

Abstract
Study Objectives
A negative intrathoracic pressure threshold is one commonly proposed mechanism for triggering respiratory-induced arousals in obstructive sleep apnea (OSA). If so, they should occur during inspiration, shortly after maximal negative pressure swings. Alternatively, respiratory-induced arousals may occur throughout the respiratory cycle if other mechanisms also contribute. However, arousal timing has been minimally investigated. This study aimed to (1) determine the temporal relationship between respiratory-induced arousals and breathing phase and (2) characterize neuromuscular and load compensation responses prior to arousal.
Methods
Fifty-one CPAP-treated OSA patients underwent a sleep physiology study with genioglossus and tensor palatini EMG, nasal mask/pneumotachograph, and epiglottic pressure. Transient CPAP reductions were delivered to induce respiratory-related arousals.
Results
Of 354 arousals, 65(60–70)%[mean(CI)] occurred during inspiration, 35(30–40)% during expiration. Nadir epiglottic pressure occurred 68(66–69)% into inspiration while inspiratory arousals had a uniform distribution throughout inspiration. Expiratory arousals occurred predominantly in early expiration. CPAP reductions initially reduced minute ventilation by ~2.5 liter/min, which was restored immediately prior to expiratory but not inspiratory arousals. Duty cycle just prior to arousal was greater for inspiratory versus expiratory arousals [0.20(0.18–0.21) vs. 0.13(0.11–0.15)Δbaseline, p = 0.001]. Peak tensor palatini EMG was higher for expiratory versus inspiratory arousals during prearousal breaths [7.6(5.8–9.6) vs. 3.7(3.0–4.5)%Δbaseline, p = 0.001], whereas genioglossus and tonic tensor palatini EMG were similar between arousal types.
Conclusions
Over one third of respiratory-induced arousals occur during expiration. These findings highlight the importance of nonpressure threshold mechanisms of respiratory-induced arousals in OSA and suggest that expiratory arousals may be a novel marker of enhanced tensor palatini neuromuscular compensation.
Keywords: arousal threshold, upper airway, sleep-disordered breathing, respiratory load compensation, pharyngeal muscles, phenotyping, EEG, lung
Statement of Significance
Respiratory disturbances (apneas/hypopneas) in adults with obstructive sleep apnea (OSA) frequently elicit cortical arousals from sleep. A negative intrathoracic pressure threshold is one proposed mechanism for triggering respiratory-induced arousals. Accordingly, they should occur during inspiration when maximal negative pressure swings occur. Alternatively, if other mechanisms contribute, they may occur throughout the respiratory cycle. However, arousal timing, neuromuscular, and load compensation responses associated with respiratory arousals have not been systematically assessed in OSA. We found that a substantial proportion (35%) of respiratory-induced arousals occur during expiration. The neuromuscular and load compensation responses immediately prior to inspiratory versus expiratory arousals were markedly different. These findings provide insight into the mechanisms of respiratory-induced arousals and have important implications for OSA pathogenesis and potential treatment targets.
Introduction
Cortical arousals from sleep are a characteristic feature of adult obstructive sleep apnea (OSA). Historically, arousals were assumed vital to restore airflow following airway narrowing and closure in OSA [1]. However, evidence now indicates that frequent arousals contribute to OSA pathogenesis by destabilizing ventilatory control and sleep continuity [2, 3]. Thus, to gain insight into disease pathogenesis and develop targeted therapies for individuals in whom arousals are a major contributor to their OSA (~30–50% of patients) [4, 5], it is important to identify the mechanisms leading to arousal, which are currently only partially understood.
A threshold level of negative intrathoracic pressure, known as the arousal threshold [2, 6–8], is one commonly proposed mechanism responsible for triggering respiratory-induced arousals. In humans, this concept was supported by a landmark study by Gleeson and colleagues [6]. Here, arousals were induced during nonrapid eye movement (NREM) sleep in eight healthy males using different respiratory stimuli (brief externally applied resistive loads, hypoxia, and hypercapnia). Negative intrathoracic pressure just prior to arousal was measured using an esophageal pressure catheter [6]. On average, regardless of the source of the respiratory stimulus, the nadir esophageal pressure immediately prior to arousal was similar. Thus, negative intrathoracic pressure appears to be a key trigger for respiratory-induced arousal. However, not all respiratory-related arousals occur at nadir intrathoracic pressure. Indeed, although not specifically highlighted, some occur during expiration (for example, see Younes [3], Eckert et al. [4], and Carter et al. [9]). This suggests that other mechanisms may also be involved. Indeed, numerous studies have subsequently highlighted the multiple respiratory afferents (chemical and mechanical) triggered in response to respiratory stimuli and the complexity of the neuroanatomical pathways and mechanisms that contribute to respiratory-related cortical arousals [2, 8, 10–15].
Consistent with multiple contributing mechanisms, Younes [3] showed that during respiratory events the timing of cortical arousals relative to resumption of airflow can differ considerably. Indeed, in many cases, people with OSA can restore airflow without arousal using neuromuscular and respiratory load compensation mechanisms [3, 16–18]. Respiratory load compensation involves prolongation of inspiratory time (while maintaining respiratory rate) to help them prevent reductions in airflow during airway narrowing and stabilize ventilation. Neuromuscular and load compensation responses vary between individuals. Thus, poor compensation during sleep may contribute, at least in part, to OSA pathogenesis in certain patients [4, 17–19]. Changes in respiratory load compensation during airway occlusion may also contribute to respiratory-induced arousals in OSA [20]. Specifically, increased PCO2 and chemical drive may contribute to arousal beyond negative pressure. A last-minute successful neuromuscular improvement in airflow would act to partially mitigate the negative epiglottic pressure but would not influence an independent brain PCO2 (chemical drive) predisposition to arousal. If true, one would expect to see that a late increase in airflow would reduce the likelihood of an inspiratory arousal. Secondly, a failure to reach the intended tidal volume, e.g. detected by pulmonary stretch-receptors, rather than negative pressure per se, may contribute to respiratory arousal. If true, one would expect to see that a failure to increase respiratory duty cycle would reduce the likelihood of an inspiratory arousal.
Accordingly, given the uncertainty regarding the mechanisms mediating respiratory-induced arousals and the potential importance of neuromuscular and respiratory load compensation in OSA pathogenesis, we conducted a detailed physiological investigation into the timing of cortical arousals and the preceding neuromuscular and respiratory load compensation responses during experimentally induced airway narrowing in people with OSA. We hypothesized that, consistent with the Gleeson model, if the primary trigger for respiratory-induced arousals is a threshold level of negative intrathoracic pressure, arousals should consistently occur during inspiration and have a temporal relationship with negative intrathoracic pressure (allowing for delays in the arrival/processing of respiratory afferent information at the cortex of ~0.5 s) [21, 22]. Alternatively, the presence of respiratory-induced arousals throughout the respiratory cycle would be consistent with contributions from other mechanisms beyond negative intrathoracic pressure.
Methods
Data for the current study were acquired during a larger study to quantify the key pathophysiological traits causing OSA [4]. The methodological details below focus on elements pivotal for the current novel investigations and only briefly outline the general experimental methodology described previously [4].
Participants
Data were acquired from 51 otherwise healthy individuals with OSA who were not taking any medications known to affect sleep or breathing. Participants had been compliant with continuous positive airway pressure (CPAP) therapy assessed via machine download for at least 3 months prior to enrollment (Table 1). Seven patients were excluded from the original OSA cohort [4] as they did not meet the criteria for one or more of the current analyses (see Data analysis). All participants provided informed written consent. The protocol was approved by the Partners’ Healthcare Institutional Review Board.
Table 1.
Anthropometric and sleep parameters
| n = 51 | |
|---|---|
| Gender (M/F) | 37/14 |
| Age (years) | 48 ± 11 |
| BMI (kg/m2) | 35 ± 6 |
| Mean AHI (events/hr sleep) | 47 ± 29 |
| Range AHI (events/hr sleep) | 11–112 |
| CPAP compliance (hr/night) | 6.5 ± 1.7 |
Data are mean ± SD, where applicable.
n = number of participants; BMI = body mass index; AHI = apnea–hypopnea index; CPAP = continuous positive airway pressure.
Experimental design and measurements
Participants completed a detailed sleep physiology study. Electroencephalograms (EEGs; C3-A2/O2-A1), electroculograms (EOGs), and chin electromyogram (EMG) were acquired for sleep stage and arousal scoring. Genioglossus and tensor palatini EMG (EMGgg/EMGtp) were measured using fine-wire intramuscular electrodes (Cooner Wire Company, Chatsworth, CA) [4, 23]. Epiglottic pressure was acquired using a transducer-tipped pressure catheter (Millar Instruments, Houston, TX) [4]. A nasal CPAP mask was fitted and pressure/airflow measured with pressure transducers (Validyne Corporation, Northbridge, CA) and pneumotachograph (Hans Rudolf Inc., Kansas City, MO) [4].
Protocol
Participants were studied supine on CPAP sufficient to eliminate inspiratory flow limitation. Transient reductions in CPAP were applied for ≤3 min during stable NREM sleep to cause varying degrees of upper airway collapse to induce arousals [4].
Data analysis
Included in the analyses were reductions in CPAP during NREM sleep that (1) lasted >10 s prior to terminating in an arousal and (2) were associated with an increase in nadir negative Pepi > 2 cmH2O within the 30 s preceding arousal. Participants were included in the analysis if they had at least two CPAP reductions that met these criteria.
Arousal and nadir epiglottic pressure timing
Arousals were scored by an experienced sleep technician according to American Academy of Sleep Medicine (AASM) criteria [24] as a sudden increase in EEG frequency lasting >3 s preceded by ≥10 s of stable sleep. The technician was blinded to the respiratory and upper airway muscle EMG channels and paid careful attention to accurately define onset/offset of arousals. Arousals were categorized as inspiratory or expiratory depending on where in the respiratory cycle they occurred (Figure 1).
Figure 1.

Schematic illustrating calculation of relative inspiratory (A) and expiratory (B) arousal time. Thick vertical dashed line indicates location of arousal onset. Tar = time to arousal from beginning of inspiration (A) or expiration (B). Ti = duration of first complete inspiratory cycle immediately prior to arousal. Te = duration of first complete expiratory cycle immediately prior to arousal. Arousal onset time relative to the start of the corresponding respiratory phase was determined and expressed as a per cent of the prearousal inspiratory or expiratory time. Note that the prearousal breath was used for relative respiratory timing of arousal since the arousal breath is disrupted by the arousal itself, hence altering inspiratory/expiratory durations.
Respiratory and muscle parameters
Respiratory and pharyngeal muscle parameters were quantified for two breaths immediately following each CPAP reduction (breaths 1 and 2) and, where available, for three breaths prior to arousal (breaths −3 to −1). Figure 2 shows a representative CPAP reduction and quantification of key variables. Respiratory-related stimuli for arousal, including the respiratory arousal threshold, were quantified (Figure 2).
Figure 2.

Raw data examples showing a transient reduction in CPAP (Pmask) that induced a respiratory-related (A) inspiratory arousal and (B) expiratory arousal. Raw genioglossus (GG) and tensor palatini (TP) EMG (EMGgg and EMGtp) were rectified, moving-time-averaged (100 ms window) and expressed as a %maximum for each participant [4, 23], i.e. GG MTA and TP MTA. Some of the key study parameters quantified are shown, including respiratory arousal threshold, the nadir epiglottic pressure (Pepi), or NadirPepi, immediately preceding arousal [2, 4]; ∆NadirPepi/∆tDrop, rate of change of NadirPepi during the CPAP drop, measured from the first breath following each CPAP reduction to the breath immediately prior to arousal; ∆Pepi/∆tPreArB, rate of change of Pepi during the breath immediately preceding arousal, measured from the breath start to NadirPepi within that breath; time to arousal; and NadirSpO2, the minimum blood arterial oxygen saturation (SpO2) caused by the reduction in CPAP. Respiratory and pharyngeal muscle parameters were quantified for the two breaths following each CPAP reduction (breaths 1 and 2) and where available for three breaths prior to arousal (breaths −3 to −1), including (not all indicated on Figure): PIF = peak inspiratory flow; tidal volume and minute ventilation; Ti = inspiratory time; Te = expiratory time; Ttot = Ti + Te; Ti/Ttot = duty cycle; RR = respiratory rate; EMGgg Peak and Tonic muscle activity; EMGtp Peak and Tonic muscle activity. Note the greater increase in tensor palatini muscle activity and flow prior to the expiratory arousal (B) compared with inspiratory arousal (A).
EEG analysis
EEG (C3/A2) spectral analysis and K-complex frequency detection were performed over a 10 s window preceding the onset of arousal, and EEG spectral power within different frequency bands determined, as described previously [25].
Statistical approach
A z-test of proportion against the null hypothesis (H0: P = 50%) was used to test the difference in the number of individual arousals during inspiration and expiration. Histograms were used to illustrate distribution of arousals separated according to respiratory phase. Sturger’s rule of thumb was used to select the appropriate number of bins used in the histograms [26]. Data are presented as percentages and 95% confidence intervals. Chi-square test statistics were used to determine whether the distribution of arousal differs from chance and were therefore tested against the null hypothesis that they were equally distributed in each bin (H0: P = 9.09%). Individual z-test of proportions was then performed to identify which individual bins were different from chance.
To compare parameters between inspiratory and expiratory arousals (i.e. respiratory and muscle parameters during stable sleep on therapeutic CPAP, respiratory related arousal stimulus factors, average EEG power/K-complex frequency prior to arousal), the correlation from the within-individual measurements was taken into account using a mixed linear model with random intercept for each individual. Measurements were found not to meet normality assumptions. Thus, the mixed linear model results were bootstrapped using 1000 bootstrap samples and Mersenne Twister seed (number = 200,000). For the respiratory and muscle parameters during CPAP reductions, data were represented as a change from stable sleep on therapeutic CPAP for each breath analyzed. Similar to above, bootstrapped mixed linear models were used with arousal category (inspiratory vs. expiratory), breath number (5 breaths), and their interaction included in the model. Data from mixed linear models are presented as estimated marginal means (95% bootstrapped confidence intervals).
Statistical significance was inferred at p < 0.05 for all analyses, except for inspiratory vs. expiratory breath-by-breath comparisons for respiratory and muscle parameters during CPAP reduction where correction for multiple comparisons was undertaken based on the number of primary outcome comparisons (i.e. 5 breaths) such that p < 0.01 was considered significant. All statistical analyses were performed in IBM SPSS Statistics (version 25, IBM Corp.). Figures were produced using Graphpad Prism (v7.04, Graphpad Software Inc.).
Results
Anthropometric, sleep characteristics and objective CPAP compliance of the study participants are summarized in Table 1. A total of 354 CPAP reductions with respiratory-induced arousals that met the study criteria were analyzed (93% from stable N2 sleep) with a median of 7 (6–8) arousals analyzed per participant.
