

Abstract

We report new SSTR5 antagonists with enhanced potency, subtype selectivity, and minimal off-target activities as compared to previously reported compounds. Starting from the reported SSTR5 antagonist 1, we systematically surveyed changes in the central core and head piece while maintaining the diphenyl tail group constant. From this study the azaspirodecanone 10 emerged as a new highly potent and selective SSTR5 antagonist. Compound 10 lowered glucose excursion by 94% in an oral glucose tolerance test (OGTT) in mice following a 3 mg/kg oral dose. The compound increased both total and active circulating incretin hormone GLP-1 levels in mice at a dose of 10 mg/kg. A synergistic effect was also demonstrated when compound 10 was coadministered with a DPP-4 inhibitor, substantially increasing circulating active GLP-1[7–36] amide and insulin in response to a glucose challenge.
Keywords: Somatostatin, SSTR5 antagonists, type 2 diabetes mellitus (T2DM), glucose-dependent insulin secretion (GDIS)
Somatostatin receptor subtype 5 (SSTR5) is one of five G-protein coupled receptors that sense the peptide hormone somatostatin (SST), also called somatotropin release-inhibiting factor. SSTR1–5 are widely distributed throughout the body.1 Their endogenous ligands exist in two isoforms with 14 and 28 amino acids (SST-14 and SST-28), respectively.2 While SST-14 is mainly secreted from pancreatic δ cells, neurons, and the stomach, SST-28 mostly exists in the enteroendocrine δ cells in the small intestine, colon, and pancreas.3 The SST-mediated biological effects involve a broad range of functional regulation and hormonal secretion control, including inhibiting the secretion of both insulin and the insulinotropic glucagon-like peptide 1 (GLP-1), which have been the subject of numerous studies and publications.4
Selective antagonism of endogenous SST effects on SSTR5 should be beneficial for maintaining glucose homeostasis. The target SSTR5 is prominently expressed in pancreatic islet β cells as well as some δ and α cells.5 It is also highly expressed in enteroendocrine cells of the gastrointestinal (GI) tract. SST inhibits GLP-1 secretion via primarily SSTR5,4 while its inhibitory effect on insulin secretion is mediated by multiple SST receptors, including SSTR5.6
Evidence for the role of SSTR5 in glucose homeostasis comes from SSTR5 knockout (KO) mice. Increased glucose-dependent insulin secretion (GDIS) was demonstrated in the perfused pancreas of SSTR5 KO mice as compared with wild type (WT) mice.7 Pancreatic islets isolated from SSTR5 KO mice displayed increased total insulin content as compared with islets obtained from WT mice. As a result, the KO mice exhibited decreased blood glucose and plasma insulin levels and increased leptin and glucagon concentrations compared with WT mice. In addition, the SSTR5 KO mice displayed decreased susceptibility to high fat diet (HFD)-induced insulin resistance.8
SSTR5 selective small molecule antagonists have been reported to lower glucose and insulin excursion during an oral glucose tolerance test (OGTT) in both mice and rats.9−12 Based on these cellular and preclinical pharmacology studies, SSTR5 is an attractive investigational target for treatment of type 2 diabetes mellitus (T2DM). Although a range of therapeutic agents are available for the treatment of T2DM, such as sulfonylureas, metformin, PPARγ-selective agonists, DPP-4 inhibitors, and recently SGLT2 inhibitors, a high proportion of diabetic patients still fail to achieve or maintain glycemic targets due to limitations and/or liabilities associated with these agents.13 An SSTR5 antagonist offers the possible dual effects of increasing GLP-1 release from the GI tract and promoting insulin secretion from the pancreatic islets in response to changes in blood glucose levels. Therefore, an SSTR5 antagonist could serve as monotherapy for the treatment of T2DM and may also be synergistic in combination with antidiabetic GLP-1 protective agents such as DPP-4 inhibitors. To this end, we have investigated the small molecule SSTR5 selective antagonists as therapeutic agents for treatment of T2DM and their potential for synergistic effects with a DPP-4 inhibitor. This study involved optimization of a previously reported SSTR5 selective antagonist, and full preclinical pharmacology characterization of the optimized compound.
