

Abstract

The multifunctional cytokine TGFβ plays a central role in regulating antitumor immunity. It has been postulated that inhibition of TGFβ signaling in concert with checkpoint blockade will provide improved and durable immune response against tumors. Herein, we describe a novel series of 4-azaindole TGFβ receptor kinase inhibitors with excellent selectivity for TGFβ receptor 1 kinase. The combination of compound 3f and an antimouse-PD-1 antibody demonstrated significantly improved antitumor efficacy compared to either treatment alone in a murine tumor model.
Keywords: Selective kinase inhibitor, immuno-oncology, TGFβ inhibitor, structure-based drug design, water-mediated protein−ligand interaction, antitumor efficacy
The development of immuno-oncology (IO) therapies that reset and strengthen immune response against tumors holds great promise for the treatment of cancer.1−3 Antibody IO therapies targeting immune checkpoint receptors CTLA-4 and PD-1 have provided durable responses in patients with melanoma, nonsmall cell lung carcinoma, and many other types of cancers. However, as tumors have evolved to evade immune surveillance through multiple mechanisms, combination treatment strategies against several immune pathways are likely needed to optimize antitumor immunity and provide durable response in a majority of cancer patients. An increasing number of such treatments has been devised and begun to enter clinical trials, often as combinations with anti-CTLA4 or anti-PD-1 antibodies.
The transforming growth factor β (TGFβ) is a pleiotropic growth factor and cytokine that regulates a wide variety of biological processes, including cell proliferation, differentiation and development, extracellular matrix (ECM) generation, angiogenesis, and immune response.4−6 TGFβ signaling begins with ligand binding to cell-surface serine and threonine kinase receptors TGFβ receptor type I (TGFβRI) and type II (TGFβRII), leading to formation of the heterodimeric TGFβRI and TGFβRII complex, and phosphorylation and activation of TGFβRI. Activated TGFβRI recruits and phosphorylates SMAD2 and SMAD3 proteins, which translocate into the nucleus as a heterocomplex to initiate target gene transcription. Dysregulation of TGFβ signaling is frequently observed in human tumors and plays a vital role in promoting tumor cell growth and differentiation, extracellular matrix (ECM) modulation, and epithelial to mesenchymal transition (EMT). Importantly, in the context of IO, TGFβ has been demonstrated to have a significant role in regulating antitumor immune response. More specifically, TGFβ has a strong inhibitory effect on native T cell proliferation and negatively impacts the functions of dendritic cells, CD8+ T cells, and natural killer (NK) cells while enhancing the activity of immunosuppressive Treg and myeloid-derived suppressor cells (MDSC), thus contributing to a favorable tumor microenvironment for tumor growth and metastasis. TGFβ signaling may also prevent CD8+ T cells from infiltrating the tumor and thus contribute to reduced response and resistance to anti-PD-1-PD-L1 treatment in cancer patients.7−10 Therefore, targeting the TGFβ pathway in combination with anti-PD1 or anti-PD-L1 antibodies may help overcome resistance and produce a more effective antitumor response.
A number of pharmacological strategies to target the TGFβ pathway have been reported,11−13 including antisense oligonucleotides, vaccines, ligand traps, monoclonal antibodies, and small molecule receptor kinase inhibitors.14−21 Of these, small molecule TGFβRI kinase inhibitor Galunisertib (LY-2157299) is under evaluation in combination with anti-PD1 (PD-L1) antibodies in clinical trials for a variety of cancers.22 Notably, it was demonstrated that combination of LY-2157299 with checkpoint blockade provided durable, complete responses in mouse tumor models.21 Furthermore, while continuous long-term dosing of LY-2157299 caused target-related cardiovascular toxicities in animals, an intermittent dosing schedule (2 weeks on/2 weeks off during a 28-day cycle) provided a therapeutic window and allows for the compound to be safely administered to humans.23
Due to the potential clinical significance of TGFβ biology, we initiated a program to identify novel TGFβ receptor kinase inhibitors with an optimized profile to combine with other IO agents for the treatment of cancer. We decided at the outset to focus on developing selective TGFβRI kinase inhibitor because it was demonstrated that inhibition of TGFβRI by LY-2157299 is sufficient to inhibit phosphorylation and signaling of downstream SMAD proteins. In this Letter, we describe the synthesis and SAR of a novel series of 4-azaindoles as TGFβ inhibitors and the identification of compounds with excellent kinase selectivity and PK properties that are efficacious in a murine tumor model when combined with anti-PD1 antibody.24
A high throughput screening of a focused 12,000 small-molecule Bristol-Myers Squibb kinase deck using a homogeneous time-resolved fluorescence (HTRF) assay25 successfully identified a novel pyrrololactam chemotype as potent inhibitors of TGFβRI and RII kinases. A high resolution crystal structure of a representative compound 1 bound to the ATP binding site of TGFβRI kinase domain was obtained and served as the basis for compound design (Figure 1). As shown in the structure, compound 1 forms hydrogen bonds directly from the pyridinylacetamide group to His283 within the hinge region. Similar to the binding mode of other reported TGFβRI kinase inhibitors, compound 1 also forms a water-mediated hydrogen bond network between the fluoropyridine nitrogen atom, the side chains of Tyr249 and Glu245, and the backbone of Asp351. In addition, a hydrogen bonding interaction is observed between the lactam amide and Lys232 and Asp351 residues. From molecular modeling and published SAR for other TGFβ kinase inhibitors, we understood that the binding interaction between the protein and the fluoropyridine through the conserved water molecule is likely vital for cellular potency.22,25,26 We hypothesized that the binding site has some level of structural flexibility that may allow for various ring replacements and substitutions while maintaining these interactions. Based on this analysis, we examined several heterocyclic ring replacements of the pyrrololactam and were encouraged that a more synthetically tractable and cell permeable 4-azaindole scaffold (i.e., 2a) demonstrated comparable activities to 1 in both biochemical (TGFβRI IC50 = 22 nM, RII IC50 = 7 nM) and functional SMAD nuclear translocation assays (IC50 = 1.8 μM).27 Additionally, 2a displayed significantly improved kinase selectivity against a panel of 240 off-target kinases compared to 1 (Table S2).
Figure 1.
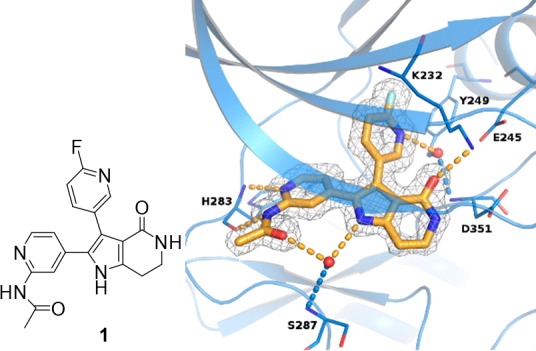
X-ray crystal structure of compound 1 bound to the TGFβRI kinase domain (T204D mutant; 1.58 A). PDB accession code is 5QIK.
With compound 2a as the new starting point for SAR exploration, a library of 3-pyridyl azaindoles was prepared to explore the hydrophobic back pocket region where the pyridine ring resides. As expected, a variety of nonpolar substitutions on the pyridine ring were tolerated and retained activity in TGFβRI biochemical assay (Table 1), but there were several unexpected observations. Replacement of the fluorine with a methoxy group (2b) led to an improvement in both TGFβRI biochemical potency and cellular activity, but reduced potency against TGFβRII. In contrast to its pyridine regioisomer 2a, compound 3a unexpectedly displayed a substantial selectivity for the RI isoform (>100 fold vs RII). Introduction of slightly bulkier substituents on the 2 or 3 position of the pyridine ring (3b–3i) resulted in further enhancement of RI-selectivity (>1000-fold). Excellent cellular activities were preserved in all cases despite significant loss of RII biochemical potency, suggesting that inhibition of the RII receptor in addition to the RI is not required for cellular activity.28
Table 1. Biochemical and Cellular Activities of 3-Pyridyl Substituted 4-Azaindolesa.
| compd | R1 | TGFβRI IC50 (μM) | TGFβRII IC50 (μM) | MINK SMAD translocation IC50 (μM) |
|---|---|---|---|---|
| 2a | F | 0.022 | 0.007 | 1.8 |
| 2b | OMe | 0.003 | 0.04 | 0.31 |
| 3a | 3-F | 0.003 | 0.39 | 0.32 |
| 3b | 3-Cl | 0.002 | 14 | 0.90 |
| 3c | 3-Me | 0.005 | >15 | NA |
| 3d | 3-OCF2H | 0.011 | 14 | 1.2 |
| 3e | 3-OMe | 0.006 | >15 | 0.79 |
| 3f | 3-OCD3 | 0.002 | 7.6 | 0.46 |
| 3g | 2-Me | 0.002 | 0.73 | 0.76 |
| 3h | 2-CF2H | 0.001 | 4.3 | 0.26 |
| 3i | 2-CF3 | 0.002 | >15 | 1.4 |
Standard deviation and number of replicates are provided in Table S3.
