

Abstract
The spliceosome has been shown to be a promising target for the development of new anticancer therapeutics. Synthetic and chemical biological efforts directed toward the development of natural product-based splice modulators (SPLMs) have shown that the potency of these compounds derives from their ability to selectively affect the alternate splicing of apoptotic genes in tumor cells. However, questions remain regarding the mechanistic understanding of splice modulation as well as the selectivity with which SPLMs impact certain genes.
Keywords: Splicing, medicinal chemistry, natural products, chemotherapeutics, drug discovery and polyketides
The spliceosome is a complex molecular machine comprising five small nuclear RNAs (snRNAs) and a number of associated proteins and cofactors that excise introns from pre-RNA to form mature RNA.1 Proper function of the spliceosome is essential to the life of higher organisms,2 and aberrant splicing is known to play a role in cancer, including clear-cell renal cell carcinoma, lung cancer, acute myeloid leukemia (AML), and chronic myeloid leukemia (CML).3 For these reasons, the spliceosome has been identified as a promising target for the design of next generation cancer therapeutics. Herein, we highlight recent efforts to drug the spliceosome through the advance of medicinally chemically optimized splice modulators (SPLMs).
Three classes of natural products have been shown to target the SF3B component of the spliceosome (Figure 1),4−6 the first of which includes the 12-membered macrolide, FD-895 (1a, Figure 1). SPLMs belonging to this class have been referred to as the pladienolides. Their structures consist of a central diene that joins a macrolide core with an acyclic side chain unit (Figure 1). The second class of SF3B-targeting SPLMs resembles 1a, differing in the ring linked to the side chain moiety. This difference is illustrated in the structure of spliceostatin A (2), which features a six-membered ring. The third class, illustrated with herboxidiene (3), contains a six-membered core like the spliceostatins and a tail that shares similarities to that contained in the pladienolides.
Figure 1.
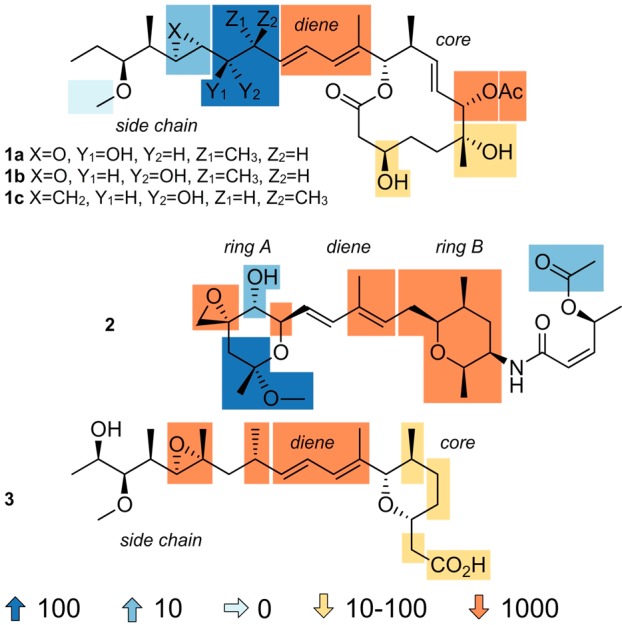
Maps of the structure–activity relationship (SAR) data derived from cytotoxicity analyses (tumor cell GI50 values) on synthetic or semisynthetic derived SPLMs displayed with FD-895 (1a), 17S-FD-895 (1b), cyclopropane 1c, spliceostatin A (2), and herboxidiene (3). Chemical modifications either enhance (blue) or attenuate (orange) activity.
To date, two compounds, E7107 (4)7 and H3B-8800 (5),8 have been explored as potential therapeutics in clinical trials (Figure 2), and a third, 17S-FD-895 (1b, Figure 1) is currently approaching IND filing. All three compounds are derived from the common 12-membered core macrolide observed in 1a–1c. The first, E7107 (4), launched by Eisai Co. Ltd. was evaluated in Phase I trials for application to solid tumors. However, the trials were halted due to patient vision loss caused by presumed side or off-target effects.7
Figure 2.
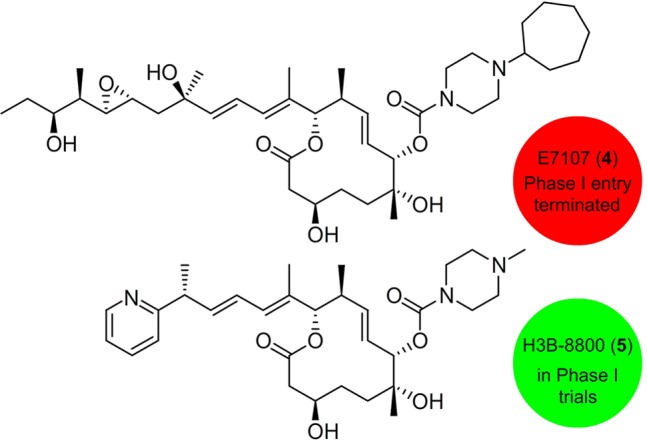
Clinical leads. Structures of analogs that entered (red) or are ongoing (green) clinical trials.
