

Abstract
Methods to identify signatures of selective sweeps in population genomics data have been actively developed, but mostly do not identify the specific mutation favored by selection. We present a method, iSAFE, that uses a statistic derived solely from population genetics signals to accurately pinpoint the favored mutation in a large region (~5 Mbp). iSAFE does not require any knowledge of demography, specific phenotype under selection, or functional annotations of mutations.
Human genetic data have revealed a multitude of genomic regions believed to be evolving under positive selection. Methods for detecting regions under selection from genetic variations exploit a variety of genomic signatures. Allele frequency based methods analyze the distortion in site frequency spectrums; Linkage Disequilibrium (LD) based methods use extended homozygosity in haplotypes; other methods use differences in allele frequency between populations; and finally, composite methods combine multiple test scores to improve the resolution1–3. Recently, a lack of rare (singleton) mutations has been used to detect very recent selection4. The signature of a selective sweep can be captured even when standing variation or multiple de novo mutations create a ‘soft’ sweep of distinct haplotypes carrying the favored mutation. Paired with deep sequencing data, these methods have identified multiple regions believed to be under selection, and provide a window into genetic adaptation and evolution.
In contrast, little work has been done to identify the favored mutation in a selective sweep. Grossman et al.5 note that different selection signals identify overlapping but different regions, and a composite of multiple signals (CMS) can localize the site of the favored mutation. An alternative strategy is to use rank SNPs based on their functional annotations. However, the signal of selection is often spread over regions up to 1–2 Mbp on either side3, and the high LD makes it difficult to pinpoint the favored mutation. Here, we propose a method, iSAFE (integrated Selection of Allele Favored by Evolution), that exploits coalescent-based signals in ‘shoulders’3 of the selective sweep (genomic regions proximal to the region under selection, but carrying the selection signal) to rank all mutations within a large (5 Mbp) region based on their contribution to the selection signal.
Haplotype Allele Frequency (HAF) score is a haplotypic score that aims to separate carrier haplotypes from non-carriers without knowing the favored mutation6. Based on properties of the HAF-score, we develop a SAFE-score (see Online Methods and Supplementary Note 1; Fig. 1a,b and Supplementary Fig. 1) that tends to be maximized for the favored mutation in a small region (50 kbp), but the performance decays when larger regions are investigated. To address the more general case of large regions (~5 Mbp) under selection, we developed the iSAFE-score, which uses a 2-step procedure to identify the favored variant. In the first step, it finds the best candidate mutations in small (low recombination) windows using the SAFE-score. Then, it combines SAFE-scores of all variants over all windows to give an iSAFE-score to each variant in the large region (see Online Methods and Supplementary Note 1; Fig. 2a,b).