Timing of arousal and nadir epiglottic pressure
Arousals occurred during inspiration and expiration (Figure 2). For all arousals examined, arousals were more likely to occur during inspiration compared with expiration (p < 0.001, Figure 3A). Although the majority of participants experienced both inspiratory and expiratory arousals, 8/51 participants had solely inspiratory arousals and one had exclusively expiratory arousals (Figure 4). The characteristics (age, apnea–hypopnea index [AHI], and body mass index [BMI]) and arousal distribution with respect to inspiratory timing of the 8 participants who only had inspiratory arousals were not different to the group as a whole (P≥0.40; see Supplementary Tables S2 and S3).
Figure 3.
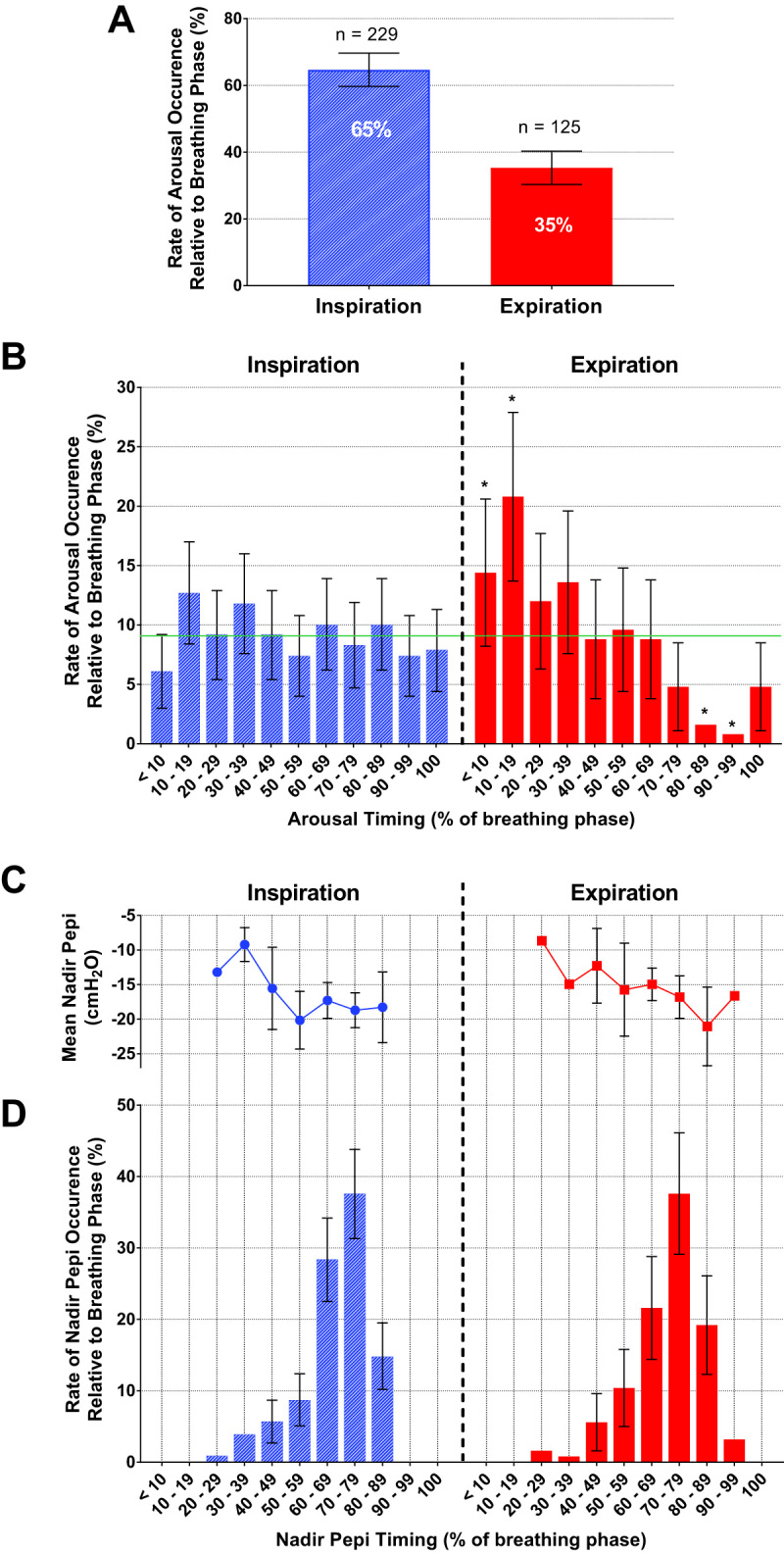
Percent and number (n) of arousals that occur during inspiration and expiration (A) and their distribution with respect to breath timing (B). Mean nadir epiglottic pressure (Pepi) calculated from the inspiratory effort just prior to the arousal breath (C), and nadir Pepi occurrence with respect to breath timing (D). Error bars represent the 95% confidence intervals. Horizontal line (green) indicates the % expected by chance (9.09%) and * indicates a significant difference in arousal probability from chance (B). See Supplementary Material for breakdown of bin and error bar values (including standard error and standard deviation for epiglottic pressure; Supplementary Table S1). Of a total of 354 arousals, 65% occurred during inspiration and 35% occurred during expiration (A). Inspiratory arousals had a uniform distribution throughout inspiration (B) and no temporal relationship with nadir epiglottic pressure, which occurred in the latter stages of inspiration (D). Expiratory arousals occurred predominantly in early expiration (B).
Figure 4.
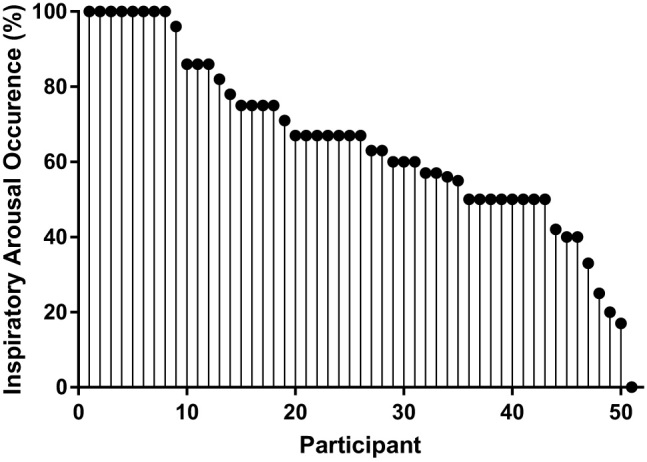
Proportion of inspiratory arousals for all 51 participants.
Inspiratory arousals occurred throughout inspiration with no temporal relationship with nadir epiglottic pressure (Figure 3, B–D; see Supplementary Material for breakdown of arousal counts and epiglottic pressure values). Indeed, the probability of arousal during each segment of inspiration was not different from chance (p = 0.468). Conversely, expiratory arousals had a biphasic pattern that was different from chance (p < 0.001) with a greater concentration of arousals occurring during early expiration compared with late expiration where arousals were rare (Figure 3B).
Nadir epiglottic pressure just prior to arousal occurred predominately during late inspiration [68(66–69)% of inspiratory time] after the onset of most inspiratory arousals (Figure 3D). Similarly, nadir epiglottic pressure occurred during late inspiration on the breath prior to expiratory arousals (Figure 3D; 70(68–72)% of inspiratory time).
Baseline respiratory and muscle activity parameters
Baseline respiratory and muscle activity parameters on therapeutic CPAP were similar for inspiratory and expiratory arousals (Table 2). There were also no differences in therapeutic CPAP holding levels (Table 2). The breakdown of parameters according to OSA severity is also presented in Table 2.
Table 2.