Since the initiation of these trials, natural product semisyntheses and total syntheses carried out by research groups across the world have revealed many of the key SARs within the three classes of natural products. As illustrated in Figure 1, only a few modifications have been found to enhance their antitumor activity.4 Based in part on these findings, H3 Biomedicine advanced an orally active analogue H3B-8800 (5) in which the pladienolide side chain, a region that mapped favorably for optimization (Figure 1), has been replaced with a pyridyl moiety (Figure 2). Through a series of rigorous preclinical analyses, this material has been systematically tested for the treatment of patients with myelodysplastic syndromes, AML, and CML.8
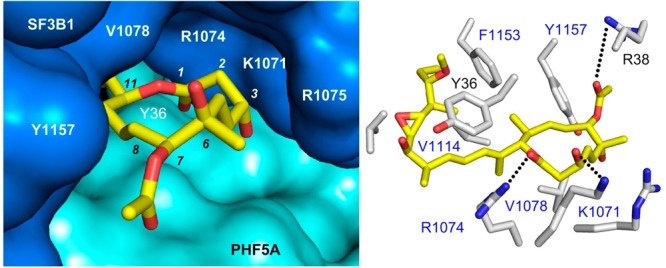
Recent structural studies have revealed the molecular details of the SPLM binding pocket.9−11 These structures, key advances brought forward by the Pena laboratory,10 demonstrate that pladienolide B binds to the branch point adenine (BPA) binding pocket of SF3B. Recently, co-crystal structures of 1a (Figure 3a–c) and 1c (Figure 3d–f) with the SF3B core were determined, confirming the same binding motif as pladienolide B. As depicted in Figure 3, two distinct pockets that accommodate either the side chain or macrolide ring define the SPLM binding cavity.10,11 The side chains of 1a (Figure 3b) and 1c (Figure 3e) occupy a hydrophobic pocket in which the diene moieties interact with Y36 (PHF5A), while their alkyl termini form a hydrophobic contact with F1153 (SF3B1). This region of the binding pocket is relatively plastic in terms of its structure, as demonstrated by V1110’s ability to reposition itself in response to different ligands, as evidenced by comparing the structure of epoxide in 1a·SF3B versus cyclopropane in 1c·SF3B. In contrast, the contacts between the macrolide rings of 1a (Figure 3c) and 1c (Figure 3f) and SF3B are largely polar in nature, with residues R38 (PHF5A) or K1071 (SF3B1) forming hydrogen bonds with the core.
Figure 3.
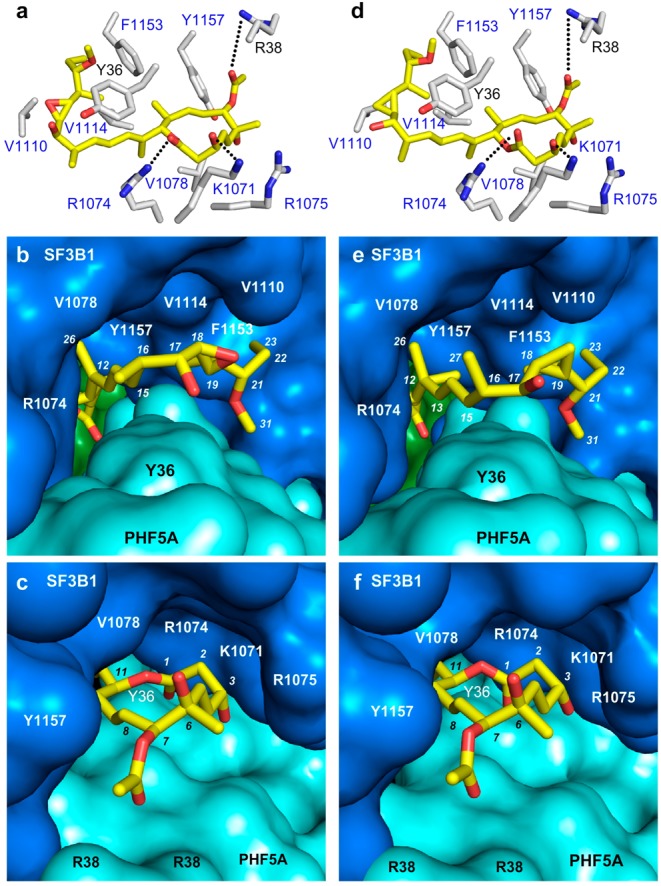
Co-crystal structures of (a–c) FD-895 (1a) and (d–f) cyclopropane 1c within the SF3B core. (a,d) Side chains of residues within 6 Å of the SPLM (yellow) are labeled in gray corresponding to SF3B1 (blue labels) and PHF5A (black labels). (b,e) “Connelly” surfaces of the SPLM binding site showing side chains of 1a and 1c occupying a pocket formed at the interface between SF3B1 (blue), PHF5A (cyan), and SF3B3 (green). (c,f) “Connelly” surfaces in the SPLM binding site depicting the macrolides of 1a and 1c positioned at the other end of this tunnel.
Ongoing studies have found that RNA splicing is altered in cancer cells, and the ability of SPLMs to regulate aberrant splicing in tumors makes them attractive clinical candidates.4−6 While these data were influential in guiding the indications clinically evaluated for E7107 (4)7 and H3B-8800 (5),8 the intricate complexity associated with RNA splicing has made it challenging to understand the comprehensive antitumor mode of action (MOA) of SPLMs.4 As shown in Figure 4, natural and small molecule-induced splice modulation can occur through multiple and often parallel pathways, yielding different gene products that can influence cell phenotypes. While systems-wide tools such as RNA-seq analyses12 can be used to gather a global perspective of the effects of SPLMs in cells, the fact that these cells are often not synchronized and hence represent a population of cells in disparate stages of cell life further complicates efforts to decipher the discrete mechanistic responses to specific modulated splicing events.4
Figure 4.
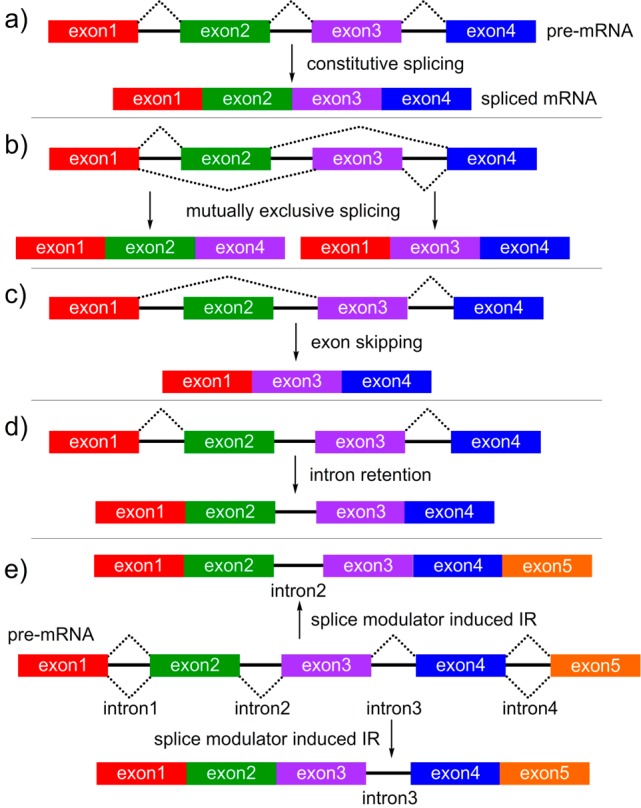
Types of RNA splicing: (a) constitutive, (b) mutually exclusive, (c) exon skipping (ES), or (d) intron retention (IR). (e) Schematic representation of SPLM-induced alternate splicing (AS). In this example, two IR products bearing intron2 (top) and intron3 (bottom) can arise from the same pre-mRNA.
The development of predictive models that reveal the links between the genetic, temporal, and cellular selectivity of splice modulation is a critical next step for future clinical translation of SPLMs. Such models play a key role in the development of validation assays that can be used to guide lead identification. Such assays also provide the tools to identify the specific cellular responses associated with SPLM activity, thereby facilitating the design of leads with minimal off-target splicing effects. Most critically, the information provided by these models can offer a therapeutic tool that may one day allow physicians to predict the efficacy of a given therapeutic on a patient-by-patient basis. The fusion of molecular and cellular biological data with home-accessible biomarker development should play a role in the broader implementation of SPLM-based and related RNA modulatory therapies as commercially viable cancer treatments.
Ongoing clinical and translational processes are focused on the advance of synthetic methods to evaluate analogs of the naturally occurring SPLMs. It is likely that computer-based drug discovery (CADD) approaches will identify alternate core and side chain motifs that reproduce the interactions between SPLMs and SF3B, as revealed by X-ray crystal structures such as 1c·SF3b (Figure 3a) and 1d·SF3b (Figure 3b). This, complemented with new screening tools, opens a new window toward the future clinical translation of splice modulators. The key to this advance lies in the unique balance between medicinal chemistry and chemical biology to provide a robust forum to advance the central principals of drug discovery.