Figure 1.
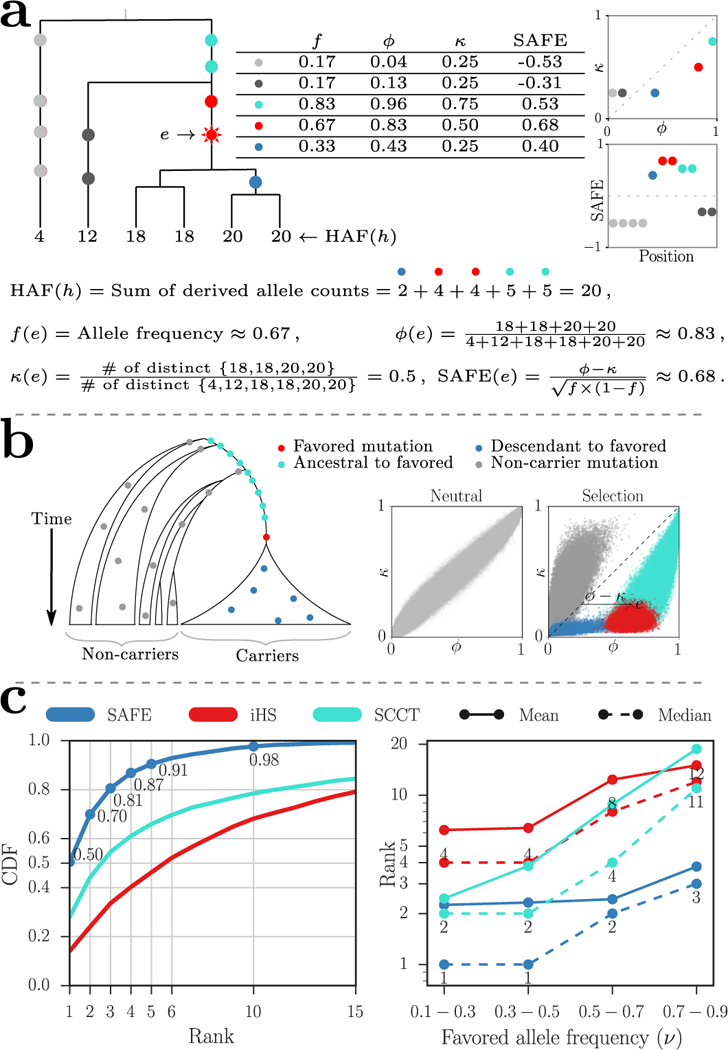
Illustration and performance of the SAFE method. (a) The HAF score for haplotype h is the sum of the derived allele counts of the mutations on h. Carriers of the favored mutation have higher fraction of the total HAF score of the sample (high ϕ)6, and lower number of distinct haplotypes compared to non-carriers (low κ). (b) Schematic of a no-recombination (for exposition purposes) genealogy under a selective sweep. The mutations can be categorized as ‘non-carrier’ (gray), ‘ancestral to favored’ (turquoise) arising prior to the favored mutation, and ‘descendant to favored’ (blue) that arise on haplotypes carrying the favored mutations but after the favored mutation, and the favored mutation itself (red). In the right panels, simulations showing ϕ versus κ values for each variant in a neutral evolution and a selective sweep for 1000 simulations with favored allele frequency (ν0 = 0.5) and default values for other simulation parameters (see Online Methods). The joint-distribution of ϕ and κ, in a selective sweep, changes in a dramatic but predictable manner that separates out non-carrier (gray), descendant (blue), and ancestral (turquoise) mutations from the favored (red) mutations. The SAFE score computes a normalized difference of the two statistics, ϕ and κ. (c) Performance (favored mutation rank) of SAFE compared to iHS and SCCT on 50 kbp windows with 1000 simulations per frequency bin. The simulations were performed with default parameter values (see Online Methods) for a fixed population size with ongoing selective sweeps. The left panel combines all allele frequencies while the right panel shows median and mean ranks for replicates divided into four bins.
Figure 2.
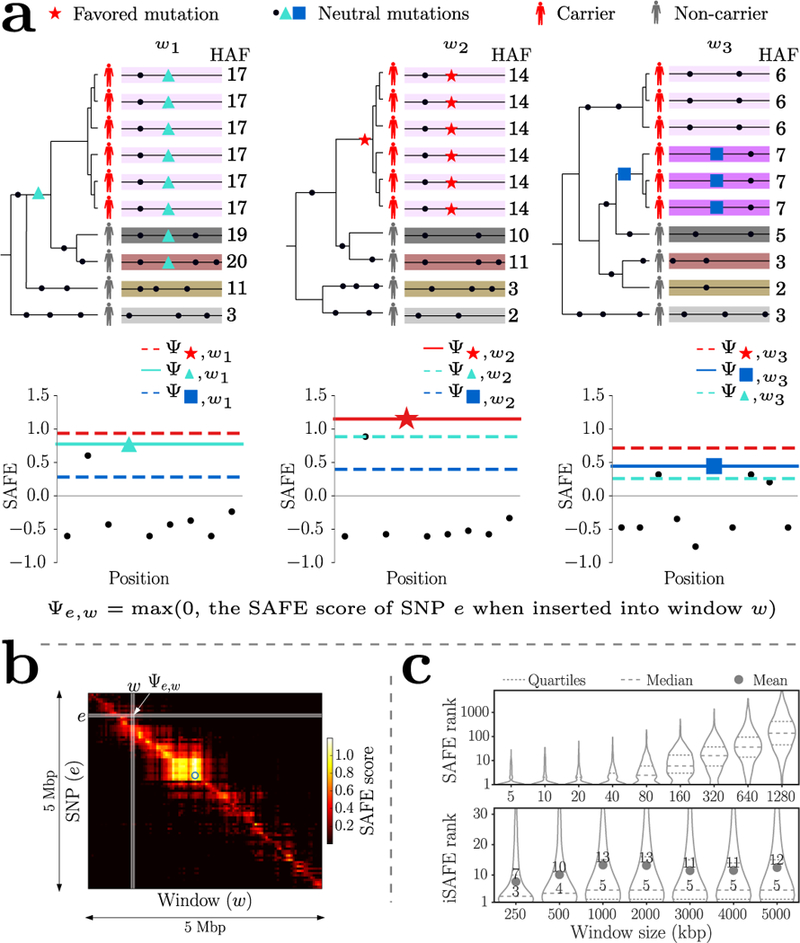
Illustration of the iSAFE method. (a) The red-star, turquoise-triangle, and blue-square denote the favored, ancestral, and descendant mutations, respectively. As different windows have different genealogies due to recombination, the SAFE-score of a non-favored mutation e is relatively low when inserted in other windows. In contrast, the SAFE-score of the favored mutation is likely to dominate other mutations (Supplementary Note 1). (b) The Ψe,w matrix for a 5 Mbp region around LCT gene in FIN population shows that the ‘shoulder’ of selection can extend for a few Mbp. The blue circle shows the location of the putative favored mutation rs4988235. (c) SAFE and iSAFE performance (rank distribution of favored mutation) as a function of window size with 1000 simulations per bin. The dashed (dotted) line represents median (quartile), and decays for large windows while iSAFE is robust to increase in window size.
The main alternatives to iSAFE are Composite of Multiple Signals (CMS)5, and Selection detection by Conditional Coalescent Tree (SCCT)7. CMS combines statistics from different selection tests, including the integrated Haplotype Score (iHS)8, so as to localize the signal. In order to develop a unified probabilistic model, CMS expects control populations as input, as well as demographic models, and cannot be run using only the SNP matrix. Therefore, we first compared SAFE against iHS and SCCT in simulations. The median SAFE rank of the favored mutation in a 50 kbp region was 1 out of ∼250 variants (Fig. 1c, left), and the favored mutation ranked among the top 5 in 91% of simulations. In comparison, the median ranks of iHS and SCCT were 6 and 3, respectively. The SAFE-score performance remained robust to a large range of parameter choices (Supplementary Fig. 2). However, in testing with increasing window sizes, we observed that the median rank increases beyond 80 kbp, perhaps because of the confounding signal at the shoulders of the selective sweep (Fig. 2c).
iSAFE, unlike SAFE, is specifically designed to exploit signal from the shoulders of the sweep (see Online Methods and Fig. 2a,b). iSAFE showed consistently high performance as the window size was increased from 250 kbp all the way to 5 Mbp (Fig. 2c). The median rank remained between 3 and 5 up to 5 Mbp, and the performance remained robust to a large range of parameter choices (Supplementary Fig. 3–6). iSAFE greatly improved upon iHS and SCCT, placing the favored mutation within top 20 in 88% of the cases, in contrast to iHS (39%), and SCCT (34%), for an ongoing selective sweep with fixed population size (Supplementary Fig. 3).
Not surprisingly, iSAFE performance deteriorates when the favored mutation is fixed, or near fixation (favored allele frequency (ν) > 0.9 in Supplementary Fig. 3). To handle this special case, we include individuals from non-target populations using a specific protocol (see Online Methods). Subsequently, the performance remained unchanged for ν < 0.9 and dramatically improved for high frequencies, including when the favored mutation was fixed in the target population (Supplementary Fig. 3). We also tested iSAFE against CMS using a model of human demography. While CMS showed excellent performance in localizing the favored mutation, iSAFE scoring greatly improved the ranking. For example, iSAFE ranked the favored mutation within the top 20 in 94% of the simulations of a 5 Mbp region (Fig. 3a and Supplementary Fig. 4), in contrast to CMS, which had a top 20 ranking in 35% of cases.
Figure 3.
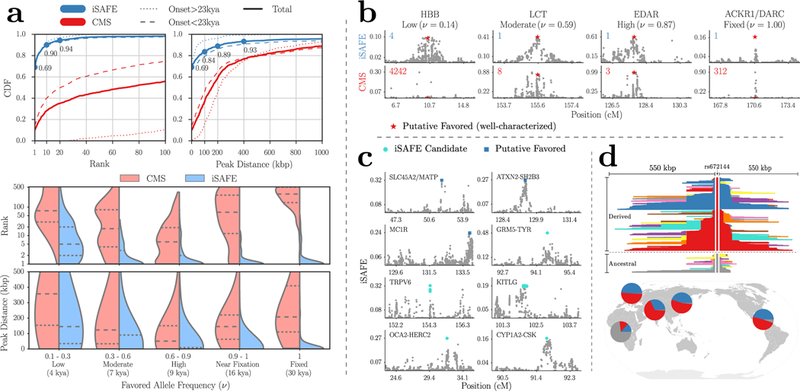
iSAFE performance. (a) The top left (right) panel is the Cumulative Distribution Function (CDF) of favored mutation rank (peak distance) for iSAFE and CMS scores. The lower panel shows the iSAFE performance (rank and peak distance distributions of favored mutation) as a function of favored allele frequency (ν ) in the target population (EUR). The dashed (dotted) line represents median (quartiles). All data is based on 1000 simulations of 5 Mbp genomic regions simulated using a model of human genome based on the human demography (Supplementary Fig. 14). The time of onset of selection was chosen at random (using the distribution in Supplementary Fig. 14) after the out of Africa event, in the lineage of EUR population (as the target population). When the onset of selection is before split of EUR and EAS (>23kya), both (EUR and EAS) are under selection. (b) iSAFE and CMS scores (top and bottom panels, respectively) on 4 well-characterized selective sweeps (Supplementary Fig 8; Supplementary Table 1). The rank of the putative favored mutation (red star) in 5 Mbp region is shown in top left corner. (c) iSAFE-scores on regions under selection. Top ranked iSAFE candidates are marked by blue squares when they match putative favored mutations, while turquoise circles represent new favored mutations suggested by iSAFE. All data-sets were chosen by taking a 5 Mbp window around the putative selected region, unless one side reached the telomere or centromere. (d) The GRM5-TYR region. The mutation rs672144 is ranked first by iSAFE and very well separated from rest of the mutations in 5 Mbp around it, in all non-African populations with high confidence (iSAFE > 0.5, P ≪ 1.3e-8; Supplementary Fig. 10). The upper panel is haplotype plot with core mutation rs672144 on all 5008 haplotypes (2504 samples) of 1000GP. This plot shows carrier haplotypes of mutation rs672144 are conserved over a longer span than haplotypes in non-carriers which is a signal of selection8. Lower panel shows global frequencies of carrier haplotypes of mutation rs672144 (red, blue) and non-carrier haplotypes (gray). The evidence is consistent with an out of Africa selection on standing variation (soft sweep) with mutation rs672144 as the favored variant.