Based on the overlay of the hinge binding pockets of the two TGFβ isoforms, we hypothesized that the slightly larger gatekeeper pocket of the R1 isoform due to presence of several smaller residues29 may contribute to the selectivity of compounds with bulkier pyridine substitution. However, it is not clear which interactions are responsible for the high level of RI-selectivity of pyridine regioisomer 3 vs 2. We have recently published several mutated constructs TGFβRII that are amenable to crystallization.30 Application of these mutant constructs as well as the known T204D TGFβRI mutant construct for compounds 2b and 3e led to several high resolution crystal structures (Figure 2). Examination of the structures revealed that compound 2b binds in the expected manner to both TGFβRI and TGFβRII kinase domains, with the pyridine acetamide group interacting with the hinge, the azaindole with the conserved lysine, and the nitrogen of the 3-pyridyl group forming hydrogen bond network through the conserved water molecule to the protein. Compound 3e binds to TGFβRI in an almost identical manner. However, because the 3-pyridyl nitrogen in compound 3e is farther away from the conserved water (HOH1), the water-mediated interaction occurs through a second water molecule (HOH2) that is located next to Ala350. The position of this second water molecule would be occupied by the side chain of Cys396 in TGFβRII, and presumably, this amino acid difference confers the selectivity of compound 3e for TGFβRI over TGFβRII.
Figure 2.

X-ray crystal structures of (a) compound 2b bound to the TGFβRI kinase domain (T204D mutant); (b) compound 2b bound to the TGFβRII kinase domain (E431A, R433A, E485A, K488A, R493A, R495A mutant); (c) compound 3e bound to the TGFβRI kinase domain (T204D mutant). PDB accession codes are 5QIL, 5QIM, and 5QIN, respectively.
With the goal of identifying a selective and orally bioavailable compound for in vivo evaluation, several potent TGFβRI-selective compounds in Table 1 (3e–3h) were further evaluated for their metabolic stability, CYP450 profiles, binding selectivity in the Bristol-Myers Squibb panel of 240 kinases, and activity in additional cellular assays (Table S2). Compound 3e benefited from excellent metabolic stability in mouse, rat, and human liver microsomal stability assays but suffered from potent CYP450 inhibition (3A4, IC50 = 4.5 μM). Fortunately, the deuterated analog 3f and a 2-difluoromethyl substituted analog 3h both demonstrated improved CYP450 profile (IC50 > 20 μM, Table 2) while maintaining excellent metabolic stability and kinase selectivity. Both compounds also potently inhibited phosphorylation of SMAD protein in normal human lung fibroblast (NHLF) cellular assay (IC50 = 0.45 μM and 0.068 μM for 3f and 3h, respectively) and primary human T cell assay (IC50 = 0.017 μM and 0.094 μM for 3f and 3h, respectively). Furthermore, both compounds inhibited TGFβ-mediated induction of regulatory T cell (Treg) by downregulation of FOXP3 expression and a repression of CD25 expression (IC50 = 0.11 μM and 0.05 μM), indicating that the azaindole inhibitors may be valuable to overcome Treg-mediated immune suppression and improve antitumor immunity. Although compound 3f displayed slightly less potent cellular activities, it benefited from lower potential for QT prolongation based on hERG channel patch clamp assay (18% and 73% inhibition for 3f and 3h at 10 μM, respectively). Furthermore, the mouse PK profile of compound 3f was more promising, providing higher exposure at one-fifth of the dose and a flatter oral PK curve compared to 3h (Figure 3). The PK profile of 3f supported a once daily dose schedule, while 3h likely would need to be administrated twice daily at a higher dose to achieve the desired PD effect.