Respiratory and muscle parameters during stable sleep on CPAP
| Mild OSA (n participants= 5) |
Moderate OSA (n participants= 11) |
Severe OSA (n participants= 35) |
All (n participants= 51) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Inspiratory Arousals (n arousals= 16) |
Expiratory Arousals (n arousals= 12) |
Inspiratory Arousals (n arousals= 39) |
Expiratory Arousals (n arousals= 26) |
Inspiratory Arousals (n arousals= 174) |
Expiratory Arousals (n arousals= 87) |
Inspiratory Arousals (n arousals= 229) |
Expiratory Arousals (n arousals= 125) |
P | |
| CPAP (cmH2O) | 9.2 (9.2–9.5) | 9.2 (9.2–9.5) | 10.7 (10.4–10.8) | 10.6 (10.2–10.8) | 10.7 (10.4–10.8) | 10.6 (10.2–10.8) | 11.3 (11.1–11.4) | 11.4 (11.2–11.5) | 0.532 |
| Ti (s) | 1.7 (1.6–1.7) | 1.7 (1.6–1.7) | 1.7 (1.7–1.8) | 1.7 (1.7–1.8) | 1.7 (1.7–1.8) | 1.7 (1.7–1.8) | 1.8 (1.7–1.8) | 1.8 (1.7–1.8) | 0.231 |
| Te (s) | 2.9 (2.8–3.0) | 2.9 (2.9–3.0) | 2.6 (2.6–2.7) | 2.5 (2.4–2.6) | 2.6 (2.6–2.7) | 2.5 (2.4–2.6) | 2.5 (2.5– 2.6) | 2.5 (2.5- 2.6) | 0.747 |
| Ttot (s) | 4.6 (4.5–4.6) | 4.6 (4.5–4.7) | 4.3 (4.3–4.4) | 4.2 (4.1–4.3) | 4.3 (4.3–4.4) | 4.2 (4.1–4.3) | 4.3 (4.2–4.3) | 4.3 (4.2–4.3) | 0.788 |
| Ti/Ttot | 0.47 (0.36–0.38) | 0.37 (0.36–0.38) | 0.40 (0.39–0.42) | 0.41 (0.40–0.43) | 0.40 (0.39–0.42) | 0.41 (0.40–0.43) | 0.42 (0.41–0.43) | 0.42 (0.40–0.43) | 0.848 |
| RR (breaths/min) |
13.5 (13.3–13.7) | 13.5 (13.2–13.7) | 14.4 (14.1–14.8) | 14.8 (14.3–15.1) | 14.4 (14.1–14.8) | 14.8 (14.3–15.1) | 14.8 (14.6–15.1) | 14.5 (14.3–14.8) | 0.174 |
| VT (l) | 0.43 (0.42–0.44) | 0.44 (0.43–0.46) | 0.46 (0.44–0.47) | 0.47 (0.46–0.48) | 0.46 (0.44–0.47) | 0.47 (0.46–0.48) | 0.49 (0.48–0.5) | 0.5 (0.49–0.51) | 0.236 |
| Vi (L/min) | 5.8 (5.7–5.9) | 5.9 (5.7–6.1) | 6.8 (6.4–6.9) | 7.0 (6.8–7.2) | 6.7 (6.4–6.9) | 7.0 (6.8–7.2) | 7.1 (7.0–7.3) | 7.2 (7.0–7.4) | 0.332 |
| PIF (L/s) | 0.40 (0.39–0.41) | 0.41 (0.40–0.43) | 0.45 (0.44–0.47) | 0.47 (0.45–0.49) | 0.45 (0.44–0.47) | 0.47 (0.45–0.49) | 0.46 (0.45–0.46) | 0.47 (0.46–0.48) | 0.119 |
| NadirPepi (cmH2O) | −2.3 (−2.9 to −1.3) | −1.7 (−2.2 to −0.9) | −2.1 (−2.6 to −1.6) | −2.7 (−3.8 to −1.5) | −2.1 (−2.6 to −1.6) | −2.7 (−3.8 to −1.5) | −2.0 (−2.3 to −1.7) | −2.3 (−2.8 to −1.8) | 0.266 |
| SpO2 (%) | 95.6 (95.4–95.9) | 95.7 (95.4–96.0) | 95.5 (94.1–95.0) | 95.2 (94.6–95.7) | 94.5 (94.1–95.0) | 95.2 (94.6–95.7) | 94.8 (94.7–94.9) | 94.9 (94.8–95.0) | 0.272 |
| EMGgg Peak (%max) | 3.0 (1.8–4.0) | 2.5 (1.4–3.5) | 3.4 (2.8–3.9) | 3.6 (2.9–4.3) | 3.4 (2.8–3.9) | 3.6 (2.9–4.3) | 3.40 (3.1–3.8) | 3.4 (2.9–3.9) | 0.930 |
| EMGgg Tonic (%max) | 1.3 (0.7–1.7) | 1.0 (0.5–1.5) | 1.7 (1.4–1.9) | 1.7 (1.5–1.9) | 1.7 (1.4–1.9) | 1.7 (1.5–1.9) | 2.6 (2.4–3.2) | 1.4 (1.1–1.8) | 0.852 |
| EMGtp Peak (%max) | 1.7 (1.3–2.0)* | 1.7 (1.5–1.9)* | 1.9 (1.6–2.3) | 2.7 (1.9–3.7) | 1.9 (1.6–2.3) | 2.7 (1.9–3.7) | 2.6 (2.2–2.9) | 2.8 (2.4–3.2) | 0.539 |
| EMGtp Tonic (%max) | 1.1 (0.9–1.3) | 1.0 (0.9–1.2) | 1.3 (1.1–1.5) | 1.7 (1.2–2.4) | 1.3 (1.1–1.5) | 1.7 (1.2–2.4) | 1.8 (1.4–2.0) | 1.9 (1.5–2.2) | 0.807 |
Respiratory and muscle parameters during stable CPAP (60 s immediately prior to CPAP reduction) for inspiratory and expiratory arousals for all of the group and according to OSA severity: mild (AHI = 10–15 events/hr), moderate (AHI = 15–30 events/hr), and severe OSA (AHI ≥ 30 events/hr). Data are presented as estimated marginal means and confidence intervals [mean (CI)] from bootstrapped mixed linear models (1000 bootstrap samples, Mersenne Twister seed number = 200,000). p Values are from comparisons between inspiratory and expiratory arousal parameters for all of the group. There were no differences in any of the parameters between inspiratory and expiratory arousals.
Ti = inspiratory time; Te = expiratory time; Ttot = Ti+Te; Ti/Ttot = duty cycle; RR = respiratory rate; VT = tidal volume; Vi = minute ventilation; PIF = peak inspiratory flow; NadirPepi = nadir epiglottic pressure; SpO2 = blood arterial oxygen saturation; EMGgg Peak and Tonic = peak inspiratory and tonic genioglossus muscle activity; EMGtp Peak and Tonic = peak and tonic tensor palatini muscle activity.
*Results from a fixed-effect linear model.
Respiratory-related arousal stimuli
CPAP reduction (respiratory load) magnitude, time to arousal (from the beginning of the reduction in CPAP), rate of change in nadir epiglottic pressure during the prearousal breath, and arousal threshold were all not different between inspiratory and expiratory arousals (Table 3). However, the rate of change in nadir epiglottic pressure from the initial reduction in CPAP to arousal was less and nadir SpO2 (minimum blood arterial oxygen saturation caused by the reduction in CPAP) was higher during expiratory versus inspiratory arousals (Table 3).
Table 3.
Respiratory-related arousal stimulus factors
| Inspiratory arousals (n = 229) |
Expiratory arousals (n = 125) |
P | |
|---|---|---|---|
| ∆CPAP (cmH2O) | 7.2 (7.0–7.46) | 7.2 (6.8–7.5) | 0.826 |
| Arousal threshold (cmH2O) | −19.0 (−20.1 to −18.0) | −17.5 (−19.1 to −16.0) | 0.135 |
| Time to arousal (s) | 37.4 (33.8–41.8) | 43.8 (38.2–50.8) | 0.086 |
| ∆NadirPepi/∆tDrop (cmH2O/s) | −0.61 (−0.67 to −0.56) | −0.49 (−0.55 to −0.42) | 0.004 |
| ∆Pepi/∆tPreArB (cmH2O/s) | −12.8 (−13.6 to −12.1) | −12.6 (−13.5 to −11.6) | 0.698 |
| Nadir SpO2 (%) | 88.3 (87.9–88.8) | 89.3 (88.7–89.8) | 0.011 |
The table reports bootstrapped outcomes from mixed linear model (1000 bootstrap samples, Mersenne Twister seed number = 200,000) and includes estimated marginal means (confidence intervals) and p values comparing inspiratory and expiratory arousal parameter values.
n = number of measurements analyzed; Time to Arousal = duration taken from the beginning of the transient CPAP reduction to arousal; ∆NadirPepi/∆tDrop = rate of change in nadir epiglottic pressure (Pepi) during the drop (nadir Pepi of breath 1 to prearousal breath); ∆Pepi/∆tPreArb = rate of change in Pepi to reach nadir for the breath immediately prior to arousal; Nadir SpO2 = minimum blood arterial oxygen saturation caused by the reduction in CPAP.
Respiratory and pharyngeal muscle responses to reductions in CPAP
Figures 5 and 6 show breath-by-breath changes in respiratory and EMG parameters during the CPAP reductions, respectively, for inspiratory and expiratory arousals.
Figure 5.

Respiratory timing and ventilatory parameters for inspiratory (Insp) arousals (closed symbols) and expiratory (Exp) arousals (open symbols) for the first two breaths following CPAP reduction (Breaths 1 and 2), and the three breaths prior to arousal during the CPAP reduction (Breaths −3 to −1; Breath −1 is immediately prior to arousal), expressed as a change (∆) from baseline therapeutic CPAP (Breath 0; average of 60 s immediately prior to CPAP reduction). (A) Inspiratory time (Ti), (B) expiratory time (Te), (C) duty cycle (Ti/Ttot; Ttot = Ti + Te), (D) respiratory rate (RR), (E) peak inspiratory flow (PIF), (F) minute ventilation (Vi), and (G) nadir epiglottic pressure (NadirPepi). Data are estimated marginal mean (confidence interval). *p < 0.05 for Insp vs. Exp arousals. Between breath comparisons are shown for Insp and Exp arousals: # p < 0.05 vs. Breath 0, + p < 0.05 vs. Breath 1, ^ p < 0.05 vs. Breath 2, ϕ p < 0.05 vs. Breath −3, – p < 0.05 vs. Breath −2. For the breath immediately prior to arousal following CPAP reduction (Breath −1), Exp arousals have lower increases in Ti and Ti/Ttot and lower reductions in Te compared with Insp arousals. RR increases are immediate following CPAP reduction for both Insp and Exp arousals and are mostly maintained at that level for the duration of the CPAP drop. PIF and Vi return to baseline levels on Breath −1 for Exp arousals, though remain reduced for Insp arousals. There were no differences between Insp and Exp arousals on Breaths 1 and 2 for any of the parameters. Negative NadirPepi progressively increases from Breaths 1 to -1. There are no differences in NadirPepi changes between Insp and Exp arousals for all analyzed breaths (G).
Figure 6.

Peak and tonic muscle activity for the genioglossus (EMGgg Peak and EMGgg Tonic) (A and B) and tensor palatini (EMGtp Peak and EMGtp Tonic) (C and D) muscles for inspiratory (Insp) (closed symbols) and expiratory (Exp) (open symbols) arousals. Data [expressed as estimated marginal mean (confidence interval)] are for the first two breaths following CPAP reduction (Breaths 1 and 2), and the three breaths prior to arousal (Breaths −3 to −1; Breath −1 is immediately prior to arousal), expressed as a change (∆) from baseline therapeutic CPAP (Breath 0). There were greater increases in EMGtp Peak for Exp arousals compared with Insp arousals. *p < 0.05 for Insp vs. Exp arousals. # p < 0.05 vs. Breath 0, + p < 0.05 vs. Breath 1, ^ p < 0.05 vs. Breath 2, ϕ p < 0.05 vs. Breath −3, – p < 0.05 vs. Breath −2.
Respiratory timing
Inspiratory time (Figure 5A) did not change from the preceding therapeutic CPAP level during breath 1 following CPAP reductions for inspiratory and expiratory arousals (p > 0.54). There was an increase in inspiratory time on breath 2 (~0.06 s) for inspiratory arousals (p = 0.002) and inspiratory time was prolonged by ~0.3–0.5 s for the three breaths leading up to arousal for inspiratory and expiratory arousals (breaths −3 to −1; p = 0.001). The increase in inspiratory time on breath −1 was greater for inspiratory compared with expiratory arousals (p = 0.001), but did not differ between inspiratory and expiratory arousals for breath −2 or −3 (p > 0.014). Expiratory time (Figure 5B) immediately decreased on breath 1 following CPAP reductions by ~0.5 s and continued to decrease further for subsequent breaths for inspiratory arousals and by breath −2 for expiratory arousals. Decreases in expiratory time were larger for inspiratory compared with expiratory arousals during breaths −3 to −1 (p < 0.004). Similar to expiratory time, duty cycle (Figure 5C) and respiratory rate (Figure 5D) increases were present on breath 1 (p = 0.001). That is, respiratory load compensation responses were immediate following transient CPAP reductions. Duty cycle increased further during the prearousal breaths and was higher for inspiratory versus expiratory arousals during breaths −3 to −1 (p < 0.003; Figure 5C). Respiratory rate remained at the elevated breath 1 level during the CPAP reduction periods for all breaths (except on breaths −3 and −2 for expiratory and inspiratory arousals where respiratory rate decreased, p = 0.009 and 0.005, respectively). Respiratory rate did not differ between arousal types (p > 0.17; Figure 5D).
Flow, ventilation, and epiglottic pressure
Peak inspiratory flow (Figure 5E) decreased immediately with CPAP reductions for inspiratory and expiratory arousals (∆peak inspiratory flow = ~−0.2 L/s). Peak inspiratory flow decreased further during the breaths preceding inspiratory arousal (breath −3 vs. 1; p = 0.001). However, there were no further decreases in peak inspiratory flow for expiratory arousals. Rather, peak inspiratory flow was restored to baseline levels on the breath preceding expiratory arousals (Figure 5E). Accordingly, peak inspiratory flow was different between inspiratory and expiratory arousals for breaths −2 and −1 (p < 0.003; Figure 5E). Minute ventilation changes (Figure 5F) followed a similar pattern to peak inspiratory flow and were also significantly different between inspiratory and expiratory arousals during breaths −2 and −1 (p < 0.006; Figure 5F).
Negative epiglottic pressure swings increased by a similar magnitude for breaths 1 and 2, with further progressive increases from breaths −3 to −1 (Figure 5G). Breath-by-breath changes in nadir epiglottic pressure were similar for inspiratory and expiratory arousals (p > 0.069; Figure 5G).
