

Abstract
In this Viewpoint, we discuss the current progress in applications of machine learning (ML) and artificial intelligence (AI) to meet the challenges of computational drug discovery. We identify several areas where existing methods have the potential to accelerate pharmaceutical research and disrupt more traditional approaches.
Keywords: Deep learning, artificial intelligence, drug discovery, machine learning, molecular potentials, force field, neural network
“Computers are useless. They can only give you answers.” Pablo Picasso
Computational methods play a key role in the design of therapeutically important molecules for modern drug development. These methods can be broadly classified as structure-based or ligand-based. Structure-based methods require knowledge of the structure of both the target and ligand. They include molecular dynamics, protein–ligand docking, and methods for calculating the free energy of binding. Ligand-based methods use only information about the ligand to predict the biological response depending on historical data about known active and inactive ligands. These methods typically include quantitative structure–activity relationships, activity cliffs analysis, and similarity search.
In terms of energy evaluation for molecular systems, both types of method still rely on force fields or empirical scoring functions. Parametrizing accurate and transferable force fields is a very difficult task especially if one needs to screen millions of possible ligands. This parametrization requires embedding very high-dimensional quantum physics behavior into a small number of parameters using a very simple analytic functional form. For this reason, a “zoo” of force fields has been developed over the last quarter century for applications in various domains, such as nucleic acids, proteins, carbohydrates, various materials, and small drug-like molecules. Understanding exactly where these system-specific force fields work, and where they fail, is an extremely challenging task. This fact boils down to the following two simple statements: (1) force-fields are fast, but can perform poorly outside of their fitting set, and (2) quantum mechanical methods can be extremely accurate, but at a prohibitive computational cost. This age-old gap between the speed of classical force-fields and the accuracy of quantum mechanical approaches has plagued academic and industrial computational chemists for generations.
We opened this Viewpoint with a quote by Pablo Picasso because it promotes deep thought about how computers have impacted our world. It could be interpreted in many ways. In computational chemistry, much of the effort has focused on methods for producing answers. Traditionally, computers have not been creative, that is, they can only do what humans tell them to do. Perhaps, by saying “they only give answers”, Picasso implies that for computers to truly have an impact, then they should be able do more than we expect.
Modern artificial intelligence (AI) has the potential to significantly enhance the role of computers and computational methods in science and engineering.1 The World Economic Forum refers to the combination of big data and AI as both the fourth paradigm of science and the fourth industrial revolution. With machines able to learn and offer solutions to some of the most complex chemistry problems, drug discovery is well positioned to be the next frontier for a potential breakthrough.
Learning Quantum Mechanical Properties
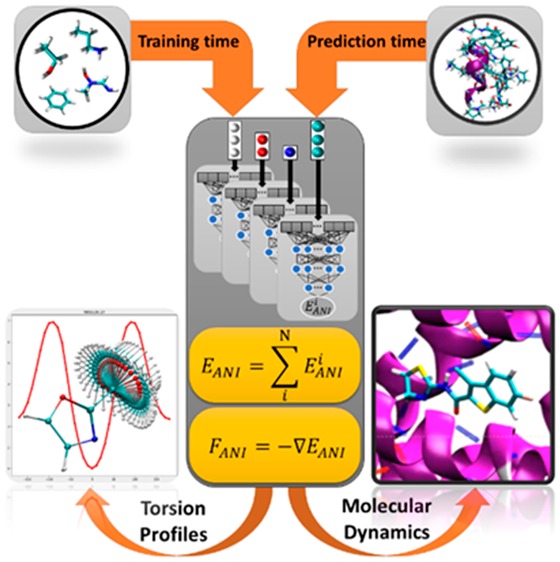
A lot of progress has been made in the development of atomistic potentials (and other properties) using machine learning (ML) with methods such as kernel ridge regression (KRR), neural networks (NN), and Gaussian process regression (GPR). The low numerical complexity and high accuracy of machine learning algorithms make them very attractive as a pragmatic substitute for ab initio and DFT methods. Due to their remarkable ability to find complex relationships among data, in most cases these “machine learned” models outperform more physically constrained approximations like force fields and semiempirical QM. However, these models are critically dependent on the quality and quantity of data used in their training. Neural networks, for example, are highly efficient and effective at modeling reference training data, due to their flexible functional form. This flexibility comes at a cost: a vast amount of reference data is required to properly train these models (especially general-purpose models) in a way that provides accurate predictions over a wide range of inputs.
In our recent work, we published the ANI-1 potential.2 ANI-1 is the first example of a universal (general-purpose) NN molecular potential, where we show that training to a large data set of 22 million small molecule conformations3 yields a potential capable of predicting energies on much larger systems and on systems wildly different from those in the training set. We show its applicability to systems with up to 70 atoms, including well-known drug molecules. ANI-1 shows exceptional predictive power on an external molecular size extensibility test set, with RMSE versus DFT relative energies as low as 0.57 kcal/mol when considering molecular conformations that are relevant at room temperature. More recently, this concept of training an ML model with small fragments of organic molecules to predict on larger systems has been verified independently. Gastegger et al.6 showed results for large organic systems that were fragmented into smaller molecules, and DFT data was generated on the fly for training. These and many other studies back up the argument that information about the physics of large systems can be learned from data sets of small molecules.
“Smart” Data Sampling and Feedback Loop
In drug discovery, projects operate via feedback loops, the process known as the design, make, test, and analyze (DMTA) cycle. This feedback loop is crucial to adapt, recover, and learn from mistakes. Traditional computational methods are static. A particular approximation is predefined; all data is generated up front prior to fitting. They are not concerned with issues of biased or redundant data.
In contrast, ML methods combined with active learning (AL) are a natural analogy to the concept of feedback loops. AL is the process of intelligently searching the problem space in an iterative way to generate the minimal data set required to achieve the same, or better, results as “random” static learning. Recently, we published improvements to ANI-1 based on AL.7 We used an ensemble of models (a process known as query by committee) to drive diverse sampling methods and automatically select new molecules and conformations to add to the training set. This process resulted in the ANI-1x potential. ANI-1x was shown to provide a very high-level of potential energy and force accuracy on large drug-like molecules and even protein sized systems from the COMP6 benchmark. The level of accuracy provided by an ANI model trained on the ANI-1x data set is shown to be better than MP2/6-311+G** and the current gold standard small molecule force field (OPLS3) on the Genentech benchmark4 of small organic molecule torsion profiles (Figure 1a). In some cases, like molecule 36 from the Genentech benchmark (Figure 1b), force fields have a hard time reproducing the shape of the dihedral potential and even produce artificial minima. Both DFT and ANI-1x closely mimic the high level coupled-cluster result. This result might be fortuitous, but it is clear from Figure 1a that ANI-1x performs better over the entire torsion benchmark, yielding credence to the claim that ANI-1x is more accurate than the best small molecule force fields, while being several orders of magnitude cheaper than advanced QM methods. Also, it should be considered that the accuracy of the ANI-1x potential is tied to that of the DFT functional used to generate training data. It has been noted in recent literature that neural network type potentials will not be able to do better than DFT because of large data requirements and the relatively low cost of DFT compared to very accurate ab initio methods. Such claims are likely true if neural network-based potentials continued to require the same amount of data from accurate ab initio methods as was required from DFT methods.
Figure 1.

Examples of ANI potential relaxed torsion scan accuracy vs various levels of theory on the Sellers et al. torsion scan benchmark.4 (a) Accuracy of ANI-1x and ANI-1ccx (as presented in the ANI-1ccx5 work) compared against results from the complete torsion benchmark for the molecules containing the chemical elements C, H, N, and O. (b) Select torsional scan (represented by bold bonds) where OPLS3 experiences a large error in the barrier height and a false minima, but DFT and the DFT trained ANI-1x potential both perform well vs CCSD(T)/CBS. (c) Select torsion scan where DFT and the DFT trained ANI-1x potential perform with similar accuracy, but the transfer learning-based ANI-1ccx potential greatly outperforms the DFT-based models.
There are various techniques from the field of ML to reduce data requirements. Some ML-based methods (more specifically neural networks) can take advantage of information from multiple sources. The key concept is to train a model to some large data set of medium accuracy, and then retrain the model to less data from a more accurate and difficult to obtain data source. This process is called transfer learning and relies on the assumption that the less accurate data source contains some information that makes it easier to learn correlations in the smaller and more accurate data set. For ML potentials, we recently employed this process by taking a deep learning model that was pretrained to medium-fidelity DFT, holding some number of parameters in the model constant, and then retraining the remaining parameters to a much smaller, high-fidelity CCSD(T)/CBS accurate data set.5 This resulted in the ANI-1ccx potential. The ANI-1ccx potential is shown to be an attractive alternative to density functional theory approaches and standard force fields for conformational searches (Figure 1a), molecular dynamics, and the calculation of reaction energies. The reaction energy results show that the transfer learning-based ANI-1ccx outperforms (Figure 2) DFT on test cases, including cases where DFT fails to capture reaction thermochemistry. As part of this research, we develop a new extrapolation scheme to CCSD(T)/CBS for high-throughput and very accurate QM data generation. The availability of such high-quality QM reference data allowed us to use these transfer learning techniques to build the chemically accurate and universal ANI-1ccx potential.
Figure 2.

Comparison of the original ANI-1x reference DFT (ωB97X-D3/6-31g*), ANI-1x, ANI-1ccx, and our high-level reference CCSD(T)*/CBS on the HC7/11 and ISOL6 benchmarks. (a) Individual HC7/11 reaction and isomerization energy differences from reference calculations. ISOL6 (C, H, N, and O atoms only) isomerization energy differences from reference. The top panel provides the HC7 reactions numbered 1, 2, 6, and 7 and bottom panel shows the ISOL6 reactions numbered 1–5. This figure appeared in Smith et al.5
Generative Methods and de Novo Molecular Design
The field of de novo or inverse molecular design has benefited tremendously from recent advances in ML. Within a very short time, numerous exciting approaches have been suggested. Notably, methods like recurrent neural networks (RNN), generative adversarial networks (GANs), and autoencoders were adapted to problems of rational design of organic and inorganic materials, synthesis planning, and device optimization.8
Popova et al. proposed a method called ReLeaSE for generating chemical compounds and focused chemical libraries with desired physical, chemical, and/or bioactivity properties that are based on deep reinforcement learning (RL).9 The general workflow for the ReLeaSE method includes generative (G) and predictive (P) neural networks. In this system, the generative model G is used to produce novel chemically feasible molecules, that is, it plays the role of an agent, whereas the predictive model P plays the role of a critic. P estimates the agent’s behavior by assigning a numerical reward (or penalty) to every generated molecule. The reward is a flexible function of the numerical property/activity of a generated molecule, and the generative model is trained to maximize the expected reward.
Synthesis Planning
Recently, ML-based methods have been employed in breakthroughs involving synthesis planning. Chematica, a computer program, was used to plan a complete syntheses of medicinally important molecules without human help.10 Chematica by Grzybowski et al. implemented about 50,000 rules of synthesis into decision trees. The reaction rules are combined into graphs connecting millions of possible molecules and different synthetic routes from which the viable synthetic pathways are extracted.
Segler et al. demonstrated that retrosynthetic routes can be discovered using Monte Carlo tree search and symbolic AI without using human expert rules.11 This neural network was trained on essentially all reactions published in organic chemistry. In a double-blind test, synthetic chemists considered computer-generated routes to be on par with methods reported in the literature.
Conclusions
The continued improvement of ML methods in chemistry, which compete with standard approaches or expert skill, are poised to become a force for change in modern computational medicinal chemistry. Machine learning potentials capable of carrying out high-throughput calculations in millisecond time scales with DFT accuracy or better will accelerate ligand conformational searching and help to avoid false positives and false negatives. Further, continued progress toward bulk phase protein/ligand simulation will allow QM accurate free energy of binding calculations that can be performed with minimal human intervention for parametrization. De novo molecular design can potentially supply accurate predictions of lead compounds to target for simulation, effectively shrinking the search space for high-throughput screening applications. Automated synthesis planning promises to provide better, higher yield, and more cost-effective synthetic routes. The combination of the methods discussed in this opinion, and other methods not discussed, such as robotic synthesis,12 could eventually provide a fully automated drug discovery pipeline driven by AI.
There are various routes researchers could take to bring these game-changing tools into reality. Research in the development of ML-based potentials should move from a benchmark/model centric culture to a culture of improved application. This can be done in a similar way that modern force-field developers have done for decades, by aiming to reproduce experimental observables. For maximum impact, researchers should also aim to make their codes more accessible to the scientific community, while not sacrificing the speed and accuracy gained through these ground-breaking methods. Further, humanity’s vast knowledge of physics should be utilized to improve model performance and further constrain possible models while also not sacrificing speed and accuracy.
In Pablo Picasso’s quote, he could be saying that it is not the answers that are important. He might be saying that answers are easy to supply once you know the question. It is just a matter of looking for the right answer. However, coming up with new and meaningful questions is hard. Then, asking those questions pushes the frontiers of our creativity and understanding of the world to the limit. Humans have always enjoyed reasoning and creative capabilities that far exceed those of machines. Human intuition currently drives methods and the experiment design. However, our experience, performance, and the pressure to publish tend to favor inclusion of “successful” points and often to forget “failed” ones. As AI erodes the barrier of human capabilities, the chemical sciences must not only adopt these new and powerful AI driven tools but also push the frontier of AI to mimic and then surpass the chemical intuition and decision making of expert scientists. Therefore, the immediate frontiers lie in (i) elevating ML from generating data models to generating human-understandable explanations and conclusions and (ii) enabling autonomous reasoning about these outcomes and developing an actionable research plan. These accomplishments are critical for conducting basic scientific research where knowledge and understanding take precedence over quantitative results. So perhaps, one day, machines can not only give you answers, but also ask the right questions.
