

Abstract
Lung cancer is the second most common cancer in the United States and the leading cause of mortality in cancer patients. Biomarkers predicting survival of patients with lung cancer have a profound effect on patient prognosis and treatment. However, predictive biomarkers for survival and their relevance for lung cancer are not been well known yet. The objective of this study was to perform machine learning with data from The Cancer Genome Atlas of patients with lung adenocarcinoma (LUAD) to find survival-specific gene mutations that could be used as survival-predicting biomarkers. To identify survival-specific mutations according to various clinical factors, four feature selection methods (information gain, chi-squared test, minimum redundancy maximum relevance, and correlation) were used. Extracted survival-specific mutations of LUAD were applied individually or as a group for Kaplan-Meier survival analysis. Mutations in MMRN2 and GMPPA were significantly associated with patient mortality while those in ZNF560 and SETX were associated with patient survival. Mutations in DNAJC2 and MMRN2 showed significant negative association with overall survival while mutations in ZNF560 showed significant positive association with overall survival. Mutations in MMRN2 showed significant negative association with disease-free survival while mutations in DRD3 and ZNF560 showed positive associated with disease-free survival. Mutations in DRD3, SETX, and ZNF560 showed significant positive association with survival in patients with LUAD while the opposite was true for mutations in DNAJC2, GMPPA, and MMRN2. These gene mutations were also found in other cohorts of LUAD, lung squamous cell carcinoma, and small cell lung cancer. In LUAD of Pan-Lung Cancer cohort, mutations in GMPPA, DNAJC2, and MMRN2 showed significant negative associations with survival of patients while mutations in DRD3 and SETX showed significant positive association with survival. In this study, machine learning was conducted to obtain information necessary to discover specific gene mutations associated with the survival of patients with LUAD. Mutations in the above six genes could predict survival rate and disease-free survival rate in patients with LUAD. Thus, they are important biomarker candidates for prognosis.
Introduction
Lung cancer is the leading cause of death in patients with cancer. It is the second most common cancer in men and women to date in the United States, following prostate cancer in men and breast cancer in women, respectively [1–3]. In the 1990s, stomach and lung cancers were leading causes of death among cancer patients in Korea, with stomach cancer accounting for 25% of cases while lung cancer accounting for 17% of cases. In 2016, 17,963 people died from lung cancer in Korea, accounting for 23% of all cancer-related deaths. Although lung cancer has the highest mortality rate, few biomarkers for predicting overall survival or disease-free survival have been reported. Accurately predicting survival rate in patients with cancer has a significant impact on their prognosis and treatment [4–6].
Machine learning methods have been used in a variety of ways in cancer research [7–10]. These methods can be used to identify breast cancer patients by genetic mutations as a result of applying gene learning methods to breast cancer samples [7]. One prostate cancer study has combined machine learning methods with National Institute for Health and Care Excellence features to observe the association between the prognosis of prostate cancer and genetic mutation profile [8]. In addition, previous studies have applied machine learning using healthy eating index scores to predict the interaction between colorectal cancer and overweight status [9]. In another study, modeling was used to demonstrate the benefit of exact binomial test for analyzing genome-wide somatic gene mutation through performance comparisons among different machine learning models [10].
In this study, machine learning methods with data from The Cancer Genome Atlas (TCGA) of patients with lung adenocarcinoma (LUAD) were utilized to discover gene mutations associated with patient survival. Results suggested that mutations in six genes, DRD3, SETX, ZNF560, DNAJC2, GMPPA, and MMRN2, were significantly associated with survival and overall survival time of patients with LUAD. These gene mutations could be used as survival-predicting biomarkers. Machine learning can be a useful tool to discover important biomarkers for predicting prognosis and survival in patients with lung cancer.
Materials and methods
Ethics statement
All patient data were acquired from previously published studies where written informed consents were obtained [1,11–15]. The TCGA-LUAD cohort, Pan-Lung Cancer cohort, Lung Squamous Cell Carcinoma TCGA cohort, Small Cell Lung Cancer cohort, Lung Adenocarcinoma Broad cohort, and Lung Adenocarcinoma MSKCC cohort in their methods stated that “Specimens were obtained from patients, with appropriate consent from institutional review boards” [1], “All specimens were obtained from patients with appropriate consent and with approval from the relevant institutional review boards” [11], “All specimens were obtained from patients with appropriate consent from the relevant Institutional Review Board” [12], “Human tumour samples were obtained from patients under IRB-approved protocols following written informed consent” [13], “Informed consent (Institutional Review Board) was obtained for each sample using protocols approved by the Broad Institute of Harvard and MIT and each originating tissue source site” [14], and “All patients had consented to Institutional Review Board-approved protocols permitting tissue collection and sequencing” [15], respectively.