iSAFE-scores are not based upon likelihood computations, and we use them primarily to rank order the mutations. However, iSAFE-scores are normalized and can be compared across samples. Empirically computed P values (see Online Methods) on iSAFE indicate good performance when P < 1e-4, (iSAFE ≥ 0.1; Supplementary Fig. 7).
We tested iSAFE performance on 22 human loci previously characterized as containing signatures of a selective sweep (Supplementary Note 2), with some evidence for the favored mutation. The list included 8 ‘well characterized’ cases with additional support for the favored mutation (Supplementary Table 1). Using genotype data from phase 3 of 1000 Genomes Project (1000GP)9 sub-populations, we used iSAFE to rank all variants (∼21,000) in a 5 Mbp region surrounding each locus. Among the 8 well characterized cases5, 10–14 (Fig. 3b and Supplementary Fig. 8), iSAFE ranked the candidate mutations as 1 in 5 cases: SLC24A5, LCT, EDAR, ACKR1, TLR1, and ranked the remaining as 2 (ABCC1), 4 (HBB), and 13 (G6PD).
We checked whether the other 14 loci5, 15–18 under selection showed a strong iSAFE signal (Supplementary Note 2). In 3 of the 14 loci (FUT2, F12, ASPM; Supplementary Fig. 9), we observed weak signals and did not make a prediction (peak iSAFE < 0.027). In other loci, iSAFE ranked the candidate mutations as 1 in the SLC45A2/MATP (CEU), MC1R (CHB+JPT), and ATXN2-SHB3 (GBR) loci (Fig. 3c), and 7, 8, and 12 in PSCA (YRI), ADH1B (CHB+JPT), and PCDH15 (CHB+JPT) loci, respectively. In each case, the iSAFE-scores were high with the exception of PSCA (peak iSAFE = 0.04, Supplementary Fig. 9).
The other 5 putative selected loci are interesting in that the top-ranked iSAFE mutations had high scores, but were distinct from the reported candidate mutations (Fig. 3c; Supplementary Note 2). Many of these loci are involved in pigmentation, determining, skin, eye, and hair color. For example, the Tyrosinase (TYR) gene, encoding an enzyme involved in the first step of melanin production, is considered to be under positive selection with a nonsynonymous mutation rs1042602 as a candidate favored variant15. A second intronic variant, rs10831496, in GRM5, 396 kbp upstream of TYR, has been shown to have a strong association with skin color19. In contrast, iSAFE ranks mutation rs672144 at the top. Interestingly, this variant was the top ranked mutation not only in CEU (iSAFE = 0.48, P ≪ 1.3e-8), but also in EUR, EAS, AMR, and SAS (iSAFE > 0.5, P ≪ 1.3e-8; Supplementary Fig. 10). The result is consistent with a signal of selection present in all populations except AFR. It may not have been previously reported because it is near fixation in all populations of 1000GP except for AFR (Supplementary Fig. 10). We found that two distinct haplotypes carry the rs672144 mutation, both of which have remained at high frequency, maintained across a large stretch of the region, suggestive of a soft sweep with standing variation (Fig. 3d). A similar analysis applied to loci TRPV6, KITLG, OCA-HERC2 (see Supplementary Note 2; Fig. 3c and Supplementary Fig. 11–13), where in each case, the top iSAFE mutations were identical across all non-African populations, and supported an out-of-Africa onset of selection. In the one remaining gene (CYP1A2/CSK; see Supplementary Note 2; Fig. 3c), the top ranked iSAFE mutation rs2470893 was previously found significant in a genome wide association study20, and was tightly linked to the candidate mutation. To summarize, iSAFE analysis ranked the candidate mutation among the top 13 in 14 of the 22 loci, did not show a strong signal in 3, and identified plausible alternatives in the remaining 5 (Supplementary Note 2).