Table 2. Profiling of Compound 3h and 3f.
| assay | 3f | 3h |
|---|---|---|
| TGFβRI IC50 (nM) | 1.6 ± 0.5 | 0.8 ± 0.1 |
| TGFβRII IC50 (μM) | 7.6 ± 1.3 | 4.3 ± 2.3 |
| MINK TSMAD IC50 (μM) | 0.46 ± 0.02 | 0.26 ± 0.16 |
| NHLF TSMAD IC50 (μM) | 0.45 ± 0.12 | 0.068 ± 0.03 |
| Hu T-cell PSMAD3 IC50 (μM) | 0.017 ± 0.01 | 0.094 ± 0.02 |
| Hu Treg FoxP3+ IC50 (μM) | 0.11 | 0.05 |
| Met Stab (h, r, m; % rem) | 96, 89, 74 | 60, 85, 66 |
| CYP inhibition (1A2, 2C9, 2C19, 2D6, 3A4, 2C8) | >20 μM | >20 μM |
| hERG PC % inh at 3, 10 μM | 3.9, 18 | 42, 73 |
| protein binding (h, r, m; % free) | 24, 25, 12 | 9, 18, 5 |
| kinase selectivity number of tested kinases with IC50 < 0.1 μM | 1 (ALK4) | 2 (ALK4, p38α) |
| number of tested kinases with IC50 < 1 μM | 4 | 13 |
| number of kinases tested | 244 | 246 |
| PK parameters in mice [dose (mg/kg), Cmax (μM) Tmax (h), AUC0–24 (μM·h)] | 10, 5.3, 1, 19.5 | 50, 7.4, 1, 14.1 |
Figure 3.
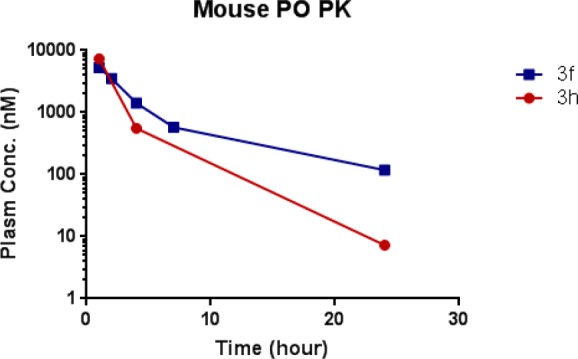
Plasma levels and pharmacokinetic parameters of compounds 3f and 3h following oral administration to mice (3f at 10 mg/kg, 3h at 50 mg/kg).
A representative azaindole synthesis is illustrated for compound 3f in Scheme 1. Sonogashira coupling between pyridinylacetamide 4 and bromopyridine 5 provided the intermediate alkyne 6. Acylation with trifluoroacetic anhydride was followed by a modified Cacchi palladium-mediated domino alkylation-cyclization reaction31 to yield the final compound in a short sequence. It is worth noting that the Cacchi reaction tolerated a variety pyridine substitutions and allowed both pyridine isomers to be prepared with high yields (2 and 3).
Scheme 1.

Reagents and conditions: (a) Pd(PPh3)2Cl2, CuI, Et3N, DMF, 70 °C, 80%, (b) trifluoroacetic anhydride, Et3N, DCM, 0 °C, 71%, (c) Pd(PPh3)4, Cs2CO3, acetonitrile, 110 °C, 44%.
On the basis of its attractive in vitro profile and desirable PK properties, compound 3f was advanced to a preclinical efficacy study to determine if TGFβ inhibition enhances the antitumor activity of PD-1 blockade. The MC38 syngeneic mouse colon adenocarcinoma tumor model, which is modestly sensitive to anti-PD-1 antibody treatment, was used to assess the effect of dose escalation of 3f alone or in combination with a surrogate antimouse-PD-1 antibody.32 As shown in Figure 4, monotherapy of 3f provided minimal tumor growth inhibition at all three dose levels tested. Administration of anti-PD-1 antibody alone was as expected partially efficacious. Combination of 3f with anti-PD-1 antibody, however, provided robust dose-dependent antitumor activity and, at the 25 mg/kg and 50 mg/kg doses, significantly greater efficacy compared to anti-PD-1 treatment alone. Furthermore, the antitumor immunity was durable with majority of the responding mice in the combination groups staying tumor-free for at least 45 days.33 No significant weight loss (>5%) was observed for any of the treatment groups.
Figure 4.
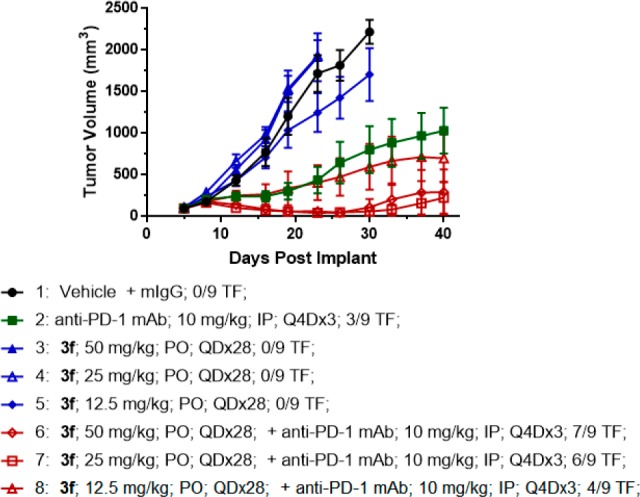
Antitumor activity of 3f alone and in combination with anti-PD-1 antibody in the MC38 syngeneic tumor model. Groups of nine C57/BL6 mice were subcutaneously injected with 1 × 106 MC38 cells. After tumors were measured on day 6, mice were randomized and then treated with the designated compounds. Compound 3f was administered orally (PO) every day for 28 days. Anti-PD-1 antibody was administered intraperitoneally (IP) every 4 days for three doses. Tumor volumes were measured twice weekly. The number of mice per group that stays tumor-free (TF) for at least 10 tumor volume doubling time (45 days) is shown in the legend for each group.
In conclusion, novel 3-pyridyl substituted 4-azaindoles were identified as potent inhibitors of TGFβRI kinase. Optimization of the SAR led to the identification of a potent and orally bioavailable analog 3f. Oral administration of 3f synergized with anti-PD-1 antibody and provided durable antitumor activity in MC38 tumor model with no observed toxicity. Combined, these results supported further advancement of compound 3f into preclinical toxicology studies.
Acknowledgments
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. This research used resources at the Industrial Macromolecular Crystallography Association Collaborative Access Team (IMCA-CAT) beamline 17-ID, supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute.
Glossary
ABBREVIATIONS
- TGFβ
transforming growth factor β
- IO
immuno-oncology
- CTLA-4
cytotoxic T-lymphocyte associated protein 4
- PD-1
programmed death 1
- PD-L1
programmed death ligand 1
- SMAD
mothers against decapentaplegic homologue
- ECM
extracellular matrix
- EMT
epithelial to mesenchymal transition
- NK cell
natural killer cell
- MDSC
myeloid-derived suppressor cells
- HTRF
homogeneous time-resolved fluorescence
- SAR
structure–activity relationship
- CYP450
cytochrome P450
- Treg
regulatory T cell
- FOXP3
foxhead box P3
- met stab
metabolic stability
- hERG PC
human ether-a-go-go-related gene patch clamp assay
- PK
pharmacokinetics
- PO
orally
- QD
once a day
- IP
intraperitoneally
- TF
tumor free
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00357.
Experimental procedures for the synthesis of all new compounds; procedures for TGFβ binding assays, MINK assay, pharmacokinetic experiments, and in vivo efficacy experiments (PDF)
Accession Codes
Coordinates and data were deposited in the PDB with following IDs: TGFβRI with compounds 1 (5QIK), 2b (5QIL), and 3e (5QIM), and TGFβRII with compound 2b (5QIN).