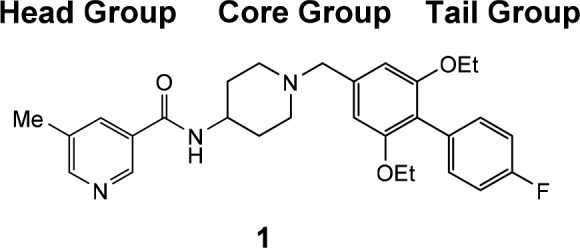
Literature reports of small molecule SSTR5 selective antagonists are relatively few. Martin reported the first nonpeptidic selective SSTR5 antagonists,14 which was followed by additional reports detailing the structure–activity relationship (SAR) around the aminopiperidine based core structures.15,16 These SSTR5 selective antagonists, such as 1 (Table 1), generally consist of three components; a heterocyclic headgroup, an aminopiperidine core group, and a substituted aromatic tail group. Recently, additional reports have appeared showing variation is tolerated in all three regions of this lead series, notably with the discovery of a spirocyclic core subunit.10−12 These accounts prompted us to detail our optimization of this lead series and report our finding of preclinical pharmacologic synergy in promoting circulation of active GLP-1 by oral coadministration of an SSTR5 antagonist with a DPP-4 inhibitor.
Table 1. Lead Series of SSTR5 Antagonists.

All IC50 values calculated are the mean of at least duplicate experiments within a maximal 3-fold range. N.D. = not determined.
Potency against human (h) and mouse (m) SSTR5.
hERG inhibition measured by competitive binding with MK-499, a known hERG blocker.
Decrease in oral glucose AUC vs vehicle in male C57BL/6 mice fed with HFD (D12492) for 21 days, n = 3, t = 0–120 min, compound dosed 1 h before glucose.
In our studies, 1 is a potent SSTR5 antagonist with good efficacy in our acute mouse oral glucose tolerance test (OGTT) with 87% lowering of plasma glucose following a 10 mg/kg oral dose with good residual whole blood exposure (C2.5h = 0.11 μM). We modified the class of SSTR5 antagonists represented by 1 in an attempt to identify new lead structures with diminished potency against cardiac ion-channels such as hERG, while maintaining SSTR5 antagonist potency and SSTR subtype selectivity. Compounds were tested in an SSTR5 filtration binding assay, which measures the competitive displacement of radiolabeled SST-28 from cell membranes expressing the human or mouse SSTR5 receptor. Potent compounds were also tested in a functional assay in Chinese hamster ovary (CHO-K1) cells expressing the human or mouse SSTR5 receptor. This whole cell assay measures the compound’s ability to inhibit the SST-28 mediated reduction of cAMP accumulation induced by forskolin. Potent compounds were also counter screened against the related membrane binding assays for SSTR1–4.
For this optimization, we maintained the reported biaryl tail piece (R) in 1 and synthesized clusters of individual analogs to survey changes in the central core and head piece. The design principle centered on replacing the 4-aminopiperidine core in 1 with alternative scaffolds while conserving the overall architecture for activity. Representative lead compounds from each of these clusters are listed in Table 1, and additional SAR tables within each series and physicochemical data for 1–10 are provided in the Supporting Information. A small set of analogs with fused bicyclic core structures represented by 2 were prepared. The fused cyclopentylpyrole ring system yielded potent SSTR5 antagonists. After a secondary SAR screen of different head pieces (Supporting Information Table S1), compound 2 was found to be optimal and was selected for further profiling (cis-racemic, more potent C-4 diastereomer characterized as described in the Supporting Information). The compound showed reasonable oral bioavailability in a mouse PK experiment (F = 48%, t1/2 = 1.7 h) and modest efficacy in the OGTT assay with 56% lowering of plasma glucose excursion following a 30 mg/kg oral dose with high residual whole blood exposure (C2.5h = 0.76 μM). The compound was devoid of significant off-target activity except for modest potencies on hERG (MK-499 binding IC50 = 15 μM) and weak SSTR1 binding activity (IC50 = 6 μM).
The carbon-linked headgroup series represented by 3 was also explored. A variety of 2-aryl substituents provided similar potency among the 2,5-disubstituted imidazole derivatives such as 3; however, a significant loss of potency was observed for the corresponding 3-oxadiazole derivative (Table S1). Compound 3 is a potent inhibitor of hERG (MK-499 binding IC50 = 0.26 μM) and also maintained potent SSTR3 antagonist activity (IC50 = 119 nM). This ancillary activity was persistent across the entire carbon-linked imidazole core series.
The sulfonamide series represented by 4 and 5 was derived from extending the 4-aminopiperidine core in 1 to spiro- or bipiperidines and replacing the head piece with a sulfonamide. This series of sulfonamides comprises highly potent and highly selective SSTR5 antagonists with moderate bioavailability but with excellent in vivo efficacy in OGTT assays. Compound 5 (mouse PK, F = 13%) lowered glucose excursion in OGTT by 60% following an oral dose of 30 mg/kg despite moderate residual whole blood exposure (C2.5h = 0.092 μM). However, similar to the arylimidazole series characterized by 3, this class of analogs was consistently potent in inhibiting hERG. Sustained SAR investigation efforts failed to mitigate this off-target activity.
The ether linked series, such as 6 and 7, was designed by replacing the connecting NH in 1 with an O or CH2O group. Similar to the sulfonamide series, all of the ether-linked compounds afforded persistent inhibition of hERG. Although these compounds were highly selective SSTR5 antagonists, their potencies were generally moderate without significant changes in ion channel activities.
Finally, replacing the 4-aminopiperidine in 1 with spiroheterocycle variants afforded series represented by 8 and 9, which showed promise in improving both potency and off-target selectivity. Compound 8, tested as a racemic mixture, is one of the most potent binders to the SSTR5 receptor, while 9 afforded high selectivity over ion channel activities, with MK-499 IC50 = 25 μM. Compound 9 displayed good oral bioavailability and reasonable in vivo efficacy of 54% glucose lowering in OGTT assay at a 30 mg/kg oral dose with good whole blood exposure (C2.5h = 0.46 μM), despite its moderate mSSTR5 binding potency (IC50 = 32 nM) and mSSTR5 functional potency (IC50 = 7.4 nM). Examination of the emerging SAR indicated that significant SSTR5 potency was gained by the spiroheterocycle core group as in 8, whereas significant selectivity over hERG was gained by the carboxyl-containing headgroup as in 2. Combining these features (including Supporting Information compounds S25–S30) led to the optimal 10, which provided the desired profile of potency and selectivity. This compound was chosen for more extensive pharmacological profiling.
The synthesis of compound 10 is outlined in Scheme 1. Starting with Boc-protected spiropiperidine intermediate 11, Buchwald–Hartwig C–N coupling with 4-bromobenzoic methyl ester 12 followed by deprotection of the Boc group afforded 13. This intermediate was then alkylated with 14 followed by hydrolysis of the ester to give 10. The compounds discussed in this Letter were synthesized by similar sequences with starting materials that are either commercially available or can be made with simple and well-established steps.
Scheme 1. Synthesis of Spiropiperidine Analog 10.

Reagents and conditions: (a) K3PO4, Pd2(dba)3, dioxane, 100 °C, 88%; (b) HCl, dioxane, EtOAc, RT, 95%; (c) Cs2CO3, DMF, RT, 86%; (d) LiOH, MeOH, 50 °C, 79%. For synthesis of intermediate 14, see Supporting Information.
Compound 10 is an advanced tool for the investigation of SSTR5 pharmacology.17 It is a very potent and highly selective SSTR5 antagonist with hSSTR5 binding (IC50 = 1.2 nM) and cAMP antagonist (IC50 = 1.1 nM). It is inactive in the binding assay against hSSTR1–4 (IC50 > 10 μM), and this represents improved subtype selectivity as compared to 1 (hSSTR1 IC50 < 300 nM). At higher concentrations, 10 exhibited partial agonist activity in the hSSTR5 cAMP assay, achieving 44% maximal activation at 8.3 μM with an inflection point of 2.7 μM. Antagonist 10 is highly selective over hERG (MK-499 binding IC50 = 45 μM) and other cardiovascular ion channels (IKs, Cav1.2, Nav1.5 IC50 > 15 μM). Analysis of 10 in over 160 ancillary target assays (Eurofins Panlabs screen) revealed only moderate binding activity against the prostanoid DP receptor (IC50 = 3 μM).
The pharmacokinetic properties of 10 indicated that it is a suitable tool for in vivo preclinical testing. The oral bioavailability of 10 in rat, dog, and rhesus ranges from 40–72% with unbound free fractions of 2.2–5.7% across species (Table 2). The compound shows no appreciable inhibition of human P450 enzymes (CYP3A4, 2D6, 2C9 IC50 > 50 μM) and no appreciable PXR activation. The solubility of 10 was modest in neutral aqueous solution (0.3 mg/mL), but it was more soluble in a 10% Tween formulation (5.3 mg/mL). This formulation was used for pharmacokinetic studies.
Table 2. Pharmacokinetic Properties of Compound 10.
| species | dosea (iv/po) | Fub (%) | Clc | Vd (K/kg) | t1/2 (h) | F (%) |
|---|---|---|---|---|---|---|
| rat | 1/2 | 5.7 | 15.2 | 2.38 | 1.9 | 41 |
| dog | 0.25/1 | 4.4 | 2.2 | 2.82 | 16.5 | 72 |
| rhesus | 0.25/1 | 5.4 | 11.8 | 1.39 | 2.0 | 40 |
Dosed in mg/kg.
Measured at 0.1 μM concentration with [3H-10]; Fu (%) for human and mouse are 2.2 and 4.6, respectively.
Measured in mL/kg/min.
The pharmacology of 10 was investigated to more fully profile the opportunity provided by selective antagonism of SSTR5. Ex vivo, 10 was incubated with isolated human pancreatic islets from nondiabetic donors using a method we have previously reported (Figure 1).18 The compound potentiated insulin secretion in a glucose-dependent manner. In the presence of high glucose concentration (16 mM, labeled G16), insulin secretion in human islets was significantly enhanced by 10 at both 5 and 0.5 μM compound concentration, presumably by blocking the inhibitory effect of endogenous SST-14 and SST-28 secreted from these islets on insulin secretion. However, insulin secretion at low glucose concentration (2 mM, labeled as G2) was not affected by 10, consistent with the GDIS mechanism. The effect of 10 was similar to 50 nM of the GDIS peptide GLP-1, as well as linoleate, a small molecule GPR40 agonist used as a positive control.19 The secretion of SST-14 and glucagon in these islets was similarly monitored in this experiment and was unaffected by 10 (data not shown).
Figure 1.
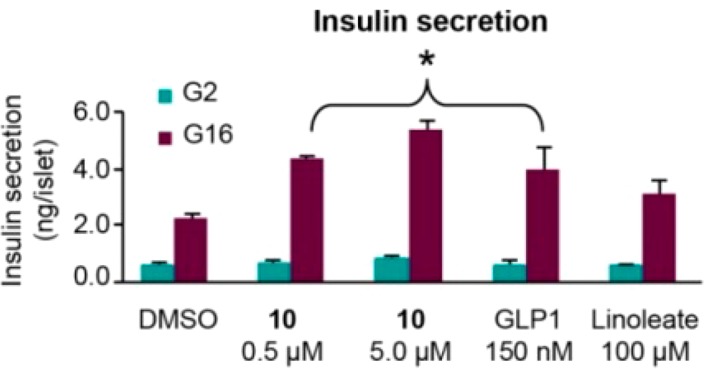
Compound 10 increased glucose-dependent insulin secretion in isolated human islets with 2 and 16 mM of glucose (G2, G16). *P ≤ 0.05 vs DMSO.
Compound 10 also lowered glucose excursions in vivo in a dose-dependent manner during OGTT in HFD mice (Table 3). Oral administration of 0.03 to 3 mg/kg of 10 dose-dependently reduced the net glucose area under the curve (AUC) in OGTT mouse ranging from 23% to 84% as compared to vehicle. Plasma exposures of the compound were measured 3 h post dose. From nine related OGTT experiments in HFD mice, we calculate the minimal exposure for maximal efficacy (EC90) = 484 nM. Furthermore, the exposure required for significant efficacy (≥50% reduction) may be calculated (EC50 ≥ 54 nM). Taking into consideration the measured free fraction in mouse plasma (Fu% = 4.6), the unbound concentration of 10 affording EC50 is 2.5 nM. This is roughly consistent with the mouse SSTR5 IC50 (0.9 nM) in the cell-based cAMP assay. The OGTT efficacy of 10 was entirely ablated in SSTR5 KO mice (Supporting Information), validating SSTR5 as the biological target.
Table 3. Compound 10 Dose Titration in HFD Mice OGTT.
| plasma exposure (nM @ 3 h) | |||
|---|---|---|---|
| oral dose (mg/kg) | blood glu AUC (change %)a | whole blood | unbound |
| 3 | –84** | 351 | 16.1 |
| 1 | –74* | 131 | 6.0 |
| 0.3 | –59* | 34 | 1.6 |
| 0.1 (MED)b | –58* | 11 | 0.5 |
| 0.03 | –23 | 4 | 0.2 |
Reduction in blood glucose AUC as compared to vehicle, n = 8.
MED = minimal dose for significant efficacy. *P < 0.01 vs veh; **P < 0.001 vs veh.
As a glucose-responsive mechanism, administration of an SSTR5 antagonist is expected to afford minimal risk of hypoglycemia. This risk was assessed in 4-h fasted lean C57BL/6N mice (Figure 2). An efficacious oral dose of 10 (3 mg/kg) and a supra-efficacious dose (30 mg/kg) administered to the mice did not decrease basal glucose levels significantly over the subsequent 5 h. Glipizide, a marketed sulfonylurea K+-ATP blocker, was utilized as a positive control in this experiment.20 Glipizide dosed orally at 10 mg/kg caused significant decreases of basal blood glucose levels starting at 60 min after dosing.
Figure 2.
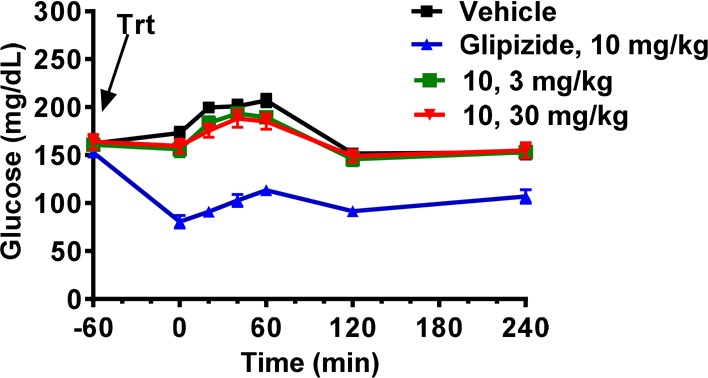
Blood glucose in 4 h fasted lean mice following administration of 10 or glipizide.
Inhibition of the DPP-4 enzyme, which is responsible for the rapid degradation of active GLP-1[7–36] amide to inactive GLP-1[9–36] amide in vivo, is a proven therapy for the treatment of T2DM. Given that a selective SSTR5 antagonist potentiates glucose-induced secretion of GLP-1, we anticipated that coadministration of a DPP-4 inhibitor and an SSTR5 antagonist should have synergistic effects on circulating active GLP-1[7–36] and therefore GDIS. As shown in the OGTT assay results in Figure 3, administration of the SSTR5 antagonist 10 to lean C57/B6N mice afforded a substantial increase in circulating total GLP-1 and plasma insulin levels and a measurable increase in circulating active GLP-1 (blood measurements taken 5 min post glucose challenge). The combination of 10 with the DPP-4 inhibitor des-fluoro sitagliptin21 resulted in significantly increased circulation of active GLP-1 and further increased plasma insulin levels, compared with either 10 or the DPP-4 inhibitor alone. Similar synergistic efficacy between SSTR5 antagonists and DPP-4 inhibitors were recently independently reported by researchers using healthy mice, DIO mice, ZDF rats, and several GDIS receptor KO mouse strains.12,22
Figure 3.
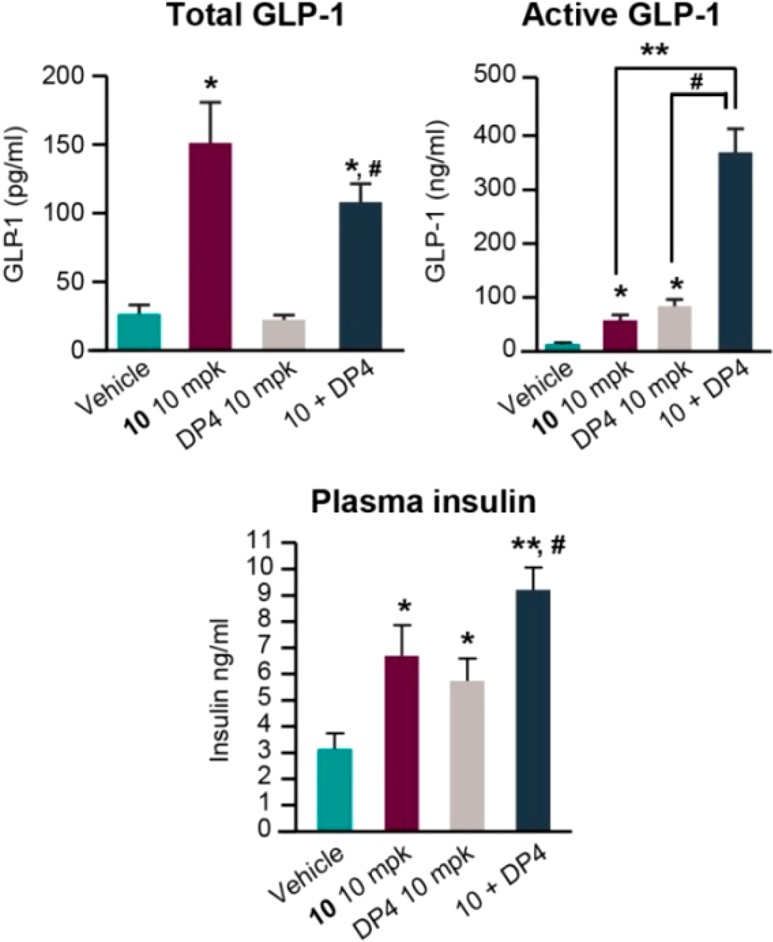
Combination of 10 with DPP-4 inhibitor des-F-sitagliptin (DP4) (10 + 10 mpk) in HFD mouse OGTT studies. *P < 0.05 vs veh; **P < 0.01 vs veh; #P < 0.001 vs 10.
In summary, we have identified multiple new lead SSTR5 antagonists through systematic lead hopping efforts based on the existing literature compound 1. Medicinal chemistry iteration and SAR optimization led to the discovery of azaspirodecanones, a novel series of highly selective and potent SSTR5 antagonists with diverse structural features and minimal off-target activity profiles regarding hERG, PXR, and CYP inhibition. We demonstrated that the exemplar SSTR5 antagonist 10 significantly lowers glucose excursions in a dose-dependent manner in a rodent diabetic model without risk of hypoglycemia. The compound increased pancreatic insulin secretion as well as total and active GLP-1 release, and demonstrates synergistic effects in combination with DPP-4 inhibitors. These results indicate that selective SSTR5 antagonists could serve as therapeutic agents for the treatment of type 2 diabetes as either monotherapy or in combination with existing antidiabetic drugs such as sitagliptin. Further optimization of this lead compound to achieve desirable physical chemical and pharmacokinetic properties are discussed in the companion paper in this issue.
Glossary
ABBREVIATIONS
- SSTR5
somatostatin receptor subtype 5
- SST
somatostatin
- GLP-1
glucagon-like peptide 1
- GI
gastrointestinal
- KO
genetic knockout
- GDIS
glucose-dependent insulin secretion
- WT
genetic wild-type
- HFD
high fat diet
- OGTT
oral glucose tolerance test
- T2DM
type 2 diabetes mellitus
- AUC
area under the curve
- DPP-4
dipeptidyl peptidase 4
- SGLT2
sodium-glucose cotransporter 2
- PK
pharmacokinetic
- mpk
milligram per kilogram
- SAR
structure–activity relationship
- CHO
Chinese hamster ovary
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00305.
Experimental procedures for the preparation of compounds 2–10. SAR table of additional representative analogs. In vitro and in vivo assay protocols. SSTR5 WTKO OGTT study results (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
All authors were employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA during the time this research was conducted. All research was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
The authors declare no competing financial interest.
Supplementary Material
References
- Selmer I.-S.; Schindler M.; Allen J. P.; Humphrey P. P. A.; Emson P. C. Advances in understanding neuronal somatostatin receptors. Regul. Pept. 2000, 90, 1–18. 10.1016/S0167-0115(00)00108-7. [DOI] [PubMed] [Google Scholar]
- Patel Y. C.; Greenwood M. T.; Warszynska A.; Panetta R.; Srikant C. B. All five cloned human somatostatin receptors (hSSTR1–5) are functionally coupled to adenyl cyclase. Biochem. Biophys. Res. Commun. 1994, 198, 605–612. 10.1006/bbrc.1994.1088. [DOI] [PubMed] [Google Scholar]
- Reisine T.; Bell G. I. Molecular biology of somatostatin receptors. Endocr. Rev. 1995, 16, 427–442. 10.1210/edrv-16-4-427. [DOI] [PubMed] [Google Scholar]
- Chisholm C.; Greenberg G. R. Somatostatin-28 regulates GLP-1 secretion via somatostatin receptor subtype 5 in rat intestinal cultures. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E311–E317. 10.1152/ajpendo.00434.2001. [DOI] [PubMed] [Google Scholar]
- Ludvigsen E.; Olsson R.; Stridsberg M.; Janson E. T.; Sandler S. Expression and distribution of somatostatin receptor subtypes in the pancreatic islets of mice and rats. J. Histochem. Cytochem. 2004, 52, 391–400. 10.1177/002215540405200310. [DOI] [PubMed] [Google Scholar]
- Fagan S. P.; Azizzadeh A.; Moldovan S.; Ray M. K.; Adrian T. E.; Ding X.; Coy D. H.; Brunicardi F. C. Insulin secretion is inhibited by subtype five somatostatin receptor in the mouse. Surgery 1998, 124, 254–259. 10.1016/S0039-6060(98)70128-X. [DOI] [PubMed] [Google Scholar]
- Strowski M. Z.; Kohler M.; Chen H. Y.; Trumbauer M. E.; Li Z.; Szalkowski D.; Gopal-Truter S.; Fisher J. K.; Schaeffer J. M.; Blake A. D. Somatostatin receptor subtype 5 regulates insulin secretion and glucose homeostasis. Mol. Endocrinol. 2003, 17, 93–106. 10.1210/me.2001-0035. [DOI] [PubMed] [Google Scholar]
- Tirone T. A.; Norman M. A.; Moldovan S.; DeMayo F. J.; Wang X. P.; Brunicardi F. C. Pancreatic somatostatin inhibits insulin secretion via SSTR-5 in the isolated perfused mouse pancreas model. Pancreas 2003, 26, e67–e73. 10.1097/00006676-200304000-00025. [DOI] [PubMed] [Google Scholar]
- Sprecher U.; Mohr P.; Martin R. E.; Maerki H. P.; Sanchez R. A.; Binggeli A.; Kuennecke B.; Christ A. D. Novel, non-peptidic somatostatin receptor subtype 5 antagonists improve glucose tolerance in rodents. Regul. Pept. 2010, 159, 19–27. 10.1016/j.regpep.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Yamasaki T.; Hirose H.; Yamashita T.; Takakura N.; Morimoto S.; Nakahata T.; Kina A.; Nakano Y.; Tamura Y. O.; Sugama J.; Odani T.; Shimizu Y.; Iwasaki S.; Watanabe M.; Maekawa T. Discovery of novel somatostatin receptor subtype 5 (SSTR5) antagonists: Pharmacological studies and design to improve pharmacokinetic profiles and human Ether-a-go-go-related gene (hERG) inhibition. Bioorg. Med. Chem. 2017, 25, 4153–4162. 10.1016/j.bmc.2017.06.003. [DOI] [PubMed] [Google Scholar]
- Hirose H.; Yamasaki T.; Ogino M.; Mizojiri R.; Tamura-Okano Y.; Yashiro H.; Muraki Y.; Nakano Y.; Sugama J.; Hata A.; Iwasaki S.; Watanabe M.; Maekawa T.; Kasai S. Discovery of novel 5-oxa-2,6-diazoaspiro[3.4]oct-6-ene derivatives as potent, selective, and orally available somatostatin receptor subtype 5 (SSTR5) antagonists for the treatment of type 2 diabetes mellitus. Bioorg. Med. Chem. 2017, 25, 4175–4193. 10.1016/j.bmc.2017.06.007. [DOI] [PubMed] [Google Scholar]