The identification of the favored allele in a selective sweep is a long-standing problem in population genomics. Our results suggest that statistics obtained from the coalescent structure of a region under a selective sweep can indeed pinpoint the favored mutation. iSAFE performance remains robust to a range of simulation parameters, including initial frequencies (standing variation) and the frequency of the favored mutation at the time of sampling. While most results in the paper are presented on human populations, iSAFE can be easily extended to other populations as it is not highly parameterized.
ONLINE METHODS
A comprehensive explanation of the method is provided in Supplementary Note 1.
Input, output and overview.
Methods to identify signatures of selective sweeps in population genomics data have been actively developed3–8, 21–33, but mostly do not identify the specific mutation favored by selection. iSAFE uses a statistic derived solely from population genetics signals to pinpoint the favored mutation in a large region (5 Mbp), without having any knowledge of demography, specific phenotype under selection, and functional annotations of mutations. iSAFE uses a 2-step procedure to identify the favored variant, given a large region (5 Mbp) under selection. In the first step, it finds the best candidate mutations in small (low recombination) windows. Finally, it combines the evidence to give an iSAFE-score to all variants in the large region. It considers only biallelic sites, taking as input a binary SNP matrix with each row corresponding to a haplotype h, each column to a site e. Entries in the matrix correspond to the allelic state, with 0 denoting the ancestral allele, and 1 denoting the derived allele.
HAF: Haplotype Allele Frequency.
A haplotype ‘contains/carries a mutation e’ if it has the derived allele at site e. Recently, we devised the HAF score to capture the dynamics of a selective sweep6. The HAF score for a haplotype h (HAF(h)) is the sum of the derived allele counts of the mutations on h (see Supplementary Note 1; Fig. 1a). It has been shown that, when h is a carrier of the favored allele, HAF(h) increases with the frequency of the favored mutation (Equation SN1.9 of Supplementary Note 2), in contrast to HAF scores of non-carriers (Equation SN1.10 of Supplementary Note 2), and this can be used to separate carrier haplotypes from non-carriers without knowing the favored mutation6.
SAFE: Selection of Allele Favored by Evolution.
Denote two haplotypes as ‘distinct’ if they have different HAF-scores. For any mutation e, let f denote the mutation frequency, or the fraction of haplotypes carrying the mutation. Let κ (e) (Fig. 1a) denote the fraction of distinct haplotypes that carry mutation e,
| 1 |
Similarly, let ϕ (e) denote the normalized sum of HAF-scores of all haplotypes carrying the mutation e,
| 2 |
We observe empirically that in a region evolving according to a neutral Wright-Fisher model, κ (e) and ϕ (e) are both estimators of f (e) (Supplementary Fig. 1). Moreover, empirical results suggest that the expected value of ϕ - κ is 0, and variance is proportional to f (1 - f ). Based on these observations, we define the SAFE-score of mutation e as
| 3 |
Empirically, SAFE(e) behaves like a Gaussian random variable, with mean 0, under neutrality (Supplementary Fig. 1), and it can be used to test departure from neutrality. However, its real power appears during positive selection, when SAFE-scores change in a dramatic, but predictable manner (Fig. 1a,b). Assuming a no recombination scenario (only for visual exposition), label mutations as ‘non-carrier’ if they are carried only by haplotypes not carrying the favored allele. The remaining mutations can be labeled as ‘ancestral’, if they arise before the favored mutation, or ‘descendant’, if they arise after (Fig. 1b). Representing each mutation as a point in a 2-dimensional plot of ϕ, κ values, these classes are clustered differentially (Fig. 1b). The selective sweep reduces the number of distinct haplotypes carrying the favored mutation (lower κ), leaving non-carrier mutations with an increased fraction of distinct haplotypes (higher κ). On the other hand, increased HAF-scores in carrier haplotypes reduces the proportion of total HAF-score contributed by non-carrier haplotypes (lower ϕ). In contrast, the favored mutation has high positive value of ϕ - κ due to high HAF-scores for carriers (higher ϕ), and the reduced number of distinct haplotypes among its descendants (lower κ). As we go up to ancestral mutations, the number of non-carrier haplotype descendants increase, and κ grows faster than ϕ. As we go down to descendant mutations, there is a reduction in the already small number of distinct haplotypes. However, ϕ decreases sharply, reducing ϕ - κ (Fig. 1a,b). Thus, we expect that the mutation with the highest SAFE-score is a strong candidate for the favored mutation.