Author Contributions
† These authors contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Lonberg N.; Korman A. J. Masterful Antibodies: Checkpoint blockade. Cancer Immunol. Res. 2017, 5, 275–281. 10.1158/2326-6066.CIR-17-0057. [DOI] [PubMed] [Google Scholar]
- Mahoney K. M.; Rennert P. D.; Freeman G. J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discovery 2015, 14, 561–584. 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- Adams J. L.; Smothers J.; Srinivasan R.; Hoos A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discovery 2015, 14, 603–622. 10.1038/nrd4596. [DOI] [PubMed] [Google Scholar]
- Massague J. How cells read TGF-β signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- Akhurst R. J.; Hata A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discovery 2012, 11, 790–811. 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup M.; Novitskiy S.; Moses H. L. The roles of TGF β in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. 10.1038/nrc3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu X.; Mahoney K. M.; Freeman G. J. Learning from PD-1 resistance: new combination strategies. Trends Mol. Med. 2016, 22, 448–451. 10.1016/j.molmed.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W.; et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 2016, 165, 35–44. 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauriello D. V. F.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- Colak S.; Dijke P. T. Targeting TGF-β signalling in cancer. Trends in Cancer 2017, 3, 56–71. 10.1016/j.trecan.2016.11.008. [DOI] [PubMed] [Google Scholar]
- de Gramont A. D.; Faivre S.; Raymond E. Novel TGF-β inhibitors ready for prime time in onco-immunology. Oncoimmunolo 2017, 6, e1257453. 10.1080/2162402X.2016.1257453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly E. C.; Freimuth J.; Akhurst R. J. Complexities of TGF-β targeted cancer therapy. Int. J. Biol. Sci. 2012, 8, 964–978. 10.7150/ijbs.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DaCosta Byfield S. D.; Major C.; Laping N. J.; Roberts A. B. SB-505124 is a selective inhibitor of transforming growth factor-β type I receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2004, 65, 744–752. 10.1124/mol.65.3.744. [DOI] [PubMed] [Google Scholar]
- Grygielko E. T.; Martin W. M.; Tweed C.; Thornton P.; Harling J.; Brooks D. P.; Laping N. J. Inhibition of gene markers of fibrosis with a novel inhibitor of transforming growth factor-β type I receptor kinase in puromycin-induced nephritis. J. Pharmacol. Exp. Ther. 2005, 313, 943–951. 10.1124/jpet.104.082099. [DOI] [PubMed] [Google Scholar]
- Gouville A. C.; Boullay V.; Krysa G.; Pilot J.; Brusq J. M.; Loriolle F.; Gauthier J. M.; Papworth S. A.; Laroze A.; Gellibert F.; Huet S. Inhibition of TGF-β signaling by an ALK5 inhibitor protects rats from dimethylnitrosamine-induced liver fibrosis. Br. J. Pharmacol. 2005, 145, 166–177. 10.1038/sj.bjp.0706172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge R.; Rajeev V.; Ray P.; Lattime E.; Rittling S.; Medicherla S.; Protter A.; Murphy A.; Chakravarty J.; Dugar S.; Schreiner G.; Barnard N.; Reiss M. Inhibition of growth and metastasis of mouse mammary carcinoma by selective inhibitor of transforming growth factor-β type I receptor kinase in vivo. Clin. Cancer Res. 2006, 12, 4315–4330. 10.1158/1078-0432.CCR-06-0162. [DOI] [PubMed] [Google Scholar]
- Kapoun A. M.; Gaspar N. J.; Wang Y.; Damm D.; Liu Y.-W.; O’Young G.; Quon D.; Lam A.; Munson K.; Tran T.-T.; Ma J. Y.; Murphy A.; Dugar S.; Chakravarty S.; Protter A. A.; Wen F.-Q.; Liu X.; Rennard S. I.; Higgins L. S. Transforming growth factor-β receptor type 1 (TGFβRI) kinase activity but not p38 activation is required for TGFβRI-induced myofibroblast differentiation and profibrotic gene expression. Mol. Pharmacol. 2006, 70, 518–531. 10.1124/mol.105.021600. [DOI] [PubMed] [Google Scholar]
- Bueno L.; de Alwis D. P.; Pitou C.; Yingling J.; Lahn M.; Glatt S.; Trocóniz I. F. Semi-mechanistic modelling of the tumour growth inhibitory effects of LY2157299, a new type I receptor TGF-β kinase antagonist, in mice. Eur. J. Cancer 2008, 44, 142–150. 10.1016/j.ejca.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Jin C. H.; Krishnaiah M.; Sreenu D.; Subrahmanyam V. B.; Rao K. S.; Lee H. J.; Park S. J.; Park H. J.; Lee K.; Sheen Y. Y.; Kim D. K. Discovery of N-((4-([1,2,4]triazolo[1,5-a]pyridin-6-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)methyl)-2-fluoroaniline(EW-7197): a highly potent, selective, and orally bioavailable inhibitor of TGF-β type I receptor kinase as cancer immunotherapeutic /antifibrotic agent. J. Med. Chem. 2014, 57, 4213–4238. 10.1021/jm500115w. [DOI] [PubMed] [Google Scholar]
- Holmgaard R. B.; Schaer D. A.; Li Y.; Castaneda S. P.; Murphy M. Y.; Xu X.; Inigo I.; Dobkin J.; Manro J. R.; Iversen P. W.; Surguladze D.; Hall G. E.; Novosiadly R. D.; Benhadji K. A.; Plowman G. D.; Kalos M.; Driscoll K. E. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immuno. Ther. Cancer 2018, 6, 47. 10.1186/s40425-018-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://clinicaltrials.gov/ct2/show/NCT02423343
- Herbertz S.; Sawyer J. S.; Stauber A. J.; Gueorguieva I.; Driscoll K. E.; Estrem S. T.; Cleverly A. L.; Desaiah D.; Guba S. C.; Benhadji K. A.; Slapak C. A.; Lahn M. M. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des., Dev. Ther. 2015, 9, 4479–4499. 10.2147/DDDT.S86621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink B. E.; Zhao Y.; Borzilleri R. M.; Zhang L.; Kim K. S.; Kamau M. G.; Tebben A. J.; Zhang Y.; Donnell A. F.. TGFβR antagonists. Granted patent US 9708316, 2017.