Data resource
TCGA-LUAD provided data for LUAD patients with somatic non-silent mutations and clinical information. These TCGA data were downloaded and then divided into clinical data matrix and mutation data matrix. In the original data, clinical information of 471 patients and mutation status of about 40,000 genes were recorded. After preprocessing, label setting and identification were given. Among 471 patients in the data set, 303 living (64.3%) and 168 dead (35.7%) patients were divided accordingly into the survival and non-survival groups, respectively. Data sets of Pan-Lung Cancer cohort (n = 954), Lung Squamous Cell Carcinoma TCGA cohort (n = 498), Small Cell Lung Cancer cohort (n = 101), Lung Adenocarcinoma Broad cohort (n = 135), and Lung Adenocarcinoma MSKCC cohort (n = 34) were also used to determine specific gene mutation frequencies. The first three data sets were used for association of specific gene mutation with survival. All data used within this study were obtained from open access data sets. They have passed the criteria for unrestricted publication with the following statement listed at https://cancergenome.nih.gov/publications/publicationguidelines “No restrictions; all data available without limitations”.
Machine learning
RapidMinor (Boston, MA, USA) was the software used for machine learning. For feature selection, information gain, Chi-squared test, minimum redundancy maximum relevance, and correlation algorithm were used. Classification algorithms included Naive Bayes, k-nearest neighbors, support vector machine, and decision trees [16]. This study concentrated on the yield and selection of gene mutations using dependent algorithms rather than improvement of algorithms (S1 Fig). Accuracy, precision, recall, classification error, and correlation are shown in S1 Table.
Data analysis
For the specificity of gene mutations, Fisher’s exact test and Kaplan-Meier analysis were applied. Frequencies of gene mutations were compared using Fisher’s exact test. Overall survival and disease-free survival were calculated using Kaplan-Meier analysis based on clinical information of mortality, survival, and observation time for patients. cBioPortal software was used to evaluate gene mutation status within the TCGA-LUAD cohort [17, 18]. Statistical significant was considered when p value was less than 0.05.
Results
In this study, to discover gene mutations predicting survival, data of LUAD patients obtained from TCGA were processed and classified using machine learning methods. Mutations in 19 genes were then selected and analyzed by frequencies, overall survival, and disease-free survival. Results suggested that specific gene mutations were associated with patient survival. Among mutations in 19 genes, mutations in GMPPA and MMRN2 were significantly associated with patient mortality while mutations in ZNF560 and SETX were significantly associated with patient survival (Fig 1 and Table 1). Mutations in DNAJC2 and MMRN2 showed significant negative association with overall survival while mutations in ZNF560 showed significant positive association with overall survival. The median survival time in patients with LUAD was about 49 months. However, the median survival time in patients with mutations in MMRN2 was about 11 months. Mutations in MMRN2 were significantly and negatively associated with disease-free survival while mutations in DRD3 and ZNF560 were significantly and positively associated with disease-free survival. The median disease-free survival time in patients with LUAD was about 36 months while that in patients with mutations in MMRN2 was about 5 months. Mutations in DNAJC2, GMPPA, MMRN2, DRD3, SETX, and ZNF560 were associated with survival in patients with LUAD.
Fig 1. Comparison of mutation frequencies in 19 genes.

Mutations in 19 genes were demonstrated as frequent and analyzed using Fisher’s extract test. * indicates p < 0.05.