We performed extensive simulations to test SAFE on samples evolving neutrally and under positive selection. We varied one parameter in each run (Supplementary Fig. 2), including window size (L = 50 kbp), number of individual haplotypes (n = 200) chosen from a larger effective population size (N = 20,000) scaled selection coefficient (Ns = 500), initial and final favored mutation frequencies (ν0 = 1/N, and ν). While standing variation, ν0 > 1/N, generally weakens the selection signal, the performance of SAFE remains relatively robust to variation in ν0. The median SAFE rank of the favored allele is at most 3 out of ∼250 variants in all cases except when ν0 > 1000/N (Supplementary Fig. 2). Similarly, the performance is robust to selection pressure, with only a slight degradation at weak selection (Ns = 50) (Supplementary Fig. 2) where the median rank goes to 9 (3.5%-ile), while for Ns ≥ 200 the median rank is at most 2. As expected, the performance improves with increasing sample size (Supplementary Fig. 2). We also tested SAFE on a model of European demography and observed similar results (Supplementary Fig. 2). These tests used L = 50 kbp, chosen so as to minimize the effects of recombination.
iSAFE: integrated Selection of Allele Favored by Evolution.
Next, we tested SAFE with increasing window sizes, and observed that the median rank of the favored mutation increases with increasing window size (Fig. 2c). The deterioration for larger windows is likely due to most haplotypes becoming unique, and κ losing its utility in pinpointing the favored mutation. However, the selective sweep signal is known to extend to large, linked regions, as far as 1 Mbp on either side of the favored allele. These ‘shoulders’3 of selective sweeps are helpful in identifying the region under selection, but make it harder to pinpoint the favored mutation. We further refined our method to exploit the signal from shoulders.
For larger regions, we considered a set of 50% overlapping windows () of fixed size (300 SNPs). For each window, we applied SAFE and chose the mutation with the highest SAFE-score. Let denote the set of selected mutations. Mutations in are likely to contain either the favored mutation itself or mutations linked to it. For mutation e in window w, let Ψe,w’ denote the larger of the SAFE-score of e, when e is ‘inserted’ into window w’, and 0 (Fig. 2a,b). As different windows have different genealogies due to recombination, Ψe,w’ is relatively high when e is the favored mutation and the genealogies of w, w’ are identical or very similar, but not otherwise. In contrast, the SAFE-score of a non-favored mutation e is relatively low when inserted in other windows (see Supplementary Note 1; Fig. 2a). Define the weight of a window w as
| 4 |
Windows that contain the favored mutation and those sharing its genealogy are expected to have high α values. We defined the iSAFE-score for all mutations e (including those not in ) as:
| 5 |
iSAFE-scores are not based upon likelihood computations, and the distribution of scores depend upon largely unknown factors including demography, time since onset of selection, selection coefficient, and other parameters. Nevertheless, they can be used to rank order the mutations. Additionally, iSAFE scores are normalized and can be compared across samples. We found distinct differences in performance below a score threshold of 0.1. The median rank of the favored mutation is 4 when peak iSAFE-score exceeds 0.1 versus a median rank of 10 along with a longer tail, when peak iSAFE-score is below 0.1 (Supplementary Fig. 7). Empirically computed P values on iSAFE indicate good performance when P < 1e-4 (Supplementary Fig. 7).
Adding outgroup samples.
Not surprisingly, iSAFE performance deteriorates when the favored mutation is fixed, or near fixation (ν > 0.9 in Supplementary Fig. 3). To handle this special case, we include individuals from non-target populations. For a mutation, define the Maximum Difference in Derived Allele Frequency score (MDDAF) as
| 6 |
Where DT is the derived allele frequency in the target population and min(DNT) is the minimum derived allele frequency over all non-target populations. Simulations of human population demography under neutral evolution shows P(MDDAF > 0.78 | DT > 0.9) = 0.001 (Supplementary Fig. 15). Therefore, when we observe the rare event of high frequency mutations in target (DT > 0.9) with MDDAF > 0.78, we add random outgroup samples to the data to constitute 10% of the data (Supplementary Note 1). In testing on the phase 3 of 1000GP data, we chose outgroup samples from non-target 1000GP populations. The addition of outgroup samples using the MDDAF criterion was tested in extensive simulations. While the performance did not change for ν<0.9, it dramatically improved for high frequencies, including when the favored mutation was fixed in the target population (Supplementary Fig. 3).
iSAFE evaluation.