Acknowledgments
J.S.S. acknowledges the Center for Nonlinear Studies (CNLS) at Los Alamos National Laboratory (LANL) and the U.S. Department of Energy (DOE) through the LANL LDRD Program for support. A.R. acknowledges NSF CHE-1802831. O.I. acknowledges support from DOD-ONR (N00014-16-1-2311) and National Science Foundation (NSF CHE-1802789) award.
Views expressed in this editorial are those of the authors and not necessarily the views of the ACS.
The authors declare no competing financial interest.
References
- Butler K. T.; Davies D. W.; Cartwright H.; Isayev O.; Walsh A. Machine Learning for Molecular and Materials Science. Nature 2018, 559 (7715), 547–555. 10.1038/s41586-018-0337-2. [DOI] [PubMed] [Google Scholar]
- Smith J. S.; Isayev O.; Roitberg A. E. ANI-1: An Extensible Neural Network Potential with DFT Accuracy at Force Field Computational Cost. Chem. Sci. 2017, 8 (4), 3192. 10.1039/C6SC05720A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. S.; Isayev O.; Roitberg A. E. Data Descriptor: ANI-1, A Data Set of 20 Million Calculated off-Equilibrium Conformations for Organic Molecules. Sci. Data 2017, 4, 170193. 10.1038/sdata.2017.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers B. D.; James N. C.; Gobbi A. A Comparison of Quantum and Molecular Mechanical Methods to Estimate Strain Energy in Druglike Fragments. J. Chem. Inf. Model. 2017, 57 (6), 1265–1275. 10.1021/acs.jcim.6b00614. [DOI] [PubMed] [Google Scholar]
- Smith J. S.; Nebgen B. T.; Zubatyuk R.; Lubbers N.; Devereux C.; Barros K.; Tretiak S.; Isayev O.; Roitberg A.. Outsmarting Quantum Chemistry Through Transfer Learning. ChemRxiv, 2018. [Google Scholar]
- Gastegger M.; Behler J.; Marquetand P. Machine Learning Molecular Dynamics for the Simulation of Infrared Spectra. Chem. Sci. 2017, 8 (10), 6924–6935. 10.1039/C7SC02267K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. S.; Nebgen B.; Lubbers N.; Isayev O.; Roitberg A. E. A. E. Less Is More: Sampling Chemical Space with Active Learning. J. Chem. Phys. 2018, 148 (24), 241733. 10.1063/1.5023802. [DOI] [PubMed] [Google Scholar]
- Sanchez-Lengeling B.; Aspuru-Guzik A. Inverse Molecular Design Using Machine Learning: Generative Models for Matter Engineering. Science 2018, 361 (6400), 360–365. 10.1126/science.aat2663. [DOI] [PubMed] [Google Scholar]
- Popova M.; Isayev O.; Tropsha A. Deep Reinforcement Learning for de Novo Drug Design. Sci. Adv. 2018, 4 (7), eaap7885. 10.1126/sciadv.aap7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klucznik T.; Mikulak-Klucznik B.; McCormack M. P.; Lima H.; Szymkuć S.; Bhowmick M.; Molga K.; Zhou Y.; Rickershauser L.; Gajewska E. P. Efficient Syntheses of Diverse, Medicinally Relevant Targets Planned by Computer and Executed in the Laboratory. Chem. 2018, 4 (3), 522–532. 10.1016/j.chempr.2018.02.002. [DOI] [Google Scholar]
- Segler M. H. S.; Preuss M.; Waller M. P. Planning Chemical Syntheses with Deep Neural Networks and Symbolic AI. Nature 2018, 555 (7698), 604–610. 10.1038/nature25978. [DOI] [PubMed] [Google Scholar]
- Granda J. M.; Donina L.; Dragone V.; Long D.-L.; Cronin L. Controlling an Organic Synthesis Robot with Machine Learning to Search for New Reactivity. Nature 2018, 559 (7714), 377–381. 10.1038/s41586-018-0307-8. [DOI] [PMC free article] [PubMed] [Google Scholar]