- Farb T. B.; Adeva M.; Beauchamp T. J.; Cabrera O.; Coates D. A.; Meredith T. D.; Droz B. A.; Efanov A.; Ficorilli J. V.; Gackenheimer S. L.; Martinez-Grau M. A.; Molero V.; Ruano G.; Statnick M. A.; Suter T. M.; Bokvist K. B.; Barrett D. G. Regulation of endogenous (male) rodent GLP-1 secretion and human islet insulin secretion by antagonism of somatostatin receptor 5. Endocrinology 2017, 158, 3859–3873. 10.1210/en.2017-00639. [DOI] [PubMed] [Google Scholar]
- Maruthur N. M.; Tseng E.; Hutfless S.; Wilson L. M.; Suarez-Cuervo C.; Berger Z.; Chu Y.; Iyoha E.; Segal J. B.; Bolen S. Diabetes medications as monotherapy or metformin-based combination therapy for type 2 diabetes: A systematic review and meta-analysis. Ann. Intern. Med. 2016, 164, 740–751. 10.7326/M15-2650. [DOI] [PubMed] [Google Scholar]
- Martin R. E.; Green L. G.; Guba W.; Kratochwil N.; Christ A. Discovery of the first nonpeptidic, small-molecule, highly selective somatostatin receptor subtype 5 antagonists: A chemogenomics approach. J. Med. Chem. 2007, 50, 6291–6294. 10.1021/jm701143p. [DOI] [PubMed] [Google Scholar]
- Martin R. E.; Mohr P.; Maerki H. P.; Guba W.; Kuratli C.; Gavelle O.; Binggeli A.; Bendels S.; Alvarez-Sanchez R.; Alker A.; Polonchuk L.; Christ A. D. Benzoxazole piperidines as selective and potent somatostatin receptor subtype 5 antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 6106–6113. 10.1016/j.bmcl.2009.09.024. [DOI] [PubMed] [Google Scholar]
- Alker A.; Binggeli A.; Christ A. D.; Green L.; Maerki H. P.; Martin R. E.; Mohr P. Piperidinyl-nicotinamides as potent and selective somatostatin receptor subtype 5 antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 4521–4525. 10.1016/j.bmcl.2010.06.026. [DOI] [PubMed] [Google Scholar]
- Aster S. D.; Duffy J. L.; Liang G.-B.; Shao P.; Ye F.. Substituted spirocyclic amines useful as antidiabetic compounds. WO 2010/129729, November 11, 2010.
- Tan C. P.; Feng Y.; Zhou Y.-P.; Eiermannm G. J.; Petrov A.; Zhou C.; Lin S.; Salituro G.; Meinke P.; Mosley R.; Akiyama T. E.; Einstein M.; Kumar S.; Berger J. P.; Mills S. G.; Thornberry N. A.; Yang L.; Howard A. D. Selective small-molecule agonists of G protein–coupled receptor 40 promote glucose-dependent insulin secretion and reduce blood glucose in mice. Diabetes 2008, 57, 2211–2219. 10.2337/db08-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G.; Alvarez-Curto E.; Hudson B. D.; Prihandoko R.; Tobin A. B. FFA4/GPR120: Pharmacology and Therapeutic Opportunities. Trends Pharmacol. Sci. 2017, 38, 809–821. 10.1016/j.tips.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard C. E.; Han X.; Brensinger C. M.; Bilker W. B.; Cardillo S.; Flory J. H.; Hennessy S. Comparative risk of serious hypoglycemia with oral antidiabetic monotherapy: A retrospective cohort study. Pharmacoepidemiol. Drug Saf. 2018, 27, 9–18. 10.1002/pds.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu J.; Woods J.; Zhou Y.-P.; Sinha Roy R.; Li Z.; Zycband E.; Feng Y.; Zhu L.; Li C.; Howard A. D.; Moller D. E.; Thornberry N. A.; Zhang B. B. Chronic inhibition of dipeptidyl peptidase-4 with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes 2006, 55, 1695–1704. 10.2337/db05-1602. [DOI] [PubMed] [Google Scholar]
- Briere D. A.; Bueno A. B.; Gunn E. J.; Michael M. D.; Sloop K. W. Mechanism to elevate endogenous GLP-1 beyond injectable GLP-1 analogs and metabolic surgery. Diabetes 2018, 67, 309–320. 10.2337/db17-0607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.