

Abstract

We report here the total synthesis of B-973 (five steps), a recently identified α7 nAChR ago-PAM, its enantiomeric resolution, and its electrophysiological characterization in Xenopus oocytes to identify (−)-B-973B as the bioactive enantiomer. The asymmetric synthesis of B-973B was accomplished in 99% ee, and X-ray crystallography studies revealed its absolute “S” stereochemistry. B-973B was effective in attenuating pain behavior and decreasing paw edema (formalin test), and its analgesic effects were mediated through α7 nAChR.
Keywords: Alpha7 nicotinic acetylcholine receptors, ligand-gated ion channels, positive allosteric modulators, Ago-PAMs, neuropathic pain, asymmetric synthesis, electrophysiological characterization
Nicotinic acetylcholine receptors (nAChRs) are members of the Cys-loop superfamily of cationic ligand-gated ion channels and are involved in the physiological responses to the neurotransmitter, acetylcholine (ACh).1−3 These receptors are distributed throughout the central and peripheral nervous systems and play important roles in several (patho)physiological processes. Compounds that selectively target the homopentameric α7 nAChR subtype are being pursued as potential pharmacotherapy agents for treating cognitive dysfunctions in schizophrenia and Alzheimer’s disease, as well as for treating neuropathic and inflammatory pain. This type of targeting is safe and is not associated with opioids-like side effects such as addiction liability.4−7 Attempts to target α7 nAChR has mainly focused on two approaches: partial agonism via the ACh binding site and positive allosteric modulation (PAM) at sites topographically distinct from ACh binding. Although α7 nAChR-selective and potent (partial) agonists have been developed and studied (pre)clinically, the rapid desensitization of α7 nAChR agents in response to high agonist concentration, together with the possibility of endogenous tone disruption, has led to some concerns regarding their utility as clinical candidates.2,4,5 PAMs of the α7 nAChR provide an alternative targeting approach that facilitates natural signaling by endogenous ACh and thus is differentiated from partial agonists.8
In addition to PAMs, allosteric agonists as well as silent agonists of α7 nAChR have been discovered.6 However, despite all of these efforts, only limited success has been achieved clinically with galantamine (Figure 1). JNJ-39393406 is an α7 PAM that is currently undergoing phase II clinical trials for depressive disorders and smoking cessation.9,10 Most of the drug candidates that were pursued as α7 nAChR (partial) agonists or PAMs, however, failed in Phase II clinical trials for cognitive disorders due to insufficient efficacy. Recently, a unique class of compounds with dual modes of action—allosteric agonism and positive allosteric modulation—have been discovered and are referred to as “ago-PAMs” (e.g., GAT107, Figure 1).11−14
Figure 1.

Representative selective positive allosteric modulators of α7 nAChR: an approved drug (1), phase II clinical candidate (2), and preclinical candidates (3–6).
Ago-PAMs of α7 nAChR activate receptors through site distinct from orthosteric sites and positively modulate orthosteric cholinergic signaling while preserving the spatiotemporal features of synaptic transmission.13,14 Such modulators that act through dual modes of action are expected to be more efficacious and potentially address the limitations observed in partial agonists and PAMs in clinical trials. Indeed, we recently demonstrated that one such dual modulator, GAT107, from the tetrahydroquinoline (THQ) class of compounds is an orally bioavailable and efficacious analgesic for treating inflammatory and neuropathic pain in animal models.15
Recently, Bristol Myers Squibb reported HTS screening of a diverse small molecule library (>106 molecules) using a fluorescence-based assay configured to detect α7 nAChR PAMs.16 Active compounds identified from the screen included a series of piperazine-containing molecules that displayed robust α7 nAChR PAM activity. One compound from this series, B-973 (Figure 1), was unique in that it was shown to be a potent ago-PAM. Although this important work led to the identification of a novel scaffold with α7 nAChR ago-PAM activity, it left several questions unanswered, thus limiting further drug optimization and developmental efforts on this scaffold. Based on the information provided, it is not clear if B-973 is a racemate or a single enantiomer. Neither its absolute stereochemistry nor the activity of the other enantiomer are known. Information on the chemical synthesis of this compound to facilitate in vivo studies and library syntheses is also not reported.