Genioglossus and tensor palatini muscle activity
Genioglossus and tensor palatini muscle activity (Figure 6) were higher for the prearousal breaths (−3 to −1) compared with baseline for inspiratory and expiratory arousals (p < 0.001), and for the first two breaths (1 and 2) following CPAP reduction for inspiratory arousals (p < 0.006, except tonic tensor palatini EMG). Changes in muscle activity were similar between inspiratory and expiratory arousals (p ≥ 0.013), except for peak tensor palatini EMG which was greater on the three breaths prior to expiratory arousals compared with inspiratory arousals (p < 0.006; Figure 6C). There also tended to be greater increases in tonic genioglossus EMG immediately prior to inspiratory arousals compared with expiratory arousals, although this difference was not statistically significant (p = 0.013; Figure 6B).
EEG power and K-complex frequency
There were no differences in total (0.5–60 Hz) EEG power between inspiratory and expiratory arousals or in δ (0.5–4 Hz), θ (4–8 Hz), α (8–12 Hz), β (12–32 Hz), and γ (32–60 Hz) frequency bands (Table 4). There were also no differences in K-complex frequency between inspiratory and expiratory arousals [6.7(5.9–7.5) vs. 6.2(5.1–7.3) K-complexes/min, p > 0.05 (bootstrapped t-test)].
Table 4.
EEG (C3/A2) power for the 10 s prior to arousal onset
| Power (µV2) | |||
| Frequency band |
Inspiratory arousals (n = 229) |
Expiratory arousals (n = 125) |
P |
|---|---|---|---|
| Total (0.5–60 Hz) | 325.0 (286.3–364.8) | 322.6 (278.6–373.6) | 0.933 |
| δ (0.5–4 Hz) | 266.2 (229.3–306.6) | 264.7 (222.0–313.6) | 0.963 |
| θ (4–8 Hz) | 32.6 (30.3–35.1) | 32.7 (30.2–35.4) | 0.962 |
| α (8–12 Hz) | 16.9 (15.3–18.4) | 16.9 (15.3–18.6) | 0.950 |
| β (12–32 Hz) | 10.6 (9.9–11.3) | 10 (9.2–10.8) | 0.247 |
| γ (32–60 Hz) | 0.29 (0.25–0.33) | 0.25 (0.21–0.30) | 0.158 |
There were no differences in EEG spectral power between inspiratory and expiratory arousals across all frequency bands. Values are estimated marginal means (confidence interval) from bootstrapped mixed linear model analysis (1000 bootstrap samples, Mersenne Twister seed number = 200,000).
n = number of measurements analyzed.
Discussion
This study provides insight into the timing of respiratory-induced cortical arousals and associated neuromuscular and load compensation responses in OSA. The major findings are that as follows: (1) respiratory-induced cortical arousals occur throughout the respiratory cycle, with over one third occurring during expiration, (2) when arousals do occur during inspiration, they typically precede the peak in negative intrathoracic pressure, and (3) preceding pharyngeal neuromuscular and respiratory load compensation responses are fundamentally different when arousals occur during inspiration versus expiration. Specifically, inspiratory arousals are associated with relatively less pharyngeal muscle activity and deterioration in airflow while the opposite occurs for expiratory arousals. As outlined below, these findings provide new understanding of how arousals occur in OSA and have implications for OSA pathogenesis and treatment.
Mechanisms of arousal
The findings of this study provide new insight into the concept that a threshold level of negative intrathoracic pressure is the predominant trigger for respiratory-induced arousals. Indeed, if this were the case, the majority of arousals in response to respiratory stimuli would be expected to occur approximately 0.5 s after maximal negative intrathoracic pressure, given this is about how long it takes for the respiratory afferent information to arrive and be processed at the cortex (based on respiratory-related evoked potential studies during sleep) [21, 27]. In the current study, this occurred only in a minority of cases. Many arousals also occurred during expiration. Thus, these findings indicate that additional mechanisms must importantly contribute to arousal during airway narrowing in OSA.
Whether an arousal occurred during inspiration or expiration was not explained by the magnitude of the reduction in CPAP used to induce airflow limitation, the time it took for arousal to occur from the initial reduction in CPAP or the rate of change in epiglottic pressure (within the prearousal breath). The magnitude of the nadir epiglottic pressure prior to arousal (arousal threshold) was also similar between inspiratory and expiratory arousals. This finding suggests that, despite different mechanisms likely triggering arousals occurring during inspiration and expiration, the arousal threshold remains an overall good approximation of an individual’s propensity for respiratory-induced arousal.
The findings that neuromuscular and respiratory load compensation responses are fundamentally different between inspiratory and expiratory arousals are intriguing and may provide clues as to what triggers the different types of arousals. Given that expiratory arousals were associated with increased pharyngeal muscle activity and restoration of minute ventilation and less pronounced O2 desaturation just prior to arousal, expiratory arousals may be a marker of enhanced tensor palatini neuromuscular recruitment and airflow compensation. Mechanistically, this could be a consequence of increased chemical drive, which stimulates the pharyngeal muscles, but can also trigger arousal [10, 28–31]. Indeed, changes in chemical drive can have slower time constants than local reflex-driven mechanical changes to the pharyngeal airway. This is also consistent with the finding of decreased rate of change in epiglottic pressure during expiratory versus inspiratory arousals during the CPAP reductions. Most previous neuromuscular compensation studies in OSA have focused on the genioglossus [17, 32, 33]. Although tonic genioglossus EMG also tended to be higher, tensor palatini showed the greatest increases preceding expiratory arousals in the current study. Thus, this finding highlights the potential importance of other pharyngeal muscles beyond genioglossus in restoring airflow prior to arousal in OSA.
Cortical arousals that occur under these circumstances (e.g. when breathing has already been restored) would appear to serve no beneficial function in OSA. Rather, their presence is anticipated to be deleterious due to subsequent destabilizing effects on breathing and sleep continuity [2]. Indeed, in accordance with knowledge that the same stimuli required to activate the pharyngeal dilator muscles can also trigger cortical arousal [2, 3], the current findings indicate that expiratory arousals, in particular, are likely to be a coincident phenomenon in OSA. Accordingly, patients with OSA with predominantly expiratory arousals may be targets for hypnotics to prevent the deleterious consequences of arousal [2, 34, 35].
Additional support for an alternate mechanism to a negative intrathoracic pressure threshold as the primary trigger for arousal comes from Younes [3] who found that ~20 per cent of arousals occur on resumption of airflow following an obstructive event. In the current study, as highlighted above, expiratory arousals tended to follow this pattern whereby recovery of minute ventilation was evident two breaths prior to arousal, reaching baseline (therapeutic CPAP) levels before arousal occurred. This is in contrast to inspiratory arousals where ventilation remained at the reduced level until arousal. The reasons for arousal occurring following airway opening are not entirely clear. Younes [3] proposed a number of possibilities, including sudden unloading of the respiratory muscles, snoring vibration of the upper airway, or the sound of snoring itself. We did not quantify snoring in the current study and therefore do not know if snoring intensity differed between inspiratory and expiratory arousals. Thus, this requires further investigation.
