

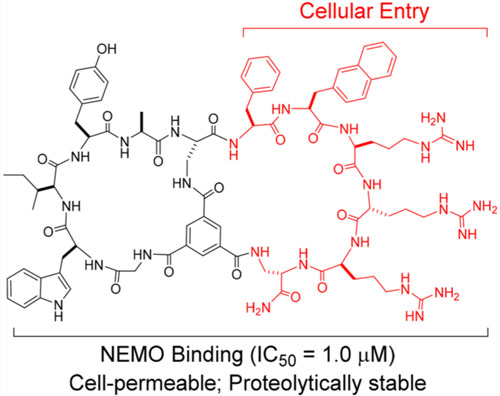
Abstract
Macrocyclic peptides are capable of binding to flat protein surfaces such as the interfaces of protein–protein interactions with antibody-like affinity and specificity, but generally lack cell permeability in order to access intracellular targets. In this work, we designed and synthesized a large combinatorial library of cell-permeable bicyclic peptides, in which the first ring consisted of randomized peptide sequences for potential binding to a target of interest, while the second ring featured a family of different cell-penetrating motifs, for both cell penetration and target binding. The library was screened against the IκB kinase α/β (IKKα/β)-binding domain of NF-κB essential modulator (NEMO), resulting in the discovery of several cell-permeable bicyclic peptides, which inhibited the NEMO-IKKβ interaction with low μM IC50 values. Further optimization of one of the hits led to a relatively potent and cell-permeable NEMO inhibitor (IC50 = 1.0 μM), which selectively inhibited canonical NF-κB signaling in mammalian cells and the proliferation of cisplatin-resistant ovarian cancer cells. The inhibitor provides a useful tool for investigating the biological functions of NEMO/NF-κB and a potential lead for further development of a novel class of anti-inflammatory and anticancer drugs.
Graphical Abstract
INTRODUCTION
Protein–protein interaction (PPI) is a fundamental aspect of biological processes. The number of PPIs in human cells has been estimated to exceed 100 000, well above the ~30 000 human genes.1 Intracellular PPIs are challenging targets for current drug modalities (i.e., small molecules and biologics) and represent the largest untapped opportunity for therapeutic development. Over the past decade or so, macrocyclic peptides have emerged as a new class of drug modalities for targeting challenging proteins such as those involved in PPIs.2–4 Several powerful platform technologies were developed to rapidly synthesize and screen large libraries of mono- and bicyclic peptides (up to 1013 different molecules) against essentially any protein target of interest.5–12 Macrocyclic peptides isolated from these libraries have demonstrated antibody-like affinity and specificity for binding to flat protein surfaces, including those found at the PPI interfaces. Proteolytic instability, a major limitation of peptide therapeutics in the past, has largely been overcome by cyclization and/or the incorporation of unnatural building blocks (e.g., D-amino acids). On the other hand, the poor membrane permeability of peptides (linear or cyclic) remains a major obstacle to their applications for intracellular targets. To overcome this barrier, we13–16 and others17–19 have previously incorporated cell-penetrating peptides (CPPs) into mono- and bicyclic peptides to render them cell-permeable and biologically active against intracellular targets. Moreover, we have demonstrated that large libraries of cell-permeable bicyclic peptides can be combinatorially synthesized in the one bead-one compound (OBOC) format20 by incorporating a fixed CPP motif (typically 5 to 7 residues) in one ring and a degenerate/random peptide sequence in the second ring.21,22 The OBOC libraries were then directly screened for binding to an intracellular protein of interest in vitro, resulting in hit peptides which are cell-permeable and capable of modulating the biological activity of the target protein in a cellular setting. We anticipated that the previous design is likely to discover bicyclic peptides in which the two rings perform separate functions, with one ring (the CPP ring) for cellular entry while the other for binding to the target protein. We envisioned that, by replacing the fixed CPP motif with a “library” of different CPP sequences, it might be possible to identify bicyclic peptides in which the CPP motif contributes to target binding in addition to ensuring cellular entry. This would greatly decrease the size of the bicyclic peptides without compromising their potency, specificity, or permeability, thereby improving the “drug-likeness” of the ligands. Herein we demonstrate the validity of this approach by discovering a moderately potent and cell-permeable bicyclic peptidyl inhibitor against a regulatory protein of the canonical NF-κB signaling pathway, NF-κB essential modulator (NEMO).
NF-κB is a family of transcription factors that play key roles in regulating immune response, inflammation, cell differentiation, and cell survival.23,24 Two different signaling pathways lead to the activation of NF-κB, known as the canonical and noncanonical pathways. During canonical NF-κB signaling, receptor activation at the cell surface results in the formation of an active inhibitor of κB (IκB)-kinase (IKK) complex consisting of IKKα, IKKβ and NEMO (which is also called IKKγ). The activated IKKα/β phosphorylates IκB, causing proteasomal degradation of IκB and release of NF-κB, which then translocates into the nucleus and turns on transcription of target genes. Aberrant activation of the canonical pathway is implicated in many inflammatory and autoimmune diseases, as well as cancer.25–27 Noncanonical NF-κB signaling, which is required for lymphoid organogenesis and B cell survival and maintenance, is mediated through a different set of cell surface receptors and cytoplasmic adaptors and does not involve NEMO.28 Inhibition of the NEMO-IKK interaction represents an attractive anti-inflammatory and anticancer strategy, as it would block the IKK activity induced by pro-inflammatory stimuli but not affect the basal NF-κB activity required for normal B and T cell function.29 In contrast, direct inhibition of NF-κB or inhibitors against IKKα/β would inhibit both signaling pathways causing greater toxicity.