Given the therapeutic potential of α7 nAChR ago-PAMs and the excitement surrounding this structurally novel scaffold, an effort was made to pursue its synthesis and enantiomeric separation and identify the stereochemical requirements for α7 nAChR ago-PAM activity through electrophysiological studies followed by in vivo evaluation of the active enantiomer. We report an efficient and scalable synthesis of the racemate as well as of the active enantiomer in 99% ee. Access to copious amounts of the active enantiomer enabled its preclinical evaluation for inflammatory pain (formalin test) in a mouse model, where it was shown to be efficacious. The five-step synthetic route reported here was optimized for producing gram-scale quantities of B-973 and is depicted in Scheme 1.
Scheme 1. Racemic Synthesis of B-973.

The synthesis begins with the formation of tricyclic bromo pyrazine derivative 9 by monosubstitution of dibromo pyrazine 7 with piperazine 8 in the presence of K2CO3 in DMF in 92% yield. To install a keto functionality on 9, we examined several reaction conditions involving BuLi-mediated lithiation followed by quenching with appropriate electrophiles. Our initial efforts were plagued by the formation of a range of undesired products, which was presumably due to the sensitivity of the pyrazine ring to these conditions. We further attempted installing the ketone moiety under mild Pd-catalyzed cross-coupling conditions.17 Stille cross coupling of compound 9 with tributyl (1-ethoxyvinyl)tin and Pd(PPh3)2Cl2 in the presence of CuI yielded the enol ether 10. The crude product was hydrolyzed with aq. HCl to give the desired ketone compound 11 in 82% yield over two steps. Thus, after obtaining the key keto intermediate 11 in multigram quantities, we carried out its conversion to an imine with NH4OAc, followed by in situ reduction with NaCNBH4 to yield racemic amine 12 in 96% yield. The conversion of acid 13 to the corresponding acid chloride 14, followed by its coupling with the amine fragment 12 in the presence of pyridine and catalytic DMAP yielded the racemic B-973, 6 in 54% yield (Scheme 1).
B-973 has one chiral center with two possible configuration. To test whether the allosteric agonist and PAM activity was attributed to a single enantiomer or was present in both compounds, we first developed conditions for the enantiomeric separation of B-973 (Scheme 2; see SI). Using chiral super fluid chromatography (SFC), baseline separation of the B-973 enantiomers was accomplished in 99% ee.
Scheme 2. Enantiomeric Resolution of Racemic B-973.

Both enantiomers were then tested in Xenopus oocytes expressing human α7 nAChR with a two-electrode voltage clamp method (Figure 2). Following the measurement of 60 μM ACh control responses, the B-973 isomers were coapplied with a control concentration at 10 or 100 μM. The average net-charge responses (±SEM) of the oocytes (n ≥ 5) to the A and B isomers, normalized to the initial ACh control responses from the same cells, are shown in Figure 2A. While there were negligible effects from the B-973A isomer, the B-973B isomer greatly increased the responses compared to those evoked by ACh alone.
Figure 2.
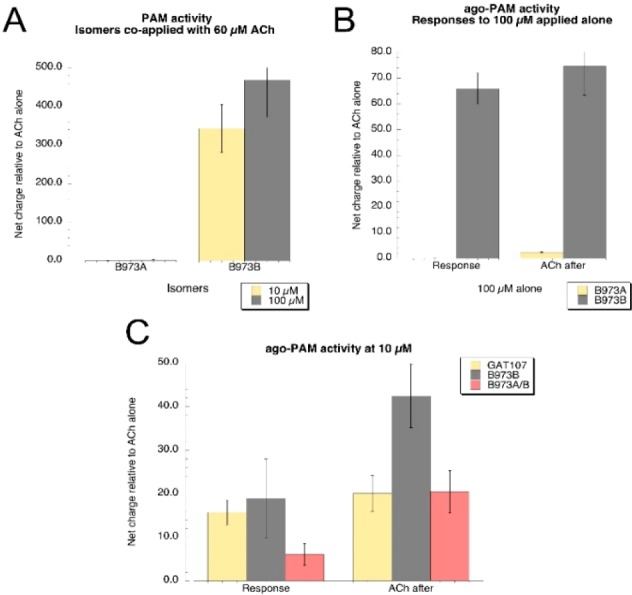
Electrophysiological characterization of the B-973 isomers on human α7 nAChR expressed in Xenopus oocytes.