In testing on models of human demography, we also compared against CMS. While CMS showed excellent performance in localizing the favored mutation, iSAFE scoring greatly improved the ranking. For example, iSAFE ranked the favored mutation within the top 20 in 94% of the simulations of a 5 Mbp region (Fig. 3a and Supplementary Fig. 4), in contrast to CMS which had a top 20 ranking in 35% of cases.
In testing instances of previously characterized sweeps in 1000GP data, we note that performance is difficult to characterize due to many complicating factors. Multiple sweeps could be occurring in response to different selection events, including background selection in the same region; or polygenic selection may also dilute the selection signal at any one locus. Moreover, the favored mutation is well-characterized in only a few instances. We looked for genes/regions that showed the signature of a selective sweep in one of the 1000GP sub-populations, and had additional evidence pointing to the favored mutation. We identified 22 genes with some evidence, but only 8 ‘well characterized’ cases with additional support for the favored mutation (see Supplementary Note 2; Supplementary Table 1).
Default simulation parameters.
Neutral and sweep samples were generated using the simulator msms34. By default, simulated populations are haploid with sample size of n = 200 haplotypes from a larger effective population of N = 20,000 haplotypes, each of length L, with default value 50 kbp for SAFE and 5 Mbp for iSAFE. For human populations, a mutation rate of approximately μ = 2.5e-8 mutations per bp per generation17, 35, and a recombination rate of approximately r = 1.25e-8 per bp per generation36 have been proposed. For SAFE simulations, we used a scaled mutation rate θ = 2Nμ = 1 mutations per kbp per generation and scaled recombination rate ρ = 2Nr = 0.5 crossovers per kbp per meosis to approximate human rates. The rates were scaled linearly by L. In the case of positive selection the default scaled selection strength of the favored allele was set to Ns = 500, with the favored mutation located at a random position uniformly distributed on the range [1, L]. The default value for favored mutation starting frequency ν0 = 1/N (hard sweep), and the frequency of the favored mutation (ν) at the time of sampling is a random value uniformly distributed on the range [0.1, 0.9]. We used the default parameters for all simulations unless otherwise stated.
A model of human demography.
We simulated demography of AFR, EUR, EAS populations with parameter shown in the Supplementary Fig. 14 based on a popular demographic model of human population37. In case of positive selection, selection coefficient was set to s = 0.05 and starting favored allele frequency ν0 = 0.001. The time of onset of selection was chosen at random (using the distribution in Supplementary Fig. 14) after the out of Africa event, in the lineage of EUR population (as the target population). When the onset of selection is before split of EUR and EAS (> 23kya), both (EUR and EAS) are under selection.
Computing iHS scores.
We used the selscan38 (v1.1.0a) software available at https://github.com/szpiech/selscan, with default settings to calculate the raw iHS8 score. Next, we normalized the iHS score by estimating the distribution of raw iHS scores on 1,000 neutral simulations with the same simulation parameters. The iHS scores were always computed on a 5 Mbp window. When comparing results with SAFE on a 50 kbp window, we used the corresponding iHS scores in the identical 50 kbp region surrounding the favored variant (Fig. 1c and Supplementary Figure 2). In considering 5 Mbp windows (Supplementary Figure 3), we compared the iHS scores on all variants for iHS against iSAFE.
Computing SCCT scores.
We used the SCCT (v1.1) software available at https://github.com/wavefancy/scct, provided by Wang et al. (2014)7, with flanking SNPs size 300, and frequency interval 0.01.
Computing CMS scores.
CMS5 requires a control population as well as a demographic model in addition to the target population under selection. All CMS comparisons on simulated data were performed using a model of human demography37 with a random onset of selection (Supplementary Figure 14). We used the CMS (v2.0) software available at https://github.com/broadinstitute/cms, disabling CMS’ default allele frequency filter in order to allow a more direct comparison with iSAFE SNP ranking.
Computing empirical P value.
We applied iSAFE on a neutrally evolving simulated population with window size 5 Mbp, based on European demography shown in Supplementary Figure 14. A P value was calculated based on empirical distribution of iSAFE on these simulated populations. We limited the number of samples to ~74,800,000 for efficiency, and this allows us to get a P value as low as 1.34e-8 for iSAFE = 0.304. Scores higher than this cut-off are considered to have P < 1.34e-8.
Putative selective sweeps in human populations.
