M.; Obuchi M.; Odagiri T.; Tashiro M. A two-year survey of the oseltamivir-resistant influenza A(H1N1) virus in Yamagata, Japan and the clinical effectiveness of oseltamivir and zanamivir. Virol. J. 2010, 7, 53. 10.1186/1743-422X-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koszalka P.; Tilmanis D.; Hurt A. C. Influenza antivirals currently in late-phase clinical trial. Influenza Other Respir. Viruses 2017, 11 (3), 240–246. 10.1111/irv.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loregian A.; Mercorelli B.; Nannetti G.; Compagnin C.; Palu G. Antiviral strategies against influenza virus: towards new therapeutic approaches. Cell. Mol. Life Sci. 2014, 71 (19), 3659–3683. 10.1007/s00018-014-1615-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Ma C.; Hu Y.; Wang Y.; Wang J. Discovery of Potent Antivirals against Amantadine-Resistant Influenza A Viruses by Targeting the M2-S31N Proton Channel. ACS Infect. Dis. 2016, 2 (10), 726–733. 10.1021/acsinfecdis.6b00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Canturk B.; Jo H.; Ma C.; Gianti E.; Klein M. L.; Pinto L. H.; Lamb R. A.; Fiorin G.; Wang J.; DeGrado W. F. Flipping in the pore: discovery of dual inhibitors that bind in different orientations to the wild-type versus the amantadine-resistant S31N mutant of the influenza A virus M2 proton channel. J. Am. Chem. Soc. 2014, 136 (52), 17987–17995. 10.1021/ja508461m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wu Y.; Ma C.; Fiorin G.; Wang J.; Pinto L. H.; Lamb R. A.; Klein M. L.; Degrado W. F. Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza A virus. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (4), 1315–1320. 10.1073/pnas.1216526110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Wang Y.; Li F.; Ma C.; Wang J. Design and expeditious synthesis of organosilanes as potent antivirals targeting multidrug-resistant influenza A viruses. Eur. J. Med. Chem. 2017, 135, 70–76. 10.1016/j.ejmech.2017.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Hu Y.; Wang Y.; Ma C.; Wang J. Expeditious Lead Optimization of Isoxazole-Containing Influenza A Virus M2-S31N Inhibitors Using the Suzuki-Miyaura Cross-Coupling Reaction. J. Med. Chem. 2017, 60 (4), 1580–1590. 10.1021/acs.jmedchem.6b01852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Ma C.; DeGrado W. F.; Wang J. Discovery of Highly Potent Inhibitors Targeting the Predominant Drug-Resistant S31N Mutant of the Influenza A Virus M2 Proton Channel. J. Med. Chem. 2016, 59 (3), 1207–1216. 10.1021/acs.jmedchem.5b01910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong G.; Peng C.; Luo J.; Wang C.; Han L.; Wu B.; Ji G.; He H. Adamantane-resistant influenza a viruses in the world (1902–2013): frequency and distribution of M2 gene mutations. PLoS One 2015, 10 (3), e0119115. 10.1371/journal.pone.0119115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Zhang J.; Wang J. Pharmacological Characterization of the Spectrum of Antiviral Activity and Genetic Barrier to Drug Resistance of M2-S31N Channel Blockers. Mol. Pharmacol. 2016, 90 (3), 188–198. 10.1124/mol.116.105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musharrafieh R.; Ma C. L.; Wang J. Profiling the in vitro drug-resistance mechanism of influenza A viruses towards the AM2-S31N proton channel blockers. Antiviral Res. 2018, 153, 10–22. 10.1016/j.antiviral.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Hu Y.; Xu S.; Zhang Y.; Musharrafieh R.; Hau R. K.; Ma C.; Wang J. Vitro Pharmacokinetic Optimizations of AM2-S31N Channel Blockers Led to the Discovery of Slow-Binding Inhibitors with Potent Antiviral Activity against Drug-Resistant Influenza A Viruses. J. Med. Chem. 2018, 61 (3), 1074–1085. 10.1021/acs.jmedchem.7b01536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Ma C.; Wang J.; Jo H.; Canturk B.; Fiorin G.; Pinto L. H.; Lamb R. A.; Klein M. L.; DeGrado W. F. Discovery of novel dual inhibitors of the wild-type and the most prevalent drug-resistant mutant, S31N, of the M2 proton channel from influenza A virus. J. Med. Chem. 2013, 56 (7), 2804–2812. 10.1021/jm301538e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Musharrafieh R.; Ma C.; Zhang J.; Smee D. F.; DeGrado W. F.; Wang J. An M2-V27A channel blocker demonstrates potent in vitro and in vivo antiviral activities against amantadine-sensitive and -resistant influenza A viruses. Antiviral Res. 2017, 140, 45–54. 10.1016/j.antiviral.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smee D.; Barnard D., Methods for Evaluation of Antiviral Efficacy Against Influenza Virus Infections in Animal Models. In Antiviral Methods and Protocols; Gong E. Y., Ed.; Humana Press: 2013; Vol. 1030, pp 407–425. [DOI] [PubMed] [Google Scholar]
- Repetto G.; del Peso A.; Zurita J. L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3 (7), 1125–1131. 10.1038/nprot.2008.75. [DOI] [PubMed] [Google Scholar]
- DiMasi J. A.; Grabowski H. G.; Hansen R. W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ 2016, 47, 20–33. 10.1016/j.jhealeco.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Scott K. A.; Njardarson J. T. Analysis of US FDA-Approved Drugs Containing Sulfur Atoms. Top Curr. Chem. (Cham) 2018, 376 (1), 5. 10.1007/s41061-018-0184-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.