Following measurement of the 60 μM ACh control responses, the B-973 isomers were applied alone at a concentration of 100 μM, followed by another application of 60 μM ACh. The average net-charge responses (±SEM) of the oocytes (n = 6) to the A and B isomers, normalized to the initial ACh control responses from the same cells, are shown in Figure 2B. The B-973A isomer failed to evoke a response, and the subsequent ACh controls were not significantly different from the initial controls. In contrast, B-973B evoked large responses, and the ACh responses greatly increased after washout compared to initial controls. The normalized net-charge responses of α7-expressing cells to 10 μM B-973B were compared to 10 μM of the previously characterized ago-PAM GAT107 (Figure 2C).12,13 While the initial allosteric activation produced by the two agents was similar, the primed potentiation of the subsequent 60 μM ACh response was greater (p < 0.05) for B-973B than for GAT107. Additionally, the responses to racemic B-973 were smaller (p < 0.05) than the response to the B-973B enantiomer. These studies thus concluded that the ago-PAM activity resided exclusively in (−)-B-973B, while (+)-B-973A was inactive at α7 nAChR.
To understand the absolute stereochemical requirements for α7 nAChR ago-PAM activity, we attempted to crystallize both enantiomers to obtain crystals suitable for X-ray studies. However, even after several crystallization attempts, neither of the B-973 enantiomers could be crystallized in an acceptable form. Furthermore, to facilitate structure–activity relationship (SAR) studies, there was a need for easy access to the homochiral version of amine 12 due to the observed high enantiospecificity for α7 nAChR activity. We therefore decided to develop an enantioselective approach that would yield the active enantiomer in high ee and allow derivatization of the chiral amine to expand the possibility of obtaining crystals suitable for X-ray studies.
Among the various methods for the preparation of optically active amines,18 we planned to synthesize chiral 12 via an approach involving imine formation with chiral tert-butanesulfinamide, followed by diastereoselective reduction with the appropriate reducing agent as a key step.19 Advantageously, both the enantiomers of tert-butanesulfinamide are inexpensive and commercially available in high optical purity. Moreover, condensation of tert-butanesulfinamide and ketone gives stable ketimine under mild reaction conditions, yields good diastereoselectivity during reduction, and allows easy removal of the N-tert-butanesulfinyl in high yield.
Accordingly, the chiral N-tert-butanesulfinyl ketimine 17 was synthesized from ketone 11 with R-(+)-2-methyl-2-propane sulfonamide in the presence of Ti(OEt)4 in dichloromethane under reflux conditions as a yellow solid in 89% yield. The chiral N-tert-butanesulfinyl ketimine 17 was stereospecifically reduced by 9-BBN20−22 in THF at −78 °C to yield sulfinamide 18 as a single diastereomer (dr ≥ 99%; determined by 1H and 13C NMR spectroscopy) in 96% yield. The hydrolysis of the sulfinamide group of 17 was accomplished using 6 M HCl in methanol and gave chiral amine 19 in 86% yield. Finally, the coupling of chiral amine 19 with acid chloride 14 yielded optically pure 15 in 94% ee (Scheme 3). By comparing the optical rotation of 15 with the enantiomers obtained from chiral SFC, 15 was identified as the inactive enantiomer.
Scheme 3. Asymmetric Synthesis of (+)-B-973A.