Table 1. Comparative analysis of mutation frequency and survival with mutations in 19 genes selected by feature selection methods.
| Chromosome | 1 | 2 | 3 | 7 | 8 | 9 | 10 | 11 | 17 | 19 | 22 | X | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutated gene | S1PR1 | GMPPA | ANKMY1 | GOLGA4 | DRD3 | CALN1 | DNAJC2 | PURG | SETX | SPAG6 | SKIDA1 | MMRN2 | FKBP2 | TNRC6C | GPR179 | ZNF560 | SIPA1L3 | PPP6R2 | SSX3 | |
| Mutation | Total | 5 | 3 | 6 | 6 | 5 | 4 | 2 | 2 | 9 | 3 | 4 | 3 | 1 | 5 | 3 | 9 | 2 | 1 | 4 |
| (1.06%) | (0.64%) | (1.27%) | (1.27%) | (1.06%) | (0.85%) | (0.42%) | (0.42%) | (1.91%) | (0.64%) | (0.85%) | (0.64%) | (0.21%) | (1.06%) | (0.64%) | (1.91%) | (0.42%) | (0.21%) | (0.85%) | ||
| Living | 1 | 0 | 6 | 6 | 5 | 4 | 0 | 0 | 9 | 3 | 4 | 0 | 0 | 5 | 3 | 9 | 1 | 0 | 2 | |
| Deceased | 4 | 3 | 0 | 0 | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 3 | 1 | 0 | 0 | 0 | 1 | 1 | 2 | |
| Fisher’s exact | 0.057 | 0.045* | 0.930 | 0.930 | 0.166 | 0.302 | 0.127 | 0.127 | 0.030* | 0.556 | 0.302 | 0.045* | 0.357 | 0.166 | 0.556 | 0.030* | 1.000 | 0.357 | 0.619 | |
| Overall Survival | P-value | 0.154 | 0.061 | 0.209 | 0.132 | 0.075 | 0.380 | 6.66E-09*** | 0.158 | 0.113 | 0.307 | 0.123 | 4.20E-05*** | 0.311 | 0.086 | 0.467 | 0.032* | 0.784 | 0.743 | 0.622 |
| Median Months | 32.82 | 28.09 | NA | NA | NA | NA | 0 | NA | NA | NA | NA | 11.27 | 29.73 | NA | NA | NA | 46.68 | 49.01 | 9.95 | |
| Disease Free Survival | P-value | 0.269 | 0.055 | 0.468 | 0.989 | 0.049* | 0.245 | NA | 0.846 | 0.062 | 0.280 | 0.843 | 4.29E-05*** | NA | 0.479 | 0.333 | 0.034* | 0.514 | 0.751 | 0.498 |
| Median Months | 13.86 | 6.87 | NA | 22.60 | NA | NA | NA | NA | NA | NA | 84.57 | 4.57 | NA | NA | NA | NA | 17.90 | 35.58 | 18.66 | |
Median month means the median survival time of patients in a given group. Fisher’s exact P-value: Log rank test, NA: Not Available,
* indicates p<0.05,
*** indicates p<0.001
As shown in Table 2, patients with LUAD lacking mutations in DNAJC2 or MMRN2 had median survival time of about 48 months while those with mutations in DNAJC2 or MMRN2 all died within 20 months (S2 Fig). About 42% of patients with LUAD lacking a mutation in MMRN2 relapsed with a median disease-free time of about 33 months while those with mutations in MMRN2 all relapsed within 10 months. Therefore, mutations in DNAJC2 and/or MMRN2 are considered to be predictors of survival or relapse of patients with LUAD since the probability of death or recurrence due to LUAD might be higher in the presence of mutations in DNAJC2 or MMRN2. In contrast, patients with LUAD without a mutation in ZNF560 had shorter survival than other patients, with a median survival time of about 45 months while those with mutations in ZNF560 all survived. Additionally, about 42% of patients with LUAD without a mutation in ZNF560 or DRD3 relapsed with a median disease-free time of about 30 months. However, those with mutations in ZNF560 or DRD3 relapsed at a rate of about 11% or 0%, respectively. Because the probability of death or relapse due to LUAD might be lower when the ZNF560 or DRD3 was mutated, mutations in ZNF560 or DRD3 were considered to be predictors of survival or relapse in patients with LUAD.
Table 2. Comparative analysis of overall survival and disease-free survival in the gene mutation group and non-mutation group.
| Survival Gene | Overall Survival | Disease Free Survival | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P-value | Mutation Negative | Mutation Positive | P-value | Mutation Negative | Mutation Positive | |||||||||||||
| Total Case | Decease Case (N) | Decease Case (%) | Median Survival (month) | Total Case | Decease Case (N) | Decease Case (%) | Median Survival (month) | