

Abstract

Optimization of the side-chain of a phenyl indole scaffold identified from a high-throughput screening campaign for inhibitors of the AAA+ ATPase p97 is reported. The addition of an N-alkyl piperazine led to high potency of this series in a biochemical assay, activity in cell-based assays, and excellent pharmaceutical properties. Molecular modeling based on a subsequently obtained cryo-EM structure of p97 in complex with a phenyl indole was used to rationalize the potency of these allosteric inhibitors.
Keywords: AAA+ ATPase, p97, allosteric inhibitor, protein homeostasis modulator, anticancer, phenyl indole
The AAA+ (ATPase associated with various cellular activities) p97 is a hexameric, multidomain protein that plays a key role in protein homeostasis. Energy from the hydrolysis of ATP is mechanically relayed by p97 for the extraction of polypeptides from membranes, the ribosome or DNA, facilitating polypeptide degradation by the proteasome. In addition, p97 is hypothesized to play a role in other protein degradation pathways, including autophagy.1,2 Its essential role in protein homeostasis and the clinical success of proteasome inhibitors suggest the potential for targeting certain cancers with p97 inhibitors. Toward this end, a number of small molecule inhibitors of p97 have been reported to show promising antiproliferative effects.3−10 One ATP-competitive active site inhibitor, CB-5083, advanced to Phase I clinical trials for multiple myeloma and advanced solid tumors but was recently discontinued due to off-target retinal toxicity related to inhibition of PDE6.10−12
As part of our efforts to identify small molecule allosteric inhibitors of p97,9,13,14 we embarked on a medicinal chemistry program to optimize a 2-phenyl indole scaffold discovered via a high-throughput screening (HTS) effort.15 At the time our work was conducted, no structural information on the compound’s binding site was available, although it was determined to bind to an allosteric site.15 A structure–activity relationship (SAR) study illustrated the importance of the indole moiety for potency.13,14 In this Letter, we disclose additional SAR data and the optimization of the flexible side-chain, as well as biological characterization of key analogs.
Compound 1 (Figure 1 and Table 1) is representative of early active compounds in the phenyl indole series. Compound 1 inhibited the ATPase activity of p97 with an IC50 = 2.4 μM at low ATP concentration (20 μM). At higher ATP concentrations (100 μM), the IC50 improved to 0.99 μM, indicating an uncompetitive binding mechanism.15 Properties (MW, CLogP, tPSA; ChemDraw) of 1 were all in the drug-like range.
Figure 1.

First-generation phenyl indole inhibitor 1 and areas of focused SAR explorations.
Table 1. p97 Inhibitory Activity of Phenyl Indolea.


Assay conditions: ADP-Glo with 20 nM WT p97 in the presence of 100 μM ATP. n denotes number of independent replicates, each assayed in duplicate or triplicate. In the same assay, the allosteric p97 inhibitor, NMS-873,8 exhibited an IC50 = 11 nM ± 4.9 (n = 13).
For further optimization, we used an approach that kept the 2-(3-(piperidin-1-yl)phenyl)-1H-indole scaffold constant and varied the amine side-chain (Scheme 1). Our synthetic strategy, which focused on late-stage diversification, relied on previously optimized Buchwald–Hartwig couplings of 3-bromo-2-phenyl indoles (2).13,14,16−18 The intermediate, 3, was used for rapid further diversification via reductive aminations19 to afford, in some cases after additional modifications, final products of general structure 4. An alternative strategy relied on direct coupling of an intact side-chain (or a related analog) with 2 to generate 4.
Scheme 1. General Synthesis of Phenyl Indoles Using Late-Stage Diversification Strategies.

The first rounds of SAR were generated from the unsubstituted 2-phenyl indole core and explored a wide array of side-chain amines that varied the terminal group as well as the linker to the piperidine. Replacement of the terminal triazole in 1 with a phenyl group, resulting in analog 5, led to a loss of potency, indicating the requirement for a polar group at this position. Replacement with a pyridine (6) restored activity to near original levels.
The length of the linker appeared to be insignificant, as the 1-carbon linker (7) possessed equivalent potency to the 2-carbon analog in this series. Replacement of the aromatic heterocycle with a piperazine group provided a first significant (∼3-fold) improvement in potency (8; IC50 = 330 nM) compared to compound 1.20 Further functionalization by capping the terminal nitrogen with an aliphatic substituent (e.g., Me, Et, iPr, tBu; 9–12), provided another boost in potency. In these examples, the general trend was that greater lipophilicity seemed to correlate with improved potency, with iPr exhibiting 160 nM potency. In contrast to compounds 6 and 7, the length of the linker between the piperidine and the terminal moiety modestly affected potency: a 3-carbon linker (13) was ∼2-fold less potent than its 2-carbon homologue 11.
Combining the preferred 5-F indole14,16 with the N-iPr piperazine afforded 14, the first analog with potency below 100 nM (IC50 = 70 nM). Previous SAR data13 indicated that the indole moiety was highly sensitive to modification, and a cryo-EM co-structure16 and extensive molecular modeling14 showed the indole deeply embedded in an allosteric binding site in p97’s D2 domain, located very near the D1–D2 interface, with the fluorine atom engaging in key interactions with surrounding residues.14,16 Given the importance of the 5-fluoroindole to binding and potency, our next iterations maintained this feature and explored additional amine groups that contained a N-iPr moiety (see SI).
The terminal amine in the piperazine moiety contributed to potency, as the truncated dimethyl amino analog 15, while maintaining some activity (IC50 = 800 nM), was >10-fold less potent than 14. Modulation of the piperazine nitrogen’s basicity by introducing a carbonyl group into a 3-carbon linker as in 16 was tolerated, but decreased potency compared to 14. While 16 (IC50 = 190 nM) was slightly more potent than 13 (IC50 = 380 nM), we attributed this difference to the presence of the potency-enhancing 5-F indole moiety rather than a preference for the modified linker. We concluded that the length of the linker was not critical to potency and that the pKa of the internal piperazine nitrogen could be manipulated without detriment to biological activity. Taking this information into account, we attempted to simultaneously constrain the conformation of the flexible side-chain and reduce the number of basic nitrogen atoms. This led to the design of analogs such as 17, containing an amine-linked bis-piperidine side-chain. This compound exhibited strong inhibition of p97 (IC50 = 50 nM) and was somewhat more potent than compounds containing a flexible linker, such as 14. While we had no data to suggest specific liabilities, the elimination of a basic nitrogen was anticipated to address potential liabilities such as hERG receptor binding and efflux. To further explore this truncated scaffold, we replaced the internal piperidine with cis- and trans-cyclohexane groups (see SI). The trans-analog 18 retained activity toward p97, albeit ∼3–4-fold less than the piperidine 17. In contrast, the cis-isomer 19 was >20-fold less potent compared to the trans-isomer and >100-fold less potent than 17, indicating a significant conformational preference. Further shortening and constraining the side-chain by eliminating the nitrogen linkage (e.g., spiro analog 20; piperidine-piperazine 21; and fused bicyclo[3.3.0] ring system 22) was not productive. These compounds inhibited p97 in the micromolar range.
As an alternate approach to conformational constraint, we incorporated small cyclic moieties as “kinks” into the flexible linker. This strategy led to some of the most potent analogs in this series. Cyclobutane 23 inhibited p97 with an IC50 = 20 nM, i.e., over ten times more potent than the parent analog 9. Based on SAR trends, this difference could not be attributed solely to the presence of the 5-F indole in 23. Unlike the flexible linker series (e.g., 9–11), within this subseries, the N-Me piperazine terminus (23) was 5-fold more potent than the N-iPr analog (24). The oxetane analog 25 (IC50 = 90 nM) was similar in potency to the cyclobutane 24.
To develop structure-based hypotheses to rationalize the biochemical activity of this series, we utilized a recently reported cryo-EM costructure of 14 bound to p97; however, while strong electron-density for the phenyl-indole component of the inhibitor was observed in this structure, there was no density detected for the conformationally flexible side-chain.16 As shown in Figure 2a, the side-chain in the cryo-EM model was positioned extending out of the allosteric binding cleft and directly into solvent, making no contacts with the protein.16 This binding mode was inconsistent with the side-chain SAR, which indicated a preference for specific, basic groups at this position (e.g., piperazine preferred over pyridine and phenyl, as in 8 vs 6, 5), as well as a preference for lipophilic groups at the piperazine terminus (e.g., ethyl, iPr, tBu: 10-12 vs H, Me: 8, 9). We therefore developed a binding model that is consistent with the available SAR (Figure 2b). Placing the piperidine ring in a chair conformation (vs the twist chair conformation in the cryo-EM costructure) allows the basic side-chain of 14 to be oriented toward the protein and to engage in multiple hydrogen bonds, instead of extending out into solvent. Favorable interactions develop between (1) the terminal piperazine nitrogen atom and the Glu534 carboxylate, (2) the internal piperazine nitrogen atom and the side-chain amide carbonyl of Gln494, (3) the secondary amine and Glu498, and (4) the piperidine ring nitrogen atom (an aniline) and the thiol of Cys535 (an interaction also observed in the twist chair conformation). Using this refined model, we were able to rationalize much of the SAR, particularly that of the side-chain.
Figure 2.

(a) Representation of cryo-EM structure of 14 (carbon = magenta; nitrogen = blue; fluorine = light blue; key hydrogens = white; yellow dashes = H-bonds) bound to p97 at an allosteric site in the D2 domain.16 The phenyl indole binds in a deep pocket, but the side-chain points directly into solvent and engages in no contacts with protein residues. (b) Molecular modeling-derived binding mode that accounts for side-chain SAR. Key inhibitor (14) side-chain–protein interactions include H-bonds (yellow dashes) with Gln494, Glu498, Glu534, and Cys535, as well as solvent shielding of the terminal iPr group by the side-chain indole of Trp476.
Figure 3 shows the proposed binding modes of trans- and cis-cyclohexanes 18 and 19. The trans-orientation in 18 results in a favorable binding mode for the side-chain component; hydrogen bonds with acidic residues Glu498 and Glu534 are maintained, and the cyclohexyl component engages in hydrophobic contacts with side-chains of Val493 and Ile531. Additionally, the iPr group orients away from the solvent toward the aromatic indole side-chain of residue Trp476. In contrast, binding of the cis-isomer 19 results in a significantly more solvent-exposed side-chain orientation that forces the cyclohexyl and iPr groups toward the solvent, an energetically unfavorable pose. Moreover, the secondary nitrogen atom engages in unfavorable hydrophobic–polar clashes with the side-chains of residues Val493 and Ile531. Finally, the constrained orientation of the cis-isomer results in the loss of a hydrogen bond with Glu498. The increased potency of 23 compared to compounds resembling 14 can be rationalized by the binding pose in Figure 4, and the observation that the kinked side-chain is embedded more deeply into the p97 helix 16–17 interface than the straight-chain analogs, and more fully occupies a hydrophobic pocket formed by Val493, Ile531 (Figure 4), and, to a lesser degree, Leu 527. We hypothesize that desolvation of the cyclobutyl group drives this deeper binding mode. Interestingly, the more buried binding mode of the side-chain of 23 results in the loss of the H-bond with Glu498 predicted for 14 and a weakening of the H-bond with Glu534 that is observed for 14 (Figure 2b). Rather, the side-chain of 23 engages in H-bonds with the amide side-chains of Gln490 and 494 and forms a favorable cation−π interaction with the side-chain of Trp476. As noted above, within this gem-disubstituted cyclobutane subseries, N-Me piperazine is favored over the N-iPr analog (23 vs 24), in contrast to other members of the class (e.g., 9 vs 11), where the larger group is preferred. We hypothesize that the presence of the smaller N-Me substituent is more sterically compatible with the formation of the cation−π interaction than larger terminal moieties.
Figure 3.
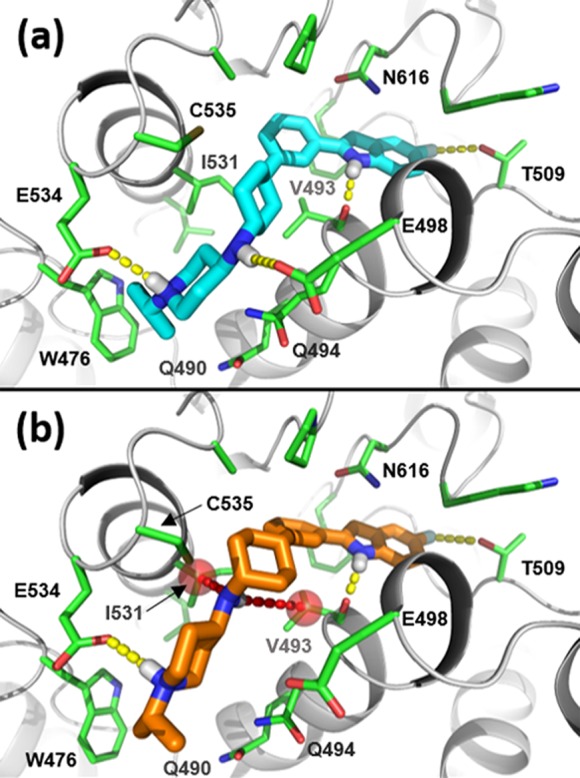
Molecular modeling comparison of the binding modes of 18 (a, carbon = cyan) and 19 (b, carbon = orange); yellow dashes = H-bond; red dashes and spheres = unfavorable interactions. (a) Side-chain of 18 retains two H-bonds to Glu498 and Glu534, the cyclohexyl group engages in hydrophobic interactions with Val493 and Ile531, and the terminal iPr group is located near, and solvent shielded by, the side-chain indole moiety of Trp476. (b) In contrast, in the model of the cis-isomer (19) the H-bond to Glu498 is lost; unfavorable interactions (red dashes and spheres) between the secondary nitrogen and hydrophobic side chains Val493 and Ile531 are observed, and the cyclohexyl and iPr groups are significantly more solvent exposed.
Figure 4.

Predicted binding mode of 23 (carbon = pink; yellow dashes = H-bonds). Like other inhibitors of this chemotype, the side-chain binds down the helix 16/17 interface and engages in multiple hydrogen bonds with side-chain residues. However, unlike the more flexible parent 14, the cyclobutyl moiety of 23 binds more deeply in a hydrophobic pocket at the helical interface. As a result, (1) new H-bond patterns are formed with the amide side-chains of Gln490 and 494 and (2) the cyclobutyl moiety engages in closer, more favorable hydrophobic contacts with the side-chains of Val493 and Ile531, and (3) the terminal piperazine nitrogen engages in a cation−π interaction with the side-chain indole of Trp476 (purple dash with terminal, semi-transparent sphere).
The biochemical potencies of compounds 17 and 23 compare favorably to another allosteric inhibitor of p97, NMS-873 (IC50= 11 nM)8 in side-by-side comparisons. However, the physical properties of the phenyl indoles are superior. Specifically, the aqueous (pH 7.4) solubility (>300 μM) and half-life in in vitro metabolic stability assays (t1/2 > 270 min) in human (HLM) and mouse liver microsomes (MLM) (Table 2) of 17 and 23 are significantly better; the corresponding value for NMS-873’s solubility is 86 μM, and its metabolic half-life is <10 min in both HLM and MLM. The improvement in properties for the phenyl indoles has facilitated biophysical studies (e.g., cryo-EM analyses) with these compounds that were not feasible with NMS-873. Another key difference between this series of inhibitors and NMS-873 appears to be the nature of the allosteric inhibition. While NMS-873 exhibits mostly noncompetitive inhibition,9 the phenyl indoles, as a class, are uncompetitive inhibitors.15,16
Table 2. Characterization of Compounds 17 and 23 and Comparison to NMS-873.
| properties | 17 | 23 | NMS-873 |
|---|---|---|---|
| p97 IC50 (nM) | 50 | 20 | 11 |
| solubility (μM) | 380 | 330 | 86 |
| HLM (t1/2, min) | 569 | 475 | <10 |
| MLM (t1/2, min) | 386 | 277 | <10 |
| UbG76 V-GFP EC50 (μM) | |||
| 1 h | 19 | 15 | 1.4 |
| 6 h | >40 | >40 | 2.1 |
| NCI-60 mean log GI50 | –5.8 | –5.7 | –6.1 |
We then evaluated whether compounds inhibited p97-dependent activities in cells. In an assay that measured accumulation of a ubiquitinated substrate,21−2317 and 23 showed modest effects at 1 h (EC50 = 19 and 15 μM, respectively), but the effect was not observed at 6 h even at 40 μM. In contrast, NMS-873 showed robust effects at both 1 and 6 h (EC50 = 1.42 and 2.06 μM, respectively) (Table 2). We considered several hypotheses to explain this lack of correlation between biochemical activity (ATPase) and cellular activity (ubiquitin-mediated degradation). MDCK permeability measurements were uninterpretable, but poor cellular penetration was considered unlikely based on the high intracellular concentrations measured for representative analogs (including 14; data not shown).24 A second possibility was that the uncompetitive mechanism of the phenyl indole inhibitors was not as efficacious as ATP-competitive (such as CB-5083) or noncompetitive (such as NMS-873) mechanisms at blocking p97’s unfoldase activity.15,25−27 Finally, an intriguing possibility is that the UbG76 V-GFP cellular assay does not recapitulate all of the diverse functions of p97 and that this phenyl indole class selectively inhibits only certain p97-dependent functions, such as autophagy. We have started to evaluate whether other p97-dependent cellular activities are affected by these phenyl indoles. Preliminary data suggests this may be the case; these results will be reported in due course.15
Consistent with blockade of p97, 17 and 23 inhibited cell growth in the NCI-60 cell line panel.28 Both demonstrated broad activity in leukemia (pGI50 = −5.7 to −6.7 for 17 and 23); NSCL cancer (pGI50 = −5.7 to −6.0 for 17; −5.7 to −5.8 for 23); colon cancer (pGI50 = 5.8 to −6.4 for 17; 5.8 to −6.3 for 23); CNS tumor (pGI50 = −5.7 to −5.8 for 17 and 23); melanoma (pGI50 = −5.7 to −5.9 for 17; −5.7 to −6.5 for 23); ovarian cancer (pGI50 = −5.7 to −5.8 for 17 and 23); renal cancer (pGI50 = −5.7 to −5.8 for 17 and 23); prostate cancer (pGI50 = −5.7 for 17; −5.7 to −5.8 for 23); and breast cancer (pGI50 = −5.8 for 17; −5.7 to −5.9 for 23) cell lines (see SI).
In conclusion, by applying systematic medicinal chemistry strategies, we optimized the side-chain region of a series of 2-phenylindole p97 inhibitors. SAR studies combined with structural information and molecular modeling were used to develop a refined binding model suitable for future analog design iterations. The most potent compounds in this series, such as 17 and 23, exhibit low nanomolar biochemical inhibition of the ATPase and excellent properties but showed variable effects in cellular assays that are commonly used to evaluate certain p97-dependent activities. However, these compounds exhibited antiproliferative effects in the NCI-60 panel in the high nanomolar to low micromolar range. Allosteric inhibitors such as 17 and 23 have the potential to be developed into novel cancer therapeutics and to be used as chemical biology tools to interrogate the function of p97 in cancer and other p97-dependent diseases. Further studies on this series of p97 inhibitors will be reported in due course.
Acknowledgments
The authors gratefully acknowledge Marina Kovaliov (University of Pittsburgh) for technical support and advice; Chaemin Lim (University of Pittsburgh) for data retrieval and collation; Mary Liang (University of Pittsburgh) for compound management and extensive assistance with manuscript preparation; Taber Maskrey (University of Pittsburgh) for analytical chemistry support; Clifford Bryant (UCSF) for preliminary chemistry; Gregory Lee (UCSF) for help with data compilation; Sean Marcsisin, CTP US Army; Jason Sousa and Brittney Potter (WRAIR) for solubility data; AMRI for microsomal stability data; Richard Gussio, CAPT USPHS (NCI) for contributions to computational and molecular modeling studies; and the NCI for access to the NCI-60 cell line. The authors also appreciate the helpful discussion and suggestions of all the CBC p97 project team members, particularly Raymond Deshaies (CalTech), Eric Baldwin (Leidos), Andrew Flint (Leidos), Barbara Mroczowski (NCI), Shizuko Sei (Leidos), Gordon Stott (Leidos), Gunda Georg (University of Minnesota), and Michael Walters (University of Minnesota).
Glossary
ABBREVIATIONS
- AAA+
ATPase associated with diverse activities
- CLogP
calculated logP
- CNS
central nervous system
- cryo-EM
cryo electron microscopy
- dba
dibenzylideneacetone
- CyJohnPhos
(2-biphenyl)dicyclohexylphosphine
- GI50
growth inhibition 50%
- HLM
human liver microsomes
- IC50
inhibitory concentration 50%
- MLM
mouse liver microsomes
- NSCL
nonsmall cell lung
- PDE6
phosphodiesterase 6
- tPSA
total polar surface area
- UbG76 V-GFP
ubiquitin Gly76 to Val-green fluorescent protein
Biographies
Michelle Arkin is a Professor in Pharmaceutical Chemistry, co-Director of the UCSF Small Molecule Discovery Center (SMDC), and an Adjunct Professor at the Buck Institute for Aging Research. Her research focuses on developing probes and drug leads that modulate allosterically regulated enzymes and protein−protein interactions (PPI). Michelle is the current President of the BOD for the Academic Drug Discovery Consortium, PI for UCSF in the NCI Chemical Biology Consortium, and editorial board member for the Assay Guidance Manual and Current Protocols in Chemical Biology. Prior to UCSF, Michelle worked at Sunesis Pharmaceuticals, where she helped discover inhibitors of PPI, including IL-2 and LFA-1 (lifitigrast; SARcode/Shire).
Peter Wipf received his Ph.D. in Chemistry from the University of Zürich, where he worked in the group of Prof. Heinz Heimgartner on synthetic transformations of 3-amino-2H-azirines. He then joined the laboratory of Prof. Robert E. Ireland at the University of Virginia as a Swiss NSF postdoctoral fellow on the total synthesis of FK-506. In 1990, he moved to the University of Pittsburgh, where, since 2004, he holds the rank of Distinguished University Professor. His group works on natural products total synthesis and also pursues a diverse but always challenging range of projects in heterocyclic, medicinal, and computational chemistry.
Donna M. Huryn received her Ph.D. in Organic Chemistry from the University of Pennsylvania and began her career as a medicinal chemist in the pharmaceutical industry (Hoffmann-La Roche & Wyeth Research), where she contributed to drug discovery efforts for HIV, cancer, asthma, and CNS disorders. In 2004, she joined academia, and is now Professor at the University of Pittsburgh School of Pharmacy and holds an adjunct appointment in the Chemistry Department at the University of Pennsylvania. Her research focuses on the design and synthesis of small molecule probes and drugs to treat cancer, acute kidney injury, and neurodegenerative disorders.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00372.
Synthetic procedures and spectral data for key compounds, computational methods, biological assay protocols, NCI-60 data, and NMR spectra of final products (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The project was funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Chemical Biology Consortium Contract No. HHSN261200800001E, Agreement No. 29XS127TO15.
The authors declare no competing financial interest.
Supplementary Material
References
- Xia D.; Tang W. K.; Ye Y. Structure and function of the AAA+ ATPase p97/Cdc48p. Gene 2016, 583 (1), 64–77. 10.1016/j.gene.2016.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boom J.; Meyer H. VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 2017, 69, 182–194. 10.1016/j.molcel.2017.10.028. [DOI] [PubMed] [Google Scholar]
- Chapman E.; Maksim N.; de la Cruz F.; La Clair J. J. Inhibitors of the AAA+ chaperone p97. Molecules 2015, 20 (2), 3027–3049. 10.3390/molecules20023027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding R.; Zhang T.; Xie J.; Williams J.; Ye Y.; Chen L. Eeyarestatin I derivatives with improved aqueous solubility. Bioorg. Med. Chem. Lett. 2016, 26 (21), 5177–5181. 10.1016/j.bmcl.2016.09.068. [DOI] [PubMed] [Google Scholar]
- Tao S.; Tillotson J.; Wijeratne E. M.; Xu Y. M.; Kang M.; Wu T.; Lau E. C.; Mesa C.; Mason D. J.; Brown R. V.; La Clair J. J.; Gunatilaka A. A.; Zhang D. D.; Chapman E. Withaferin A Analogs That Target the AAA+ Chaperone p97. ACS Chem. Biol. 2015, 10 (8), 1916–1924. 10.1021/acschembio.5b00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T. F.; Li K.; Frankowski K. J.; Schoenen F. J.; Deshaies R. J. Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase. ChemMedChem 2013, 8 (2), 297–312. 10.1002/cmdc.201200520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnaghi P.; D’Alessio R.; Valsasina B.; Avanzi N.; Rizzi S.; Asa D.; Gasparri F.; Cozzi L.; Cucchi U.; Orrenius C.; Polucci P.; Ballinari D.; Perrera C.; Leone A.; Cervi G.; Casale E.; Xiao Y.; Wong C.; Anderson D. J.; Galvani A.; Donati D.; O’Brien T.; Jackson P. K.; Isacchi A. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat. Chem. Biol. 2013, 9 (9), 548–556. 10.1038/nchembio.1313. [DOI] [PubMed] [Google Scholar]
- Polucci P.; Magnaghi P.; Angiolini M.; Asa D.; Avanzi N.; Badari A.; Bertrand J.; Casale E.; Cauteruccio S.; Cirla A.; Cozzi L.; Galvani A.; Jackson P. K.; Liu Y.; Magnuson S.; Malgesini B.; Nuvoloni S.; Orrenius C.; Sirtori F. R.; Riceputi L.; Rizzi S.; Trucchi B.; O’Brien T.; Isacchi A.; Donati D.; D’Alessio R. Alkylsulfanyl-1,2,4-triazoles, a new class of allosteric valosine containing protein inhibitors. Synthesis and structure-activity relationships. J. Med. Chem. 2013, 56 (2), 437–450. 10.1021/jm3013213. [DOI] [PubMed] [Google Scholar]
- Alverez C.; Bulfer S. L.; Chakrasali R.; Chimenti M. S.; Deshaies R. J.; Green N.; Kelly M.; LaPorte M. G.; Lewis T. S.; Liang M.; Moore W. J.; Neitz R. J.; Peshkov V. A.; Walters M. A.; Zhang F.; Arkin M. R.; Wipf P.; Huryn D. M. Allosteric Indole Amide Inhibitors of p97: Identification of a Novel Probe of the Ubiquitin Pathway. ACS Med. Chem. Lett. 2016, 7 (2), 182–187. 10.1021/acsmedchemlett.5b00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. J.; Wang J.; Yao B.; Wong S.; Djakovic S.; Kumar B.; Rice J.; Valle E.; Soriano F.; Menon M. K.; Madriaga A.; Kiss von Soly S.; Kumar A.; Parlati F.; Yakes F. M.; Shawver L.; Le Moigne R.; Anderson D. J.; Rolfe M.; Wustrow D. Discovery of a First-in-Class, Potent, Selective, and Orally Bioavailable Inhibitor of the p97 AAA ATPase (CB-5083). J. Med. Chem. 2015, 58 (24), 9480–9497. 10.1021/acs.jmedchem.5b01346. [DOI] [PubMed] [Google Scholar]
- Anderson D. J.; Le Moigne R.; Djakovic S.; Kumar B.; Rice J.; Wong S.; Wang J.; Yao B.; Valle E.; Kiss von Soly S.; Madriaga A.; Soriano F.; Menon M. K.; Wu Z. Y.; Kampmann M.; Chen Y.; Weissman J. S.; Aftab B. T.; Yakes F. M.; Shawver L.; Zhou H. J.; Wustrow D.; Rolfe M. Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell 2015, 28 (5), 653–665. 10.1016/j.ccell.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://deainfo.nci.nih.gov/advisory/fac/0518/Doroshow.pdf (accessed August 2, 2018).