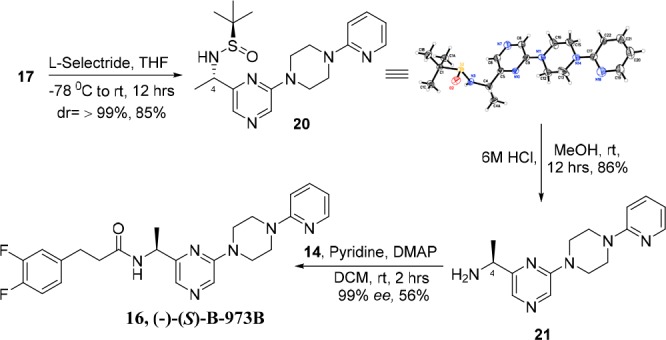
To obtain the other enantiomer, N-tert-butanesulfinyl ketimine 17 was stereospecifically reduced with l-Selectride20−22 in anhydrous THF at −78 C to yield 20 as a single diastereomer (dr ≥ 99%; determined by 1H and 13C NMR spectroscopy) in 92% yield. An X-ray crystallographic study revealed the absolute stereochemistry of 20 at C4 to be “S”. The hydrolysis of the sulfinamide group of 20 using 6 M HCl gave the chiral amine 21 in 86% yield. The absolute “S” stereochemistry of the chiral center C4 was further confirmed by a single-crystal X-ray study of p-nitrobenzoyl derivative 22 of chiral amine 21 (see SI). Finally, the coupling of 21 with acid chloride 14 yielded the optically pure bioactive isomer 16, (S)-(−)-B-973B, in 56% yield and with 99% optical purity (Scheme 4).
Scheme 4. Asymmetric Synthesis of (−)-B-973B.
We previously demonstrated the antinociceptive and anti-inflammatory efficacy of GAT107 toward pain in a series of mouse models.15 Here, we tested whether the systemic administration of B-973B would produce antinociception in mouse formalin testing. As seen in Figure 3A, B-973B did significantly reduce nociceptive behavior during phase I [F(3,28) = 3.941, P = 0.018] at the highest dose of 10 mg/kg. Moreover, B-973B (1–10 mg/kg, i.p.) dose-dependently attenuated formalin-induced paw licking behavior in Phase II [F(3,28) = 13.07, P < 0.001] with 3 and 10 mg/kg doses, differing from the vehicle group.
Figure 3.

In vivo evaluation of the active enantiomer B-973B in a mouse model of inflammatory pain and establishment of target specificity.
In addition, B-973B significantly reduced paw edema [F(3,28) = 21.05, P < 0.001; Figure 3B] in mice treated with 3 and 10 mg/kg doses, differing from the vehicle group. We next explored the possible role of α7 nAChRs as related to B-973B’s effect in the formalin test. To study this, we tested whether the α7 nAChR antagonist, MLA, blocks the antinociceptive effects of B-973B in the formalin test. As shown in Figure 3C, a significant effect of treatment was found [F(3,15) = 33.05; P < 0.0001]. MLA given alone significantly affected the nociceptive behavior (P = 0.77). MLA (10 mg/kg, s.c.) totally blocked the antinociceptive effect of B-973B in both phases I and II of the formalin test (P < 0.001).
Acute B-973B mainly attenuated pain behavior in the second phase of the formalin test, which is associated with the development of inflammation and spinal dorsal horn sensitization but was only effective in phase I at the highest dose (immediately after formalin injection), which is mediated by C-fiber activity.23,24 Moreover, B-973B decreased formalin-induced paw edema, which is consistent with the idea that it acts on the inflammatory phase of the formalin test. Furthermore, using a complementary pharmacological agent (i.e., the α7 nAChR antagonist, MLA), we confirmed that the antinociceptive effects of B-973B in the formalin test were mediated through α7 nAChRs.
Thus, in this work, we have developed the first total, gram-scale synthesis of B-973, a structurally novel α7 nAChR ago-PAM. We also developed conditions for its enantiomeric separation using chiral SFC to obtain both enantiomers in high enantiomeric excess. The (−)-B-973B enantiomer retained all α7 nAChR ago-PAM activity, while the (+)-B-973B was inactive in electrophysiological assays. Asymmetric synthesis of (−)-B-973B was accomplished in high enantiomeric excess (99% ee). When tested in vivo, B-973B was effective in attenuating pain behavior and decreased formalin-induced paw edema (formalin test). Target specificity of B-973B for analgesic activity was established using MLA. B-973B was as effective as structurally distinct GAT107 in in vitro assays and in vivo experiments, thus providing a suitable lead for future SAR optimization.
Acknowledgments
This work was supported by the National Institute of Health (NIH) RO1-EY024717 to G.A.T. and RO1-GM057481 to R.L.P., and a grant from a Pilot Project from the VCU Massey Cancer Center and R01-CA206028 to M.I.D. Oocyte experiments were conducted by Clare Stokes and Alexander den Boef.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00407.