Unlike inspiratory arousals that occur throughout inspiration with a relatively constant distribution not different from chance, expiratory arousals are more likely to occur during early versus late expiration. This is a time when peak expiratory flows and pharyngeal pressures are greatest. Although the reasons mediating this observation are unknown, substantial movement of the pharyngeal structures/cheek puffing likely occurs during this phase of the breathing cycle. Thus, factors such as expiratory velum–related narrowing may trigger arousal in some cases. Further work is clearly required to understand the mechanisms mediating expiratory arousals and their predominance in early expiration. Another potential trigger for respiratory-induced arousal is inability to sustain respiratory load compensation responses that are activated to maintain/restore airflow. This is similar to concepts raised by Vincken and colleagues [20]. The current findings show that respiratory load compensation responses occur directly following reductions in CPAP. There is also an immediate increase in respiratory rate, a change that is maintained until arousal. The rapid increase in respiratory rate suggests that vagal feedback via changes in CPAP may be more pronounced in sleeping humans than previously believed [36]. Consistent with a prior wakefulness study [37], respiratory rate decreased during wakefulness and sleep with therapeutic CPAP compared with wakefulness with no CPAP by ~3 breaths/min (15.0 ± 3.8 and 14.7 ± 2.5 respectively vs. 18.2 ± 4.1 breaths/min, p < 0.001). In addition, expiratory duration decreases further overtime for both arousal types during CPAP reductions, particularly prior to inspiratory arousals, causing duty cycle to increase. Thus, without ongoing respiratory rate modulation (to allow for expiration), there is a limit to the extent to which inspiratory time can increase to maintain/restore ventilation during airway narrowing.
In the case of the expiratory arousals, the prearousal opening of the pharyngeal airway that occurred may cause over distension of the lungs and dynamic hyperinflation to trigger chest wall afferents to arousal centers. Similar to the “neuromechanical uncoupling” hypothesis in the dyspnea literature [38], these factors may create a mismatch between respiratory pattern generator output and information received from respiratory afferents that exceeds a certain limit. Cortical activation under these conditions may serve to rapidly “re-set” the imbalance. As highlighted, physiologically, there must be a limit between what is possible to achieve in terms of changes in respiratory timing (i.e. load compensation via prolongation of inspiratory time and reductions in expiratory time to maintain minute ventilation) before the system exceeds its capacity. However, duty cycle just prior to arousal was lower and minute ventilation was higher during expiratory versus inspiratory arousals. Thus, this mechanism is unlikely to be a key contributing factor to expiratory arousals.
In the current study, the increase in peak tensor palatini activity likely contributed to the gradual increase in minute ventilation prior to expiratory arousals, which was significantly greater than for inspiratory arousals. The tensor palatini is a state-dependent muscle that is generally dormant during sleep and primarily activated during wakefulness or in response to cortical arousal [23, 39, 40]. However, it can also be stimulated by large negative pharyngeal pressures [32, 41], which were similar prior to both inspiratory and expiratory arousals. Given the state-dependence of tensor palatini activity, the increase in peak tensor palatini activity may be explained by reintroduction of wakefulness drive to this muscle not detected visually on the EEG. However, there were no differences in K-complex frequency or EEG power bands between inspiratory and expiratory arousals. Thus, the trigger for expiratory arousals remains unknown, but they do not appear to be attributable to subtle increases in cortical activation prior to American Academy of Sleep Medicine–defined arousal. Although not examined in the current study, quantification of heart-rate changes and timing with inspiratory versus expiratory arousals may provide further insight into potential differences in noncortical contributions between arousal types.
Study limitations
Despite its strengths, this study has a number of limitations that need to be considered (in addition to those discussed previously [4]). Although careful attention was paid to the scoring of arousal onset by an experienced blinded technician, accurate detection of arousal onset can be challenging and is prone to human error. However, given that over 350 arousals were included in analysis, the signal to noise ratio is high and was indeed sufficient to detect real temporal relationships during expiration. Furthermore, we did not separate NREM sleep into its constituent stages. However, 93 per cent of CPAP drops/arousals were during stable N2 sleep. Thus, we do not know if the current findings would be different in light versus deep slow-wave sleep, for example. Similarly, REM sleep was not analyzed, so outcomes cannot be extended into this sleep stage. Finally, we only studied patients with OSA who were adherent with CPAP therapy. Thus, it remains to be determined whether these findings extend to untreated patients with OSA in whom the arousal threshold is elevated.
Study implications
Recent advances in simplified measures for respiratory phenotyping of OSA [4] allow estimation of three of the four traits that contribute to an individual’s OSA from a standard diagnostic sleep study or a CPAP titration: pharyngeal collapsibility [42, 43], arousal threshold [5, 44], and loop gain [45]. These surrogate measures have been derived to translate complex respiratory phenotyping concepts to the clinic to advance tailored therapy for OSA [46]. However, a simplified method for estimating muscle responsiveness, the final trait, is lacking. The current findings raise the possibility that expiratory arousals may be a marker of enhanced neuromuscular compensation and airflow restoration. Indeed, the onset of arousal relative to the respiratory cycle, in addition to other estimates indicating effective dilator muscle responsiveness (e.g. % events resolved without cortical arousal), can be easily extracted from a standard sleep study. Hence, future investigations are required into the utility of arousal timing, including in the absence of CPAP, and other estimates of neuromuscular compensation.
Conclusions
This study has demonstrated that respiratory-induced cortical arousals occur during both inspiration and expiration. Thus, the mechanisms that trigger respiratory-induced cortical arousal are more complex than a sole negative intrathoracic pressure threshold mechanism in OSA. Arousals occurring during expiration are preceded by greater increases in tensor palatini muscle activity and minute ventilation compared with inspiratory arousals. Although arousal threshold remains a good approximation of arousal propensity, the current findings have highlighted the need to reconsider current definitions of arousal for improved understanding of OSA pathogenesis. This study also raises the possibility that identification of timing of arousals in relation to respiratory phase may be helpful in advancing respiratory phenotyping efforts in OSA.
Supplementary Material
Supplementary material is available at SLEEP online.
Funding
This work was supported by NeuroSleep, a National Health and Medical Research Council of Australia (NHMRC) Centre of Research Excellence (CRE) (1060992) and a National Institutes of Health grant (5R01HL048531). J.A. is supported by a NeuroSleep NHMRC CRE Postdoctoral Fellowship (1060992). D.J.E. is supported by a NHMRC Research Fellowship (1116942).
Conflict of interest statement. Outside the submitted work, D.J.E. has received additional grants from NHMRC and a Collaborative Research Centre (CRC) Consortium Grant between the Australian Government, Academia, and Industry (Industry partner: Oventus Medical) and personal fees from Bayer. Outside the submitted work, A.W. has received grants from Philips Respironics, grants and personal fees from Bayer, grants and personal fees from Varnum Sleep and Breathing Solutions, and grants and personal fees from Cambridge Sound Management. C.N. now works for ResMed. The other authors do not have any conflicts of interest to disclose.
Supplementary Material
Acknowledgments
The authors would like to thank the Brigham and Women’s Sleep Disorders Research Program staff for technical support.