As an intracellular PPI, the NEMO-IKK complex has been a challenging target for drug discovery. Despite more than a decade of intense efforts, small-molecule inhibitors of modest potency against the NEMO-IKK interaction (IC50 ~ 20 μM) have only recently been reported.30–32 It remains unclear whether these modest hits can be further developed into potent and selective inhibitors. A weak peptidyl inhibitor, Antp-NBD (Kd ~ 37 μM), which consists of the 11-residue NEMO-binding domain (NBD) of IKKβ covalently linked to a CPP, Drosophila Antennapedia domain (Antp), was also developed.33 Antp-NBD has been used as an NF-κB inhibitor in numerous in vitro and in vivo studies. For example, it has demonstrated in vivo efficacy for treatment of acute lung injury,34 Duchenne muscular dystrophy,35,36 and large B-cell lymphoma37 in murine and canine models. However, its low potency and poor pharmacokinetics (e.g., serum half-life of ~15 min) prevented further clinical development. We38 and others39 have attempted to improve the potency, proteolytic stability, and/or cell-permeability of Antp-NBD through cyclization, but the potency of the resulting peptides remained rather modest (IC50 in the low μM range). In this work, we have discovered a relatively potent bicyclic peptidyl inhibitor against the NEMO-IKK interaction (IC50 =1.0 μM), by screening of a combinatorial library followed by limited optimization. In cell culture, the inhibitor blocks the activation of IKKs and the transcriptional activity of NF-κB, and selectively kills cancer cells with elevated NF-κB activity but not normal cells. The inhibitor provides a powerful tool compound for investigating the biological function of NEMO and NF-κB and a potential lead for further development of a novel class of anti-inflammatory and anticancer drugs.
RESULTS AND DISCUSSION
Design, Synthesis and Screening of Bicyclic Peptide Library.
We designed a bicyclic peptide library featuring random peptide sequences of 3–6 residues in the first ring (A ring) and 12 different CPP sequences in the second ring (B ring) (Figure 1). The peptide sequences in the A ring were constructed through combinatorial synthesis using a set of 24 proteinogenic and unnatural amino acids (e.g., D-amino acids; Figure S1). The 12 CPP sequences consisted of different combinations of two or three aromatic hydrophobic residues (l-or d-Phe and l- or d-naphthylalanine) and three or four l- or d-arginine residues40 (Table S1 in Supporting Information) and were prepared by parallel synthesis. Because the NEMO surface near the IKK-binding site is negatively charged (vide infra),41 we envisioned that in addition to ensuring cell penetration, some of the positively charged CPP sequences might also interact electrostatically with the negatively charged NEMO surface. The bicyclic library has a theoretical diversity of 2.4 × 109 and was synthesized on 130-μm TentaGel beads in the one bead-two compound (OBTC) format,7 with each bead displaying a unique bicyclic peptide on its surface layer and a linear peptide of identical sequence in its inner layer as an encoding tag. The library design also included a propargylglycine-β-alaninehydroxylmethylbenzoyl ester (Pra-B-Hmb) linker, which was intended for selective on-bead fluorescent labeling and release of the bicyclic peptide for an additional round of in-solution screening,7 although the latter turned out to be unnecessary. Approximately 600 mg of the library (~600000 different compounds) was screened for binding to NEMO in two different rounds as detailed under Experimental Procedures, resulting in 12 unique hit sequences (Table S2). Ten of these 12 peptides were resynthesized and tested for inhibition of the NEMO-IKKβ interaction by using a homogeneous time-resolved fluorescence (HTRF) assay.42 All 10 peptides inhibited the NEMO-IKKβ interaction with IC50 values of 3.4–28 μM (Table S2). The three most potent compounds (hits #1, #4, and #7 in Table S2) were labeled with fluorescein and their cellular uptake into HeLa cells was quantitated by flow cytometry analysis. All three peptides were cell-permeable and had similar cell entry efficiency (Figure S2). Hit #4 (peptide 2 in Table 1) was selected for further optimization, because it showed the highest potency in the HTRF assay, with an IC50 value of 3.4 μM (Figure 2a).
Figure 1.

Structures of the bicyclic peptide library, hit #4, and peptide 7. Amino acid residues in peptide 7 are numbered from N- to C-terminus. The CPP sequence is shown in red, whereas the residues modified during optimization are shown in blue color. B, β-alanine; CPP, cell-penetrating peptide; Hmb, hydroxylmethylbenzoyl; Pra, propargylglycine; Δ, l-2,3-diaminopropionic acid (Dap).
Table 1.
Sequences, NEMO-Binding Affinities, and Cell-Permeability of Peptides in This Worka
| peptide ID | sequence | IC50 (nM) | perpermeability (%) |
|---|---|---|---|
| 1 | RQIKIWFQNRRMKWKKGGTALDWSWLQTE | >40 | 35 |
| 2 | Tm(GWIY)Δ(FΦRRRΔ)-BBK | 3.4 ± 0.6 | 76 |
| 3 | Tm(GWIY)Δ(FΦRRRRΔ)-BBK | 1.5 ± 0.2 | 55 |
| 4 | Tm(GWIY)Δ(FΦRRRΔ)-RBK | 1.8 ± 0.3 | 71 |
| 5 | Tm(GWIY)Δ(FΦRRRΔ)-rBK | 1.3 ± 0.4 | 71 |
| 6 | Tm(GWIY)Δ(FΦRrRΔ)-BBK | 5.9 ± 0.6 | 322 |
| 7 | Tm(GWIYA)Δ(FΦRrRΔ)-BBK | 1.0 ± 0.1 | 163 |
| 8 | Tm(GWIYa)Δ(FΦRrRΔ)-BBK | 2.9 ± 0.1 | |
| 9 | Tm(AGWIY)Δ(FΦRrRΔ)-BBK | 2.2 ± 0.1 | |
| 10 | Tm(aGWIY)Δ(FΦRrRΔ)-BBK | 2.3 ± 0.3 | |
| 11 | Tm(AWIYA)Δ(FΦRrRΔ)-BBK | 1.2 ± 0.2 | |
| 12 | Tm(GAIYA)Δ(FΦRrRΔ)-BBK | 3.0 ± 0.3 | |
| 13 | Tm(GWAYA)Δ(FΦRrRΔ)-BBK | 1.7 ± 0.1 | |
| 14 | Tm(GWIAA)Δ(FΦRrRΔ)-BBK | 2.8 ± 0.3 | |
| 15 | Tm(GWIYA)Δ(AΦRrRΔ)-BBK | 1.9 ± 0.2 | |
| 16 | Tm(GWIYA)Δ(FARrRΔ)-BBK | 2.8 ± 0.2 | |
| 17 | Tm(GWIYA)Δ(FΦArRΔ)-BBK | 1.1 ± 0.3 | |
| 18 | Tm(GWIYA)Δ(FΦRaRΔ)-BBK | 2.1 ± 0.2 | |
| 19 | Tm(GWIYA)Δ(FΦRrAΔ)-BBK | 1.2 ± 0.2 | |
| 20 | Tm(GAIAA)Δ(FΦRrRΔ)-BBK | 16 ± 2 | 176 |
| 21 | Tm(GAAAA)Δ(FΦRrRΔ)-BBK | >100 | 110 |
Tm, trimesic acid; Δ, l-2,3-diaminopropionic acid, Φ, l-2-naphthylalanine; B, l-β-alanine; r, d-arginine. See Figure S1 for detailed structures. Cell-permeability values are relative to that of CPP1 (100%).