- Harikrishnan L. S.; Warrier J.; Tebben A. J.; Tonukunuru G.; Maddur S. R.; Baligar V.; Mannoori R.; Seshadri B.; Rahaman H.; Dikundawr A.; Fink B. E.; Fargnoli J.; Fereshteh M.; Fan Y.; Lippy J.; Ho C.; Wautlet B.; Borzilleri R. M. Heterobicyclic inhibitors of transforming growth factor beta receptor I. Bioorg. Med. Chem. 2018, 26, 1026–1034. 10.1016/j.bmc.2018.01.014. [DOI] [PubMed] [Google Scholar]
- Goldberg F. W.; Ward R. A.; Powell S. J.; Debreczeni J. E.; Norman R. A.; Roberts N. J.; Dishington A. P.; Gingell H. J.; Wickson K. F.; Roberts A. L. Rapid generation of a high quality lead for tranforming growth factor-β (TGF-β) type I receptor (ALK5). J. Med. Chem. 2009, 52, 7901–7905. 10.1021/jm900807w. [DOI] [PubMed] [Google Scholar]
- Permeability for compound 1 and 2a are 8 and 289 nm/s, respectively (PAMPA, pH 7.4). PAMPA data for additional analogs are listed in Table S3.
- Li H. Y.; Wang Y.; Heap C. R.; King C. H.; Mundla S. R.; Voss M.; Clawson D. K.; Yan L.; Campbell R. M.; Anderson B. D.; Wagner J. R.; Britt K.; Lu K. X.; McMillen W. T.; Yingling J. M. Dihydropyrrolopyrazole transforming growth factor-beta type I receptor kinase domain inhibitors: a novel benzimidazole series with selectivity versus transforming growth factor-beta type II receptor kinase and mixed lineage kinase-7. J. Med. Chem. 2006, 49, 2138–2142. 10.1021/jm058209g. [DOI] [PubMed] [Google Scholar]
- Key changes in gatekeepter pocket residues: Ser280, Ala350, and Leu352 in TGFβR1 vs Thr325, Cys396 and Phe398 in TGFβR2.
- Tebben A. J.; Ruzanov M.; Gao M.; Xie D.; Kiefer S. E.; Yan C.; Newitt J. A.; Zhang L.; Kim K.; Lu H.; Kopcho L. M.; Sheriff S. Crystal structures of apo and inhibitor-bound TGFβR2 kinase domain: insights into TGFβR isoform selectivity. Acta Crystallographica Section D Structural Biology 2016, 72, 658–674. 10.1107/S2059798316003624. [DOI] [PubMed] [Google Scholar]
- Arcadi A.; Cacchi S.; Marinelli F. A Versatile approach to 2, 3-disubstituted indoles through the palladium-catalyzed cyclization of o-alkynyltrifluoroacetanilides with vinyl triflates and aryl halides. Tetrahedron Lett. 1992, 33, 3915–3918. 10.1016/S0040-4039(00)74818-0. [DOI] [Google Scholar]
- Selby M. J.; Engelhardt J. J.; Johnston R. J.; Lu L.; Han M.; Thudium K.; Yao D.; Quigley M.; Valle J.; Wang C.; Chen B.; Cardarelli P. M.; Blanset D.; Korman A. J. Preclinical development of ipilimumab and nivolumab combination immunotherapy: mouse tumor models, in vitro functional studies, and cynomolgus macaque toxicology. PLoS One 2016, 11, e0167251. 10.1371/journal.pone.0167251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- These results compare favorably to those of galunisertib, where at least a 75 mg/kg dose on a more frequent b.i.d. schedule was required to achieve efficacy in syngeneic mouse tumor models (see ref (21)).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.