References
- 1. Phillipson EA, et al. Arousal: the forgotten response to respiratory stimuli. Am Rev Respir Dis. 1978;118(5):807–809. [DOI] [PubMed] [Google Scholar]
- 2. Eckert DJ, et al. Arousal from sleep: implications for obstructive sleep apnea pathogenesis and treatment. J Appl Physiol (1985). 2014;116(3):302–313. [DOI] [PubMed] [Google Scholar]
- 3. Younes M. Role of arousals in the pathogenesis of obstructive sleep apnea. Am J Respir Crit Care Med. 2004;169(5):623–633. [DOI] [PubMed] [Google Scholar]
- 4. Eckert DJ, et al. Defining phenotypic causes of obstructive sleep apnea. Identification of novel therapeutic targets. Am J Respir Crit Care Med. 2013;188(8):996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Edwards BA, et al. Clinical predictors of the respiratory arousal threshold in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2014;190(11):1293–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gleeson K, et al. The influence of increasing ventilatory effort on arousal from sleep. Am Rev Respir Dis. 1990;142(2):295–300. [DOI] [PubMed] [Google Scholar]
- 7. O’Donnell CP, et al. Effect of sleep deprivation on responses to airway obstruction in the sleeping dog. J Appl Physiol. 1994;77:1811–1818. [DOI] [PubMed] [Google Scholar]
- 8. Berry RB, et al. Respiratory arousal from sleep: mechanisms and significance. Sleep. 1997;20(8):654–675. [DOI] [PubMed] [Google Scholar]
- 9. Carter SG, et al. Zopiclone increases the arousal threshold without impairing genioglossus activity in obstructive sleep apnea. Sleep. 2016;39(4):757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ayas NT, et al. Hypercapnia can induce arousal from sleep in the absence of altered respiratory mechanoreception. Am J Respir Crit Care Med. 2000;162(3 Pt 1):1004–1008. [DOI] [PubMed] [Google Scholar]
- 11. Kaur S, et al. Glutamatergic signaling from the parabrachial nucleus plays a critical role in hypercapnic arousal. J Neurosci. 2013;33(18):7627–7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaur S, et al. A genetically defined circuit for arousal from sleep during hypercapnia. Neuron. 2017;96(5):1153–1167.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yan J, et al. Role of electroencephalogram and oxygen saturation in the induction mechanism of arousal for obstructive sleep apnea-hypopnea syndrome patients. Biol Rhythm Res. 2016;47:483–495. [Google Scholar]
- 14. Xiao SC, et al. Neural respiratory drive and arousal in patients with obstructive sleep apnea hypopnea. Sleep. 2015;38(6):941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chamberlin NL. Brain circuitry mediating arousal from obstructive sleep apnea. Curr Opin Neurobiol. 2013;23(5):774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Younes M. Contributions of upper airway mechanics and control mechanisms to severity of obstructive apnea. Am J Respir Crit Care Med. 2003;168(6):645–658. [DOI] [PubMed] [Google Scholar]
- 17. Jordan AS, et al. Mechanisms used to restore ventilation after partial upper airway collapse during sleep in humans. Thorax. 2007;62(10):861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schneider H, et al. Inspiratory duty cycle responses to flow limitation predict nocturnal hypoventilation. Eur Respir J. 2009;33(5):1068–1076. [DOI] [PubMed] [Google Scholar]
- 19. Hudgel DW, et al. Neuromuscular and mechanical responses to inspiratory resistive loading during sleep. J Appl Physiol (1985). 1987;63(2):603–608. [DOI] [PubMed] [Google Scholar]
- 20. Vincken W, et al. Inspiratory muscle activity as a trigger causing the airways to open in obstructive sleep apnea. Am Rev Respir Dis. 1987;135(2):372–377. [DOI] [PubMed] [Google Scholar]
- 21. Wheatley JR, et al. Influence of NREM sleep on respiratory-related cortical evoked potentials in normal humans. J Appl Physiol (1985). 1993;74(4):1803–1810. [DOI] [PubMed] [Google Scholar]
- 22. Afifi L, et al. Sleep and respiratory stimulus specific dampening of cortical responsiveness in OSAS. Respir Physiol Neurobiol. 2003;136(2–3):221–234. [DOI] [PubMed] [Google Scholar]
- 23. Carberry JC, et al. Upper airway collapsibility (Pcrit) and pharyngeal dilator muscle activity are sleep stage dependent. Sleep. 2016;39(3):511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Berry RB, et al. ; American Academy of Sleep Medicine. Rules for scoring respiratory events in sleep: update of the 2007 AASM manual for the scoring of sleep and associated events. Deliberations of the sleep apnea definitions task force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2012;8(5):597–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nguyen CD, et al. Mild airflow limitation during N2 sleep increases K-complex frequency and slows electroencephalographic activity. Sleep. 2016;39(3):541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sturges HA. The choice of a class interval. J Am Stat Assoc. 1926;21:65–66. [Google Scholar]
- 27. Webster KE, et al. Multichannel EEG analysis of respiratory evoked-potential components during wakefulness and NREM sleep. J Appl Physiol (1985). 1998;85(5):1727–1735. [DOI] [PubMed] [Google Scholar]
- 28. Hedemark LL, et al. Ventilatory and heart rate responses to hypoxia and hypercapnia during sleep in adults. J Appl Physiol Respir Environ Exerc Physiol. 1982;53(2):307–312. [DOI] [PubMed] [Google Scholar]
- 29. Younes M, et al. Mechanisms of breathing instability in patients with obstructive sleep apnea. J Appl Physiol (1985). 2007;103(6):1929–1941. [DOI] [PubMed] [Google Scholar]
- 30. Berthon-Jones M, et al. Ventilatory and arousal responses to hypoxia in sleeping humans. Am Rev Respir Dis. 1982;125(6):632–639. [DOI] [PubMed] [Google Scholar]
- 31. Berthon-Jones M, et al. Ventilation and arousal responses to hypercapnia in normal sleeping humans. J Appl Physiol Respir Environ Exerc Physiol. 1984;57(1):59–67. [DOI] [PubMed] [Google Scholar]
- 32. Jordan AS, et al. Termination of respiratory events with and without cortical arousal in obstructive sleep apnea. Am J Respir Crit Care Med. 2011;184(10):1183–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jordan AS, et al. Airway dilator muscle activity and lung volume during stable breathing in obstructive sleep apnea. Sleep. 2009;32(3):361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jordan AS, et al. Physiology of arousal in obstructive sleep apnea and potential impacts for sedative treatment. Am J Respir Crit Care Med. 2017;196(7):814–821. [DOI] [PubMed] [Google Scholar]
- 35. Eckert DJ, et al. Eszopiclone increases the respiratory arousal threshold and lowers the apnoea/hypopnoea index in obstructive sleep apnoea patients with a low arousal threshold. Clin Sci (Lond). 2011;120(12):505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hamilton RD, et al. The effect of lung inflation on breathing in man during wakefulness and sleep. Respir Physiol. 1988;73(2):145–154. [DOI] [PubMed] [Google Scholar]
- 37. BuSha BF, et al. Identification of respiratory vagal feedback in awake normal subjects using pseudorandom unloading. J Appl Physiol (1985). 2001;90(6):2330–2340. [DOI] [PubMed] [Google Scholar]
- 38. O’Donnell DE, et al. Mechanisms of activity-related dyspnea in pulmonary diseases. Respir Physiol Neurobiol. 2009;167(1):116–132. [DOI] [PubMed] [Google Scholar]
- 39. Amatoury J, et al. Arousal intensity is a distinct pathophysiological trait in obstructive sleep apnea. Sleep. 2016;39(12):2091–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Horner RL, et al. State-dependent and reflex drives to the upper airway: basic physiology with clinical implications. J Appl Physiol (1985). 2014;116(3):325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carberry JC, et al. Mechanisms contributing to the response of upper-airway muscles to changes in airway pressure. J Appl Physiol (1985). 2015;118(10):1221–1228. [DOI] [PubMed] [Google Scholar]
- 42. Azarbarzin A, et al. Estimation of pharyngeal collapsibility during sleep by peak inspiratory airflow. Sleep. 2017;40(1):zsw005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Landry SA, et al. Therapeutic CPAP level predicts upper airway collapsibility in patients with obstructive sleep apnea. Sleep. 2017;40(6):zsx056. doi:10.1093/sleep/zsx056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sands SA, et al. Quantifying the arousal threshold using polysomnography in obstructive sleep apnea. Sleep. 2017;41(1):zsx183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Terrill PI, et al. Quantifying the ventilatory control contribution to sleep apnoea using polysomnography. Eur Respir J. 2015;45(2):408–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eckert DJ. Phenotypic approaches to obstructive sleep apnoea – new pathways for targeted therapy. Sleep Med Rev. 2018;37:45–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.