Figure 2.

Inhibition of the NEMO-IKKα/β interaction and NF-κB signaling by peptide 7 and control peptides. (a) Inhibition of the NEMO-IKKβ interaction as monitored by the HTRF assay. (b) Cellular uptake efficiency of FITC-labeled peptides into HeLa cells as determined by flow cytometry. All values are relative to that of CPP1 (100%). (c) Differential effects of peptide 7 on the basal (open bars) and TNFα-induced NF-κB activation (closed bars) in HEK293(Luc) cells. *, p < 0.001 using Student’s t test. (d) Comparison of peptides 1, 7, 20, and 21 for inhibition of TNFα-induced luciferase activity in HEK293(Luc) cells. (e) Western blot showing the effect of peptide 7 on IκBα and IKKβ levels in HT29 colon cancer cells in the absence and presence of TNFα.
Optimization of Hit Peptide.
We conducted a limited medicinal chemistry campaign to improve the NEMO-binding affinity and/or cell-permeability of peptide 2. We first modified the CPP motif in the B ring to improve cellular uptake. Addition of a fourth arginine to the CPP motif, either inside (Table 1, peptide 3) or outside the B ring (peptides 4 and 5) slightly decreased the cellular entry efficiency. Interestingly, replacement of the second arginine with D-arginine increased the uptake efficiency by 4.2-fold (peptide 6). We therefore chose FΦRrR (where Φ is l-naphthylalanine and r is d-arginine) as the CPP motif and modified the A ring to improve the NEMO-binding affinity. Insertion of an Ala immediately N-terminal to the internal Dap residue (with the intention of creating an additional site of diversification) increased the binding affinity by 5.9-fold (IC50 = 1.0 μM for peptide 7; Figure 1 and 2). On the other hand, insertion of a d-Ala at this position (N-terminal to Dap) or l/d-alanine at the N-terminus of the A ring sequence was less effective (peptides 8, 9 and 10). In a fluorescence anisotropy (FA) assay, fluorescently labeled peptide 7 bound directly to NEMO with a Kd value of 220 ± 80 nM, but only weakly to a panel of control proteins (Kd > 10 μM) (Figure S3). Next, the proteolytic stability of peptide 7 was assessed by incubation in human serum at 37 °C and quantitation of the remaining peptide as a function of time by analytical HPLC. Peptide 7 showed minimal degradation (<1%) after 20 h of incubation. Under the same conditions, Antp-NBD (Table 1, peptide 1) was rapidly degraded with a half-life of ~20 min (Figure S4). Given its excellent proteolytic stability and respectable potency and cell permeability (163% relative to CPP1,13,40 a previously reported and relatively efficient cyclic CPP; Figure 2a and 2b), we selected peptide 7 (Figure 1) for further characterization.
Structural Basis of Peptide 7 Binding to NEMO.
To gain insight into how peptide 7 interacts with NEMO, we performed in silico docking of peptide 7 to the NEMO protein, with the best-scoring binding pose of peptide 7 shown in Figure 3. As expected, peptide 7 binds to the canonical IKK-binding site on NEMO, with the peptide A ring partially inserted into a hydrophobic groove normally occupied by the NBD of IKKβ.41 The side chain of Tyr-4 is deeply inserted into a large hydrophobic pocket and excluded from the solvent (Figure 3b). Ile-3 interacts intimately with a shallow hydrophobic pocket on the NEMO surface, whereas Trp-2 makes surface contacts primarily through the pyrrole ring while the benzene ring is mostly solvent exposed. Gratifyingly, the B ring (CPP ring) of peptide 7 also makes important interactions with NEMO. As anticipated, the three arginine residues of the CPP motif interact electrostatically with the acidic patch next to the NBD binding groove (Figure 3c). Phe-7, which is a part of the CPP motif, is completely buried and makes hydrophobic interaction with NEMO. Nal-8, on the other hand, is almost completely solvent exposed, although the naphthalene ring may make hydrophobic interactions with the NEMO surface.
Figure 3.

In silico model of the NEMO-peptide 7 complex. (a) The overall complex between peptide 7 (shown as green sticks) and NEMO (PDBID: 3BRT; shown as van der Waals surface) with residues critical for the NEMO-IKKβ interaction shaded pink. (b) Close-up of the interaction between the A ring (in green) and NEMO including the insertion of Tyr-4 into a hydrophobic pocket. (c) Zoom-in view of the charge–charge interactions between the three arginine residues of peptide 7 and acidic residues on NEMO. Basic and acidic residues of NEMO are shown in blue and red, respectively.
To assess the validity of the molecular modeling results, we performed an “alanine scan” of peptide 7 by replacing each residue with an alanine (or d-alanine) and determining the IC50 values of the resulting peptides against the NEMO-IKKβ interaction (Table 1, peptides 11–19). Consistent with the modeling results, replacement of Trp-2, Ile-3, or Tyr-4 in the A ring with alanine significantly decreased the potency of the inhibitor (by 2- to 3-fold for peptides 12–14). Likewise, substitution of Ala for Phe-7 (peptide 15), Nal-8 (peptide 16), or d-Arg at position 10 (peptide 18) also significantly decreased the NEMO-binding affinity. Thus, both molecular modeling and alanine-scan results indicate that the CPP motif in the B ring also interacts with the NEMO protein and contributes to the overall binding affinity and specificity of peptide 7.
On the basis of the SAR data, we generated two negative control peptides by replacing two (Trp-2 and Tyr-4) or three of the NEMO-binding residues in the A ring (Trp-2, Ile-3, and Tyr-4) with Ala. The resulting peptides (peptides 20 and 21) have similar cellular entry efficiencies to peptide 7, but 16- and >100-fold lower NEMO-binding affinity, respectively (Figure 2a,b and Table 1).
Inhibition of NF-κB Signaling.
The ability of peptide 7 to enter the cell and block the intracellular NEMO-IKK interaction was assessed by using HEK293(Luc) cells, which harbor a luciferase gene under the transcriptional control of NF-κB.33 In the absence of any inhibitor, treatment of HEK293(Luc) cells with TNFα activated the IKK complex and nuclear translocation of NF-κB, resulting in a 15-fold increase in the luciferase activity (Figure 2c). Prior incubation of the cells with peptide 7 dose-dependently inhibited the TNFα-induced NF-κB activation with an IC50 value of 10 μM (Figure 2c,d). Antp-NBD (peptide 1) also inhibited NF-κB activation, but with an IC50 of ~41 μM, in agreement with previous reports.33 The two negative control peptides (20 and 21) decreased the luciferase activity only at high concentrations. It should be noted that at very high concentrations, cationic CPPs may cause nonspecific cytotoxicity to mammalian cells and reduce the expression of the luciferase gene. Further, at high concentrations (>10 μM), CPPs can directly cross the plasma membrane through a yet poorly defined “direct translocation” mechanism,43 in addition to endocytic uptake, resulting in much greater cytosolic entry efficiency. Both factors may cause artificially low cellular IC50 values in the luciferase assay for poorly active peptides (such as Antp-NBD, 20, and 21). As discussed earlier, NEMO is not involved in the noncanonical NF-κB signaling pathway. A specific inhibitor against NEMO should not block the basal NF-κB activity, which has important physiological functions. To test whether peptide 7 also affects the basal NF-κB activity, the luciferase assay was repeated in the absence of TNFα stimulation. As shown in Figure 2c, peptide 7 did not inhibit the basal NF-κB activity. Instead, it caused a small but statistically significant increase in the basal NF-κB activity (~1.5-fold). Such a small stimulatory effect was previously reported for Antp-NBD and attributed to increased availability of IKKα/β for the noncanonical NF-κB signaling pathway when the canonical pathway is blocked.33
To ascertain that inhibition of NF-κB signaling by peptide 7 is caused by disruption of the NEMO-IKK interaction, we treated HT29 colon cancer cells with peptide 7 and TNFα and examined the levels of phosphorylated (and activated) IKKβ and IκBα, which acts immediately downstream of the NEMOIKK complex, by Western blot analysis. As expected, peptide 7 (0–25 μM) dose-dependently inhibited TNFα-induced phosphorylation of IKKβ, while the total intracellular IKKβ level was unchanged (Figure 2e). Concomitantly, peptide 7 increased the intracellular level of IκBα, presumably by inhibiting TNFα-induced proteasomal degradation. The same effects on NF-κB signaling had previously been reported for Antp-NBD.29,33 Importantly, in agreement with the luciferase assay results (Figure 2c), peptide 7 showed no effect on the phospho-IKKβ or IκBα level when cells were not stimulated with TNFα. Taken together, the above results indicate that peptide 7 efficiently enters the cytosol of mammalian cells and selectively inhibits the canonical NF-κB signaling pathway by blocking the NEMO-IKK interaction.
Anticancer Activity.
Aberrant activation of NF-κB via the canonical signaling pathway plays critical roles during the initiation and progression of certain cancers.25 For example, NF-κB is excessively activated in ovarian cancer cell lines A2780 and CP70.44,45 Moreover, blocking canonical NF-κB activation in A2780 cells resulted in apoptosis.44 We therefore tested peptide 7 for potential anticancer activity against cell lines with hyperactivated NF-κB. We first examined the entry of peptide 7 into A2780 ovarian cancer cells by live-cell confocal microscopy. Treatment of the cells with 5 μM FITC-labeled peptide 7 for 2 h resulted in intense green fluorescence inside all cells (Figure 4a). Next, the effect of peptide 7 on the viability of A2780 and CP70 cells was assessed by a methylene blue cell viability assay. Peptide 7 dose-dependently reduced the viability of both ovarian cancer cell lines, with LD50 values of ~20 and 10 μM for A2780 and CP70 cells, respectively (Figure 4b). In contrast, peptide 7 had no significant effect on ovarian surface epithelial cells (OSE), a noncancerous ovarian cell line with normal NF-κB activity.45 The greater sensitivity of CP70 cells to peptide 7 than A2780 cells is consistent with the greater dependency on NF-κB activity by the former.46 CP70 cells were derived from A2780 cells by subjecting the latter to low, repeated doses of cisplatin and selecting for cisplatin resistant clones.46 Peptide 7 also reduced the viability of A375 melanoma cells, which have constitutively activated NF-κB (Figure S5). Ianaro et al. previously reported that treatment of A375 cells with Antp-NBD decreased the NF-κB activity and induced growth arrest and apoptosis.47
Figure 4.

Anticancer activity of peptide 7. (a) Live-cell confocal microscopic image of A2780 ovarian cancer cells after 2 h treatment with 5 μM FITC-peptide 7. I, FITC fluorescence; II, DIC. (b) Viability of ovarian cancer cells (A2780 and CP70) and noncancerous ovarian cells (OSE) in the presence of increasing concentrations of peptide 7, as determined by the methylene blue assay. (c) Comparison of peptide 7 and control peptides for their effect on the viability of A2780 cells. (d) Effect of peptide 7 and control peptides on noncancerous OSE cells. Viability tests in panel (c) and (d) were performed by the MTT assay.
To determine whether the observed anticancer activity of peptide 7 is correlated with specific inhibition of the NEMOIKK interaction, we also tested the effect of peptides 1, 20, and 21 on A2780 cells by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell viability assay. Although Antp-NBD also reduced the viability of A2780 cells in a dose-dependent manner, it was less active than peptide 7 (LD50 values of ~30 μM and >80 μM for peptide 7 and Antp-NBD, respectively; Figure 4c). This is consistent with the fact that Antp-NBD is less potent in NEMO binding, less cell-permeable, and less metabolically stable than peptide 7. As expected, peptides 20 and 21 were also much less active than peptide 7 in this assay. Under the same conditions, none of the four peptides had significant effect on the viability of noncancerous OSE cells (Figure 4d). These results strongly suggest that the observed anticancer activity of peptide 7 (and Antp-NBD) is caused by their inhibition of the NEMO-IKK interaction and NF-κB activation.
CONCLUSION
In summary, we have discovered a novel class of cell-permeable bicyclic peptidyl inhibitors against the NEMO-IKKα/β interaction. To our knowledge, peptide 7 represents the most potent NEMO inhibitor reported to date. Most excitingly, peptide 7 selectively inhibits the canonical NF-κB signaling pathway but not the noncanonical NF-κB pathway. Consequently, peptide 7 inhibits the proliferation and survival of cancer cells with elevated NF-κB activities but has minimal cytotoxicity to normal cells. Further optimization of peptide 7 may lead to a novel therapeutic agent for treatment of cancer and autoimmune/inflammatory diseases. Peptide 7 should also provide a useful chemical probe for investigating the biological function of NEMO and NF-κB in vitro and in vivo. In addition, we have demonstrated, for the first time, the feasibility of designing cell-permeable bicyclic peptides whose CPP motif performs the dual function of cell penetration and target engagement. This strategy results in biologically active macro-cycles that are relatively small in size and have more “drug-like” properties. Indeed, with a molecular mass of 1748, peptide 7 is only slightly larger than cyclosporine A, a clinically used cycloundecapeptide natural product. Finally, the bicyclic peptide library strategy developed in this work should be applicable to the discovery of cell-permeable bicyclic peptides against other intracellular targets.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification.
Escherichia coli BL21-(DE3) cells were transformed with a pGEX4T3-NEMO(1–196) plasmid and grown at 37 °C in Luria broth supplemented with 0.05 mg/mL ampicillin to an OD600 of 0.4. Expression of GST-NEMO was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (150 μM final concentration). After 5 h at 30 °C, the cells were harvested by centrifugation. The cell pellet was suspended in 40 mL of lysis buffer (50 mM Tris-HCl, 100 mM NaCl, 0.5 mM MgCl2, 5 mM β-mercaptoethanol, 0.1% Triton-X-100, pH 8.0), 100 μg/mL lysozyme, 100 μL of DNase I (New England BioLabs), and 100 μL of Halt Protease Inhibitor Cocktail (EDTA-free) (Thermo Scientific). This mixture was stirred at 4 °C for 30 min and briefly sonicated (2 × 10 s pulses). The crude lysate was centrifuged to yield a clear supernatant, which was directly loaded onto a glutathione-Sepharose 4B column (GE Healthcare). The bound protein was eluted from the column with 10 mM glutathione in 50 mM Tris-HCl (pH 8.0), concentrated to 0.5 mL with the use of Amicon Ultra-15 centrifugal filter units (MWCO 10 kDa), and dialyzed against PBS before flash freezing.
An engineered prokaryotic expression plasmid pJCC04a,48 which encodes a fusion protein containing an N-terminal six-histidine tag, thioredoxin, a TEV protease cleavage site, and the K703R/K704R mutant form of IKKβ C-terminal fragment (amino acids 701–745) [His-thx-IKKbKK/RR(701–745)], was kindly provided by Dr. Maria Pellegrini (Dartmouth College). His-thx-IKKbKK/RR(701–745) was similarly expressed in E. coli BL21 (DE3) cells and purified by affinity chromatography using a HisTrap FF column (GE Healthcare). The fusion protein was eluted with 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, 250 mM imidazole, 2 mM β-mercaptoethanol and treated with TEV protease (150 units for 1 mg of fusion protein) for 16 h at 4 °C to remove the thioredoxin (thx). The resulting protease digestion mixture was reloaded onto the HisTrap column. The flow-through fraction was collected and concentrated to ~2 mg/mL using Amicon Ultra-15 centrifugal filter units (MWCO 10 kDa). The IKKbKK/RR(701–745) peptide was biotinylated by treatment with a 10-fold molar excess of biotin-NHS at 4 °C overnight. The biotinylated IKKbKK/RR(701–745) was purified by reversed-phase HPLC equipped with a C18 column and stored frozen at –80 °C.
Library Synthesis and Screening.
The peptide library was synthesized on 2 g of TentaGel S NH2 resin (130 μm) by modifying a previously reported protocol.7 A detailed description of the library design and synthesis is included in the Supporting Information (Figure S1). Library screening was performed according to previously established protocols.7 Approximately 600 mg of the bicyclic peptide library was swollen in DCM and washed extensively with DMF, ddH2O and finally incubated overnight at 4 °C in 1.0 mL of blocking buffer (30 mM sodium phosphate, pH 7.4, 150 mM NaCl, 0.05% Tween 20, 3% BSA and 0.1% gelatin). The solution was drained and the resin was resuspended in blocking buffer containing 1.0 μM biotinylated GSTNEMO for 4 h at 4 °C. Unbound NEMO was washed away with blocking buffer and the beads were resuspended in 10 mL of blocking buffer. Twenty μL of M280 streptavidin-coated Dynabeads (Invitrogen) was added to the solution and allowed to incubate on a rotary wheel for 1 h at 4 °C. The magnetic beads were isolated from the bulk by using a TA Dynal MPC-1 magnetic particle concentrator (Invitrogen). Hit beads were transferred to a Bio-Spin column (0.8 mL, BioRad) and incubated in blocking buffer containing 1.0 μM biotinylated GST-NEMO for 4 h at 4 °C. The solution was drained and the resin was washed with blocking buffer to remove unbound protein. The resin was resuspended in 1 mL of blocking buffer and streptavidin-alkaline phosphatase (SA-AP) conjugate was added to the tube (1 mg/mL final concentration). After 10 min at 4 °C the solution was drained and the beads were quickly washed with 1 mL of blocking buffer (3×) and 1 mL of staining buffer (30 mM Tris pH 8.5, 100 mM NaCl, 5 mM MgCl2, and 20 μM ZnCl2) (3×). The resin was resuspended in 1.5 mL of staining buffer in a Petri dish and 150 μL of a 5-bromo-4-chloro-3-indolyl-phosphate (BCIP) solution (5 mg/mL) was added. After 30 min, 50 μL of 1 M HCl was added to quench the reaction and the intensely turquoise positive beads were isolated under a dissecting microscope (Figure S6). The sequences of hit beads were determined using partial Edman degradation-mass spectrometry (PED-MS)49 as previously published and representative spectra are available in Supporting Information (Figure S7).
HTRF Assay.
Recombinant GST-NEMO (20 nM), biotin-IKKβKK/RR(701–745) (50 nM), streptavidin labeled with d2 acceptor(2.5 μg/mL), anti-GST monoclonal antibody labeled with Tb donor(2.5 μg/mL), and varying concentrations of peptide (0–100 μM) were mixed in PBS containing 1 mM TCEP and 0.01% Triton X-100 (total volume 20 μL) in a 384-well plate. The plate was incubated for 2 h at room temperature to establish an equilibrium. The HTRF signals were measured on a Tecan Infinite M1000 Pro microplate reader and plotted as a function of the peptide concentration. The data was analyzed using GraphPad Prism 6.0 and IC50 values were obtained by fitting the data to the dose–response inhibition curves. Data presented were the mean ± SD of three independent experiments.
Flow Cytometry Analysis of Cellular Uptake.
HeLa cells were seeded into 12-well plates (1.5 × 105 cells per well) 24 h before treatment. The next day, 5 μM FITC-labeled peptide in DMEM containing 10% FBS was added to each well and allowed to incubate at 37 °C for 2 h. After compound treatment, the cells were washed with DPBS, detached from the plate with trypsin (0.25%), diluted in DMEM containing 10% FBS and pelleted at 250g for 5 min. This washing process was repeated once to remove any free compound and left over trypsin. The washed pellet was resuspended in DPBS with 1% FBS and analyzed on a BD LSR II flow cytometer. Data presented were the mean ± SD of three independent experiments.
Confocal Microscopy.
A2780 cells were seeded into a glass-bottomed culture dish (MatTek) and cultured overnight at 37 °C (5 × 104 cells/mL). The next day the cells were washed with DPBS (2×) and treated with 5 μM fluorescein-labeled peptide in RPMI media containing 10% FBS, and incubated at 37 °C for 2 h. The media containing the peptide was removed and the cells were washed twice with phenol-free RPMI media (2×). The cells were imaged on a Visitech Infinity 3 Hawk 2D-array live cell imaging confocal microscope equipped with a 60× oil objective.
NF-κB Luciferase Assays.
Culture media was exchanged for DMEM containing 10% FBS and 1% penicillin/streptomycin the day before seeding to remove hygromycin B. HEK293(Luc) cells were seeded in 50 μL of assay medium (DMEM, 10% FBS, and 1% penicillin/streptomycin) in an opaque 96-well microplate (3000 cells per well) and incubated overnight. The peptide inhibitors were added to the cells the next day in 5 μL of assay medium and the plate was incubated at 37 °C for 2 h. After that, 5 μL of assay media containing recombinant TNFα (final concentration 5 ng/mL) was added to the wells. The plate was then returned to the incubator for 4 h at 37 °C. Finally, 50 μL of ONE-Step luciferase assay reagent was added to each well and after 10 min the luminescence was measured on a Tecan Infinite M1000 Pro microplate reader. Data presented were the mean ± SD of at least three independent experiments (n = 6 for basal activities).
Methylene Blue Cell Viability Assay.
A2780 (2500 cells/well), CP70 (2000 cells/well) or OSE (3000 cells/well) were seeded into a 96-well microplate in 100 μL respective growth media and incubated at 37 °C overnight. The following day, the seeded cells were treated with increasing concentrations of peptide for 72 h. The treated cells were washed with DPBS and fixed to the plate by treating with 3.7% formaldehyde solution for 1 h. Fixation solution was removed and the fixed cells were treated with 1.0% methylene blue solution for 30 min. Following staining, the plate was rinsed under running water for approximately five min and left to dry. Finally, 100 μL of solubilization buffer (10% acetic acid, 50% methanol, and 40% H2O) was added to each well and the absorbance was measured at 630 nm using a Tecan Infinite M1000 Pro microplate reader. Data presented were the mean ± SD of five independent experiments.
MTT Cell Viability Assay.
HeLa, HEK293, A2780, OSE or A375 cells were seeded into a 96-well microplate (3000 cells/well) in 100 μL of respective growth media and incubated at 37 °C overnight. The next day, a serial dilution of peptide was added to each well in 10 μL of assay medium. The treated cells were incubated at 37 °C with 5% CO2 for 72h. Following compound treatment, 10 μL of MTT stock solution (Roche) was added to each well. After an additional 4 h at 37 °C, 100 μL of SDS-HCl solubilizing solution was added to each well and the plate was returned to the incubator overnight at 37 °C. A Tecan Infinite M1000 Pro microplate reader was used the following morning to measure the absorbance of the formazan product at 565 nm. Data presented were the mean ± SD of three independent experiments.
Immunoblot Analysis.
HT29 cells were grown in a 6-well plate to 80–90% confluency in standard DMEM (Life Technologies) supplemented with 10% FBS and 1% penicillin-streptomycin sulfate at 37 °C in 5% CO2. The cells were treated with 0, 2, 10, or 25 μM peptide 7 for 2 h followed by stimulation with TNFα (5 ng/mL) for 10 min. The cells were rapidly washed with cold PBS and trypsinized to detach from the plate. Following centrifugation and resuspension in PBS to remove any remaining trypsin the pelleted cells were lysed in 100 μL of Pierce RIPA Buffer (Thermo) containing protease and phosphatase inhibitors for 30 min on ice. Cell lysates were centrifuged at 15 000 rpm for 20 min, and the extracted proteins in the supernatant were collected. After measuring the concentration of the samples using the BCA Protein Assay Kit (Thermo) and adjusting the total protein concentration to be the same for each sample, SDS-PAGE loading buffer was added and the samples were boiled for 5 min. Equal amounts of protein were loaded onto a 10% SDS-PAGE gel (130 V, 2.5 h) followed by electrophoretic transfer to a nitrocellulose membrane at 4 °C (90 V, 2.5 h). The membrane was blocked using TBST buffer (20 mM Tris pH 7.5, 150 mM NaCl, 0.1% (v/v) Tween-20) containing 5% milk proteins (Bio-Rad) at room temperature for 1 h and finally the membrane was probed with the following rabbit monoclonal antibodies: p-IKKα (Ser176)/IKKβ (Ser177) (Cell Signaling Technologies, 2071), IKKβ (Cell Signaling Technologies, 8943), IκBα (Cell Signaling Technologies, 4812), and β-Actin (Sigma, A5441). Primary antibodies were diluted according to the suggestion of the manufacturer and incubated with the nitrocellulose membrane overnight at 4 °C. After washing, the appropriate HRP conjugated secondary antibody was added to each membrane and incubated at RT for 2 h. The membrane was washed with TBST solution again and the signals were detected with the Chemiluminescent HRP Antibody Detection Reagent (Denville, E-2500) by following the manufacturer’s protocol.
In Silico Docking of Peptide 7 to NEMO.