We examined 8 well characterized selective sweeps with strong candidate mutation. These genes are LCT, SLC24A5, TLR1, EDAR, ACKR1/DARC, ABCC11, HBB, and G6PD. iSAFE results for these genes are summarized in Fig. 3b, Supplementary Fig. 8 and Supplementary Table 1. We also examined 14 other regions reported to be under selection with one or more candidate favored mutations. A detailed report for each of these 14 loci is provided in Supplementary Note 2.
Supplementary Material
Acknowledgments.
This research was supported in part by grants from the NSF (DBI-1458557), and from the NIH (R01GM114362).
Footnotes
Competing financial interests. V.B. is a co-founder, has an equity interest, and receives income from Digital Proteomics, LLC. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. D.P. was not involved in the research presented here.
Code availability. The iSAFE software and the instruction are available at https://github.com/alek0991/iSAFE and as Supplementary Software.
Data availablity. For all the following datasets, the genome build is GRCh37/hg19. We downloaded the phased haplotypes of the 1000GP9 (phase 3) dataset from http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/ release/20130502 /. The ancestral allele dataset from Ensembl39 (release 75) is downloaded from http://ftp.ensembl.org/pub/release-75/fasta/ancestral_alleles/. The physical position was converted into genetic position using the HapMap II40 genetic map downloaded from http://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/working/20110106_recombination_hotspots/. A Life Sciences Reporting Summary is available.
References
- 1.Vitti JJ, Grossman SR & Sabeti PC Annual review of genetics 47, 97–120 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Fan S, Hansen MEB, Lo Y & Tishkoff SA Science 354, 54–59 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schrider DR, Mendes FK, Hahn MW & Kern AD Genetics 200, 267–284 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Field Y et al. Science 354, 760–764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grossman SR et al. Science 327, 883–886 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Ronen R et al. PLoS Genet 11, e1005527–e1005527 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang M et al. Molecular biology and evolution, msu244–msu244 (2014). [Google Scholar]
- 8.Voight BF, Kudaravalli S, Wen X & Pritchard JK PLoS Biol. 4, e72–e72 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Genomes Project C et al. Nature 526, 68–74 (2015).26432245 [Google Scholar]
- 10.Sabeti PC et al. science 312, 1614–1620 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Enattah NS et al. Nature genetics 30, 233–237 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Ohashi J, Naka I & Tsuchiya N Molecular biology and evolution 28, 849–857 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Tishkoff SA et al. Science 293, 455–462 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Heffelfinger C et al. European Journal of Human Genetics 22, 551–557 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilde S et al. Proceedings of the National Academy of Sciences 111, 4832–4837 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coop G et al. PLoS Genet 5, e1000500–e1000500 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campbell CD et al. Nature genetics 44, 1277–1281 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galinsky KJ, Loh P-R, Mallick S, Patterson NJ & Price AL The American Journal of Human Genetics 99, 1130–1139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beleza S et al. PLoS Genet 9, e1003372–e1003372 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cornelis MC et al. Molecular psychiatry 20, 647–656 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrer-Admetlla A, Liang M, Korneliussen T & Nielsen R Mol Biol Evol 31, 1275–1291 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pybus M et al. Bioinformatics 31, 3946–3952 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Garud NR, Messer PW, Buzbas EO & Petrov DA PLoS Genet 11, e1005004 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeGiorgio M, Huber CD, Hubisz MJ, Hellmann I & Nielsen R Bioinformatics 32, 1895–1897 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Ronen R, Udpa N, Halperin E & Bafna V Genetics 195, 181–193 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pavlidis P, Zivkovic D, Stamatakis A & Alachiotis N Mol Biol Evol 30, 2224–2234 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen H, Patterson N & Reich D Genome Res 20, 393–402 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sabeti PC et al. Nature 449, 913–918 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sabeti PC et al. Nature 419, 832–837 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Nielsen R et al. Genome Res 15, 1566–1575 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim Y & Nielsen R Genetics 167, 1513–1524 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alachiotis N, Stamatakis A & Pavlidis P Bioinformatics 28, 2274–2275 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Shriver MD et al. Hum Genomics 1, 274–286 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ewing G & Hermisson J Bioinformatics 26, 2064–2065 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nachman MW & Crowell SL Genetics 156, 297–304 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jensen-Seaman MI et al. Genome research 14, 528–538 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gravel S et al. Proceedings of the National Academy of Sciences 108, 11983–11988 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szpiech ZA & Hernandez RD Molecular biology and evolution 31, 2824–2827 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zerbino DR et al. Nucleic Acids Res (2017). [Google Scholar]
- 40.International HapMap C et al. Nature 449, 851–861 (2007).17943122 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.