Acknowledgments
This work was supported by funding to W.C.C. by the National Institutes of Health (NIH) Cancer Training Grant (T32 CA009523). B.L. was supported by a UC San Diego Chancellor's Postdoctoral Fellowship. K.A.K. was supported in part by a NIH Chemistry-Biology Interface Training Grant (T32 GM112584). The X-ray structures of 1a·SF3B core and 1c·SF3B core were determined by Vlad Pena (MPI BPC) and Constantin Cretu (MPI BPC) and uploaded to the PDB.
Author Contributions
† This article was written as a team and hence these authors share co-first authorship.
Views expressed in this editorial are those of the authors and not necessarily the views of the ACS.
The authors declare no competing financial interest.
References
- Shi Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat. Rev. Mol. Cell Biol. 2017, 18, 655–670. 10.1038/nrm.2017.86. [DOI] [PubMed] [Google Scholar]
- Sperling R. The nuts and bolts of the endogenous spliceosome. Wiley Interdiscip. Rev. RNA 2017, 8, e1377. 10.1002/wrna.1377. [DOI] [PubMed] [Google Scholar]
- Lee S. C.; Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. 10.1038/nm.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- León B.; Kashyap M. K.; Chan W. C.; Krug K. A.; Castro J. E.; La Clair J. J.; Burkart M. D. A challenging pie to splice: Drugging the spliceosome. Angew. Chem., Int. Ed. 2017, 56, 12052–12063. 10.1002/anie.201701065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham D.; Koide K. Discoveries, target identifications, and biological applications of natural products that inhibit splicing factor 3B subunit 1. Nat. Prod. Rep. 2016, 33, 637–647. 10.1039/C5NP00110B. [DOI] [PubMed] [Google Scholar]
- Effenberger K. A.; Urabe V. K.; Jurica M. S. Modulating splicing with small molecular inhibitors of the spliceosome. Wiley Interdiscip. Rev. RNA 2017, 8, e1381. 10.1002/wrna.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D. S.; Kurzrock R.; Naing A.; Wheler J. J.; Falchook G. S.; Schiffman J. S.; Faulkner N.; Pilat M. J.; O’Brien J.; LoRusso P. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) spliceosome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest. New Drugs 2014, 32, 436–444. 10.1007/s10637-013-0046-5. [DOI] [PubMed] [Google Scholar]
- Seiler M.; Yoshimi A.; Darman R.; Chan B.; Keaney G.; Thomas M.; Agrawal A. A.; Caleb B.; Csibi A.; Sean E.; Fekkes P.; Karr C.; Klimek V.; Lai G.; Lee L.; Kumar P.; Lee S. C.-W.; Liu X.; Mackenzie C.; Meeske C.; Mizui Y.; Padron E.; Park E.; Pazolli E.; Peng S.; Prajapati S.; Taylor J.; Teng T.; Wang J.; Warmuth M.; Yao H.; Yu L.; Zhu P.; Abdel-Wahab O.; Smith P. G.; Buonamici S. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. 10.1038/nm.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finci L. I.; Zhang X.; Huang X.; Zhou Q.; Tsai J.; Teng T.; Agrawal A.; Chan B.; Irwin S.; Karr C.; Cook A.; Zhu P.; Reynolds D.; Smith P. G.; Fekkes P.; Buonamici S.; Larsen N. A. The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev. 2018, 32, 309–320. 10.1101/gad.311043.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretu C.; Agrawal A. A.; Cook A.; Will C. L.; Fekkes P.; Smith P. G.; Lührmann R.; Larsen N.; Buonamici S.; Pena V. Structural Basis of Splicing Modulation by Antitumor Macrolide Compounds. Mol. Cell 2018, 70, 265–273. 10.1016/j.molcel.2018.03.011. [DOI] [PubMed] [Google Scholar]
- Teng T.; Tsai J. H.; Puyang X.; Seiler M.; Peng S.; Prajapati S.; Aird D.; Buonamici S.; Caleb B.; Chan B.; Corson L.; Feala J.; Fekkes P.; Gerard B.; Karr C.; Korpal M.; Liu X.; Lowe J.; Mizui Y.; Palacino J.; Park E.; Smith P. G.; Subramanian V.; Wu Z. J.; Zou J.; Yu L.; Chicas A.; Warmuth M.; Larsen N.; Zhu P. Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nat. Commun. 2017, 8, 15522. 10.1038/ncomms15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews L. A.; Balaian L.; Delos Santos N. P.; Leu H. S.; Court A. C.; Lazzari E.; Sadarangani A.; Zipeto M. A.; La Clair J. J.; Villa R.; Kulidjian A.; Storb R.; Morris S. R.; Ball E. D.; Burkart M. D.; Jamieson C. H. M. RNA Splicing Modulation Selectively Impairs Leukemia Stem Cell Maintenance in Secondary Human AML. Cell Stem Cell. 2016, 19, 599–612. 10.1016/j.stem.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]