The all-atom 3D structure of peptide 7 was prepared in Maestro from the 2D structure (Schrodinger, LLC ver. 11.1) and relaxed using Polak–Ribier conjugate gradient minimization to resolve steric and torsional strain introduced during conversion. To account for the complex conformational landscape of a bicyclic peptide, a conformational ensemble of structures was obtained for peptide 7 using molecular dynamics. In brief, peptide 7 was parametrized using the OPLS3 force-field and then solvated in an orthorhombic periodic box containing 1867 TIP3P water molecules.50 The system was neutralized with the addition of three Cl– ions and then simulated for 1 ns at 303 K and 1.01 MPa, controlled using the Nose-Hoover thermostat and Martyna–Tobias–Klein barostat, respectively, through the Desmond software package.51 Structures corresponding to every 10 ps of the simulation were extracted using the Trajectory tool in Maestro. To ensure that the ensemble contains a diverse selection of initial compound geometries, the molecular dynamics structures were passed through the Macrocycle Conformational Sampling tool in MacroModel using the OPLS3 force-field. Generalized Born/Solvent Area water electrostatics were applied, sampling across a temperature range from 300 to 1000 K using a global low-mode search over 50 simulation cycles.52 Redundant conformers (defined as a heavy-atom RMSD of ≤0.75 Å) were rejected, yielding an ensemble of 105 structures spanning an energy range of 20 kcal/mol, which were carried forward for docking.
The receptor was prepared from the reported crystal structure of a NEMO dimer (PDBID: 3 V3B),41 by removing cocrystallized solvent and reconstructing incomplete side-chains using Prime (Schrodinger LLC). The binding site was defined as a 20-Å cube centered on the geometric mean of receptor residues Leu-93, Phe-97 and Val-104. Rigid-receptor/flexible-ligand docking was performed using extra-precision mode in Glide.53 Ligand flexibility was accounted for by applying a scaling factor of 0.8 to ligand atom van der Waals radii and the generation of 100 000 poses per ligand sampled, for a theoretical total of 100 000 000 poses sampled for peptide 7 during the combined docking runs. Top scoring poses were subjected to a brief round of energy minimization using Embrace (Schrodinger LLC) to remove any steric clashes resulting from van der Waals’ radii scaling during docking. The final poses were analyzed and electrostatic potential surfaces generated using UCSF Chimera.54
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. M. Pellegrini for the IKKβ(701–745) plasmid andV. Roberts for technical assistance. Financial support from the National Institutes of Health (GM110208 and GM122459) is gratefully acknowledged. C.A.R. was supported by a Chemistry-Biology Interface Training Program (GM08512). Drug discovery work in the Lindert laboratory is supported through NIH (R01 HL137015, R03 AG054904), and a Falk Medical Research Trust Catalyst Award.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b06738.
Experimental details and additional information (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Venkatesan K; Rual J-F; Vazquez A; Stelzl U; Lemmens I; Hirozane-Kishikawa T; Hao T; Zenkner M; Xin X; Goh KI; Yildirim MA; Simonis N; Heinzmann K; Gebreab F; Sahalie JM; Cevik S; Simon C; de Smet AS; Dann E; Smolyar A; Vinayagam A; Yu H; Szeto D; Borick H; Dricot A; Klitgord N; Murray RR; Lin C; Lalowski M; Timm J; Rau K; Boone C; Braun P; Cusick ME; Roth FP; Hill DE; Tavernier J; Wanker EE; Barabási AL; Vidal M Nat. Methods 2009, 6, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Dougherty PG; Qian Z; Pei D Biochem. J 2017, 474, 1109–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Cardote TAF; Ciulli A ChemMedChem 2016, 11, 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Passioura T; Katoh T; Goto Y; Suga H Annu. Rev. Biochem 2014, 83, 727–752. [DOI] [PubMed] [Google Scholar]
- (5).Tavassoli A; Benkovic SJ Angew. Chem., Int. Ed 2005, 44, 2760–2763. [DOI] [PubMed] [Google Scholar]
- (6).Joo SH; Xiao Q; Ling Y; Gopishetty B; Pei DJ Am. Chem. Soc 2006, 128, 13000–13009. [DOI] [PubMed] [Google Scholar]
- (7).Lian W; Upadhyaya P; Rhodes CA; Liu Y; Pei DJ Am. Chem. Soc 2013, 135, 11990–11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Millward SW; Fiacco S; Austin RJ; Roberts RW ACS Chem. Biol 2007, 2, 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Heinis C; Rutherford T; Freund S; Winter G Nat. Chem. Biol 2009, 5, 502–507. [DOI] [PubMed] [Google Scholar]
- (10).Sako Y; Morimoto J; Murakami H; Suga HJ Am. Chem. Soc 2008, 130, 7232–7234. [DOI] [PubMed] [Google Scholar]
- (11).Kleiner RE; Dumelin CE; Tiu GC; Sakurai K; Liu DR J. Am. Chem. Soc 2010, 132, 11779–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Guillen Schlippe YV; Hartman MCT; Josephson K; Szostak JW J. Am. Chem. Soc 2012, 134, 10469–10477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Qian Z; Liu T; Liu Y-Y; Briesewitz R; Barrios AM; Jhiang SM; Pei D ACS Chem. Biol 2013, 8, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lian W; Jiang B; Qian Z; Pei DJ Am. Chem. Soc 2014, 136, 9830–9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Upadhyaya P; Qian Z; Selner NG; Clippinger SR; Wu Z; Briesewitz R; Pei D Angew. Chem., Int. Ed 2015, 54, 7602–7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bedewy W; Liao H; Abou-Taleb NA; Hammad SF; Nasr T; Pei D Org. Biomol. Chem 2017, 15, 4540–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Dietrich L; Rathmer B; Ewan K; Bange T; Heinrichs S; Dale TC; Schade D; Grossmann TN Cell Chem. Biol 2017, 24, 958–968. [DOI] [PubMed] [Google Scholar]
- (18).Quach K; LaRochelle J; Li X-H; Rhoades E; Schepartz A Bioorg. Med. Chem 2018, 26, 1197–1202. [DOI] [PubMed] [Google Scholar]
- (19).Speltz TE; Danes JM; Stender JD; Frasor J; Moore TW ACS Chem. Biol 2018, 13, 676–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lam KS; Salmon SE; Hersh EM; Hruby VJ; Kazmierski WM; Knapp RJ Nature 1991, 354, 82–84. [DOI] [PubMed] [Google Scholar]
- (21).Jiang B; Pei DJ Med. Chem 2015, 58, 6306–6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Trinh TB; Upadhyaya P; Qian Z; Pei D ACS Comb. Sci 2016, 18, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zhang Q; Lenardo MJ; Baltimore D Cell 2017, 168, 37–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Napetschnig J; Wu H Annu. Rev. Biophys 2013, 42, 443–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Baud V; Karin M Nat. Rev. Drug Discovery 2009, 8, 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Gasparini C; Feldmann M Curr. Pharm. Des 2012, 18, 5735–45. [DOI] [PubMed] [Google Scholar]
- (27).Herrington FD; Carmody RJ; Goodyear CS J. Biomol. Screening 2016, 21, 223–242. [DOI] [PubMed] [Google Scholar]
- (28).Cildir G; Low KC; Tergaonkar V Trends Mol. Med 2016, 22, 414–429. [DOI] [PubMed] [Google Scholar]
- (29).Baima ET; Guzova JA; Mathialagan S; Nagiec EE; Hardy MM; Song LR; Bonar SL; Weinberg RA; Selness SR; Woodard SS; Chrencik J; Hood WF; Schindler JF; Kishore N; Mbalaviele GJ Biol. Chem 2010, 285, 13498–13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Robbins P; Niedernhofer L; Kamenecka T; Wilson GM Patent Application WO2016196117A1, 2015.
- (31).De Falco F; Di Giovanni C; Cerchia C; De Stefano D; Capuozzo A; Irace C; Iuvone T; Santamaria R; Carnuccio R; Lavecchia A Biochem. Pharmacol 2016, 104, 83–94. [DOI] [PubMed] [Google Scholar]
- (32).Zhao J; Zhang L; Mu X; Doebelin C; Nguyen W; Wallace C; Clemens PR; Wilson GM; Watkins SC; Solt LA; Cameron MD; Huard J; Niedernhofer LJ; Kamenecka TM; Robbins PD PLoS Biol. 2018, 16, e2004663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).May MJ; D’Acquisto F; Madge LA; Glockner J; Pober JS; Ghosh S Science 2000, 289, 1550–1554. [DOI] [PubMed] [Google Scholar]
- (34).Huang J; Li L; Yuan W; Zheng L; Guo Z; Huang W Mediators Inflammation 2016, 2016, 7349603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Reay DP; Yang M; Watchko JF; Daood M; O’Day TL; Rehman KK; Guttridge DC; Robbins PD; Clemens PR Neurobiol. Dis 2011, 43, 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kornegay J; Peterson JM; Bogan DJ; Kline W; Bogan JR; Dow JL; Fan Z; Wang J; Ahn M; Zhu H; Styner M; Guttridge DC Skeletal Muscle 2014, 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Habineza Ndikuyeze G; Gaurnier-Hausser A; Patel R; Baldwin AS; May MJ; Food P; Krick E; Propert KJ; Masson NJ PLoS One 2014, 9, e95404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Qian Z; Rhodes CA; McCroskey LC; Wen J; Appiah-Kubi G; Wang DJ; Guttridge DC; Pei D Angew. Chem., Int. Ed 2017, 56, 1525–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bruno PA; Morriss-Andrews A; Henderson AR; Brooks CL III; Mapp AK Angew. Chem., Int. Ed 2016, 55, 14997–15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Qian Z; Martyna A; Hard RL; Wang J; Appiah-Kubi G; Coss C; Phelps MA; Rossman JS; Pei D Biochemistry 2016, 55, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Rushe M; Silvian L; Bixler S; Chen LL; Cheung A; Bowes S; Cuervo H; Berkowitz S; Zheng T; Guckian K; Pellegrini M; Lugovskoy A Structure 2008, 16, 798–808. [DOI] [PubMed] [Google Scholar]
- (42).Gotoh Y; Nagata H; Kase H; Shimonishi M; Ido M Anal. Biochem 2010, 405, 19–27. [DOI] [PubMed] [Google Scholar]
- (43).Madani F; Lindberg S; Langel Ü ; Futaki S; Graslund AJ Biophys. 2011, 2011, 414729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Hernandez L; Hsu SC; Davidson B; Birrer MJ; Kohn EC; Annunziata CM Cancer Res. 2010, 70, 4005–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Chen Y; Camacho SC; Silvers TR; Razak AR-A; Gabrail NY; Gerecitano JF; Kalir E; Pereira E; Evans BR; Ramus SJ; Huang F; Priedigkeit N; Rodriguez E; Donovan M; Khan F; Kalir T; Sebra R; Uzilov A; Chen R; Sinha R; Halpert R; Billaud J-N; Shacham S; McCauley D; Landesman Y; Rashal T; Kauffman M; Mirza MR; Mau-Sørensen M; Dottino P; Martignetti JA Clin. Cancer Res 2017, 23, 1552–1563. [DOI] [PubMed] [Google Scholar]
- (46).Johnson SW; Swiggard PA; Handel LM; Brennan JM; Godwin AK; Ozols RF; Hamilton TC Cancer Res. 1994, 54, 5911. [PubMed] [Google Scholar]
- (47).Ionaro A; Tersigni M; Belardo G; Di Martino S; Napolitano M; Palmieri G; Sini M; De Maio A; Ombra M; Gentilcore G; Capone M; Ascierto PA; Ialenti A Cancer Lett. 2009, 274, 331–336. [DOI] [PubMed] [Google Scholar]
- (48).Guo B; Audu CO; Cochran JC; Mierke DF; Pellegrini M Biochemistry 2014, 53, 6776–6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Thakkar A; Wavreille AS; Pei D Anal. Chem 2006, 78, 5935–5939. [DOI] [PubMed] [Google Scholar]
- (50).Friesner RA; Murphy RB; Peasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschargrin PC; Mainz DT J. Med. Chem 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- (51).Sindhikara D; Spronk SA; Day T; Borrelli K; Cheney DL; Posy SL J. Chem. Inf. Model 2017, 57, 1881–1894. [DOI] [PubMed] [Google Scholar]
- (52).Harder E; Damm W; Maple J; Wu C; Reboul M; Xiang JY; Wang L; Lupyan D; Dahlgren MK; Knight JL; Kaus JW; Cerutti DS; Krilov G; Jorgensen WL; Abel R; Friesner RA J. Chem. Theory Comput 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- (53).Bowers KJ; Chow E; Xu H; Dror RO; Eastwood MP; Gregersen BA; Klepeis JL; Kolossváry I; Moraes MA; Sacerdoti FD; Salmon JK; Shan Y; Shaw DE In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), 2006. [Google Scholar]
- (54).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.