- Alverez C.; Arkin M. R.; Bulfer S. L.; Colombo R.; Kovaliov M.; LaPorte M. G.; Lim C.; Liang M.; Moore W. J.; Neitz R. J.; Yan Y.; Yue Z.; Huryn D. M.; Wipf P. Structure-Activity Study of Bioisosteric Trifluoromethyl and Pentafluorosulfanyl Indole Inhibitors of the AAA ATPase p97. ACS Med. Chem. Lett. 2015, 6 (12), 1225–1230. 10.1021/acsmedchemlett.5b00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett J. C.; Lim C.; Peyser B. D.; Samankumara L. P.; Kovaliov M.; Colombo R.; Bulfer S. L.; LaPorte M. G.; Hermone A. R.; McGrath C. F.; Arkin M. R.; Gussio R.; Huryn D. M.; Wipf P. A threonine turnstile defines a dynamic amphiphilic binding motif in the AAA ATPase p97 allosteric binding site. Org. Biomol. Chem. 2017, 15 (19), 4096–4114. 10.1039/C7OB00526A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuscript in preparation.
- Banerjee S.; Bartesaghi A.; Merk A.; Rao P.; Bulfer S. L.; Yan Y.; Green N.; Mroczkowski B.; Neitz R. J.; Wipf P.; Falconieri V.; Deshaies R. J.; Milne J. L.; Huryn D.; Arkin M.; Subramaniam S. 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 2016, 351 (6275), 871–875. 10.1126/science.aad7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehne M. E.; Kitagawa T. Reactions of indoles with benzyne. J. Org. Chem. 1964, 29, 1270–1273. 10.1021/jo01028a514. [DOI] [Google Scholar]
- Ruiz-Castillo P.; Buchwald S. L. Applications of Palladium-Catalyzed C-N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. 10.1021/acs.chemrev.6b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumpaty H. J.; Bhattacharyya S.; Rehr E. W.; Gonzalez A. M. Selective Access to Secondary Amines by a Highly Controlled Reductive Mono-N-Alkylation of Primary Amines. Synthesis 2003, 2206–2210. 10.1055/s-2003-41066. [DOI] [Google Scholar]
- Recently, a series of indoles containing a basic side chain was reported to allosterically inhibit p97 at the same site; the potency of this series was in the low micromolar range.; Pöhler R.; Krahn J. H.; van den Boom J.; Dobrynin G.; Kaschani F.; Eggenweiler H. M.; Zenke F. T.; Kaiser M.; Meer H. A non-competitive inhibitor of VCP/p97 and VPS4 reveals conserved allosteric circuits in tpe I and II AAA ATPases. Angew. Chem., Int. Ed. 2018, 57 (6), 1576–1580. 10.1002/anie.201711429. [DOI] [PubMed] [Google Scholar]
- Dantuma N. P.; Lindsten K.; Glas R.; Jellne M.; Masucci M. G. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat. Biotechnol. 2000, 18, 538–543. 10.1038/75406. [DOI] [PubMed] [Google Scholar]
- Dantuma N. P.; Lindsten K.; Glas R.; Jellne M.; Masucci M. G.. Screening assay for compounds targeting the p97 AAA-ATPase complex in the ubiquitin proteasome system. WO 2010003908.
- Chou T. F.; Brown S. J.; Minond D.; Nordin B. E.; Jones A. C.; Chase P.; Porubsky P. R.; Stoltz B. M.; Schoenen F. J.; Patricelli M. P.; Hodder P.; Rosen H.; Deshaies R. J. Reersible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 4834–4839. 10.1073/pnas.1015312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assays performed by Quintara Biosciences: briefly, cells are treated with compounds at varying concentrations. After incubation and removal of supernatant, cells are lysed and compound concentrations quantified by LC/MS/MS.
- Bodnar N. O.; Rapoport T. A. Molecular mechanism of substrate processing by the Cdc48 ATPase complex. Cell 2017, 169, 722–735. 10.1016/j.cell.2017.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blythe E. E.; Olson K. C.; Chau V.; Deshaies R. J. Ubiquitin- and ATP-dependent unfoldase activity of p97/VCP-NPLOC4-UFD1L is enhanced by a mutation that causes multisystem proteinopathy. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E4380–E4388. 10.1073/pnas.1706205114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The uncompetitive mechanism of 17 and 23 is inferred from the cryo-EM costructure of 14 and preliminary data for 23 in ATPase assays.
- Shoemaker R. H. The NCI60 human tumor cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–23. 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.