Experimental details, structural characterization data, chiral SFC separation details, NMR spectra, X-ray study details, and biological studies (PDF)
Accession Codes
CCDC 1842480 and 1842483 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Bertrand D.; Terry A. V. The wonderland of neuronal nicotinic acetylcholine receptors. Biochem. Pharmacol. 2018, 151, 214. 10.1016/j.bcp.2017.12.008. [DOI] [PubMed] [Google Scholar]
- Papke R. L. Merging old and new perspectives on nicotinic acetylcholine receptors. Biochem. Pharmacol. 2014, 89, 1. 10.1016/j.bcp.2014.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes C.; Treinin M.; Papke R. L. Looking below the surface of nicotinic acetylcholine receptors. Trends Pharmacol. Sci. 2015, 36, 514. 10.1016/j.tips.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagdas D.; Gurun M. S.; Flood P.; Papke R. L.; Damaj M. I. New Insights on Neuronal Nicotinic Acetylcholine Receptors as Targets for Pain and Inflammation: A Focus on α7 nAChRs. Curr. Neuropharmacol 2018, 16, 415. 10.2174/1570159X15666170818102108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hone A. J.; McIntosh J. M. Nicotinic acetylcholine receptors in neuropathic and inflammatory pain. FEBS Lett. 2018, 592, 1045. 10.1002/1873-3468.12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manetti D.; Bellucci C.; Chiaramonte N.; Dei S.; Teodori E.; Ro manelli M. N. Designing selective modulators for the nicotinic receptor subtypes: challenges and opportunities. Future Med. Chem. 2018, 10, 433. 10.4155/fmc-2017-0169. [DOI] [PubMed] [Google Scholar]
- Hudzik T. J.; Basso A. M.; Lynch J. J.; Bracken W. M.; Mohler E. G.; Kohlhaas K. L.; Xu H.; Haig G.; Gault L. Preclinical abuse liability assessment of ABT-126, an agonist at the α7 nicotinic acetylcholine receptor (nAChR). Pharmacol., Biochem. Behav. 2017, 158, 22. 10.1016/j.pbb.2017.05.010. [DOI] [PubMed] [Google Scholar]
- Williams D. K.; Wang J.; Papke R. L. Positive allosteric modulators as an approach to nicotinic acetylcholine receptor-targeted therapeutics: advantages and limitations. Biochem. Pharmacol. 2011, 82, 915. 10.1016/j.bcp.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins K. A.; Roy Chengappa K. N.; Karelitz J. L.; Boldry M. C.; Michael V.; Herb T.; Gannon J.; Brar J.; Ford L.; Rassnick S.; Brunzell D. H. Initial Cross-Over Test of A Positive Allosteric Modulator of Alpha-7 Nicotinic Receptors to Aid Cessation in Smokers With Or Without Schizophrenia. Neuropsychopharmacology 2018, 43, 1334. 10.1038/npp.2017.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterer G.; Gallinat J.; Brinkmeyer J.; Musso F.; Kornhuber J.; Thuerauf N.; Rujescu D.; Favis R.; Sun Y.; Franc M. A.; Ouwerkerk-Mahadevan S.; Janssens L.; Timmers M.; Streffer J. R. Allosteric alpha-7 nicotinic receptor modulation and P50 sensory gating in schizophrenia: a proof-of-mechanism study. Neuropharmacology 2013, 64, 197. 10.1016/j.neuropharm.2012.06.040. [DOI] [PubMed] [Google Scholar]
- Gill J. K.; Savolainen M.; Young G. T.; Zwart R.; Sher E.; Millar N. S. Agonist activation of alpha7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 5867. 10.1073/pnas.1017975108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur G. A.; Kulkarni A. R.; Deschamps J. R.; Papke R. L. Expeditious synthesis, enantiomeric resolution, and enantiomer functional characterization of (4-(4-bromophenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide (4BP-TQS): an allosteric agonist-positive allosteric modulator of α7 nicotinic acetylcholine receptors. J. Med. Chem. 2013, 56, 8943. 10.1021/jm401267t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke R. L.; Horenstein N. A.; Kulkarni A. R.; Stokes C.; Corrie L. W.; Maeng C. Y.; Thakur G. A. The activity of GAT107, an allosteric activator and positive modulator of α7 nicotinic acetylcholine receptors (nAChR), is regulated by aromatic amino acids that span the subunit interface. J. Biol. Chem. 2014, 289, 4515. 10.1074/jbc.M113.524603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horenstein N. A.; Papke R. L.; Kulkarni A. R.; Chaturbhuj G. U.; Stokes C.; Manther K.; Thakur G. A. Critical Molecular Determinants of α7 Nicotinic Acetylcholine Receptor Allosteric Activation: Separation of Direct Allosteric Activation and Positive Allosteric Modulation. J. Biol. Chem. 2016, 291, 5049. 10.1074/jbc.M115.692392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagdas D.; Wilkerson J. L.; Kulkarni A.; Toma W.; AlSharari S.; Gul Z.; Lichtman A. H.; Papke R. L.; Thakur G. A.; Damaj M. I. The α7 nicotinic receptor dual allosteric agonist and positive allosteric modulator GAT107 reverses nociception in mouse models of inflammatory and neuropathic pain. Br. J. Pharmacol. 2016, 173, 2506. 10.1111/bph.13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post-Munson D. J.; Pieschl R. L.; Molski T. F.; Graef J. D.; Hendricson A. W.; Knox R. J.; McDonald I. M.; Olson R. E.; Macor J. E.; Weed M. R.; Bristow L. J.; Kiss L.; Ahlijanian M. K.; Herrington J. B-973, a novel piperazine positive allosteric modulator of the α7 nicotinic acetylcholine receptor. Eur. J. Pharmacol. 2017, 799, 16. 10.1016/j.ejphar.2017.01.037. [DOI] [PubMed] [Google Scholar]
- Sato N.; Narita N. Studies on Pyrazines; 38: Acylation of Bromopyrazines and 2-Bromopyridine via Copper-Cocatalytic Stille Reaction. Synthesis 2001, 2001, 1551. 10.1055/s-2001-16082. [DOI] [Google Scholar]
- Nugent T. C.; El-Shazly M. Chiral Amine Synthesis – Recent Developments and Trends for Enamide Reduction, Reductive Amination, and Imine Reduction. Adv. Synth. Catal. 2010, 352, 753. 10.1002/adsc.200900719. [DOI] [Google Scholar]
- Robak M. T.; Herbage M. A.; Ellman J. A. Synthesis and Applications of tert-Butanesulfinamide. Chem. Rev. 2010, 110, 3600. 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]
- Chelucci G.; Baldino S.; Chessa S. Diastereoselective reduction of enantiopure N-p-toluenesulfinyl ketimines derived from pyridyl ketones. Tetrahedron 2006, 62, 619. 10.1016/j.tet.2005.10.011. [DOI] [Google Scholar]
- Chelucci G.; Baldino S.; Chessa S.; Pinna G. A.; Soccolini F. An easy route to optically active 1-substituted-1-pyridyl-methylamines by diastereoselective reduction of enantiopure N-tert-butanesulfinyl ketimines. Tetrahedron: Asymmetry 2006, 17, 3163. 10.1016/j.tetasy.2006.11.026. [DOI] [Google Scholar]
- Chelucci G.; Baldino S.; Solinas R.; Baratta W. Asymmetric synthesis of 1-substituted-1-(pyridin-2-yl)methylamines by diastereoselective reduction of enantiopure N-p-toluenesulfinyl ketimines. Tetrahedron Lett. 2005, 46, 5555. 10.1016/j.tetlet.2005.06.019. [DOI] [Google Scholar]
- Abbott F. V.; Franklin K. B.; Westbrook R. F. The formalin test: scoring properties of the first and second phases of the pain response in rats. Pain 1995, 60, 91. 10.1016/0304-3959(94)00095-V. [DOI] [PubMed] [Google Scholar]
- Davidson E. M.; Carlton S. M. Intraplantar injection of dextrorphan, ketamine or memantine attenuates formalin-induced behaviors. Brain Res. 1998, 785, 136. 10.1016/S0006-8993(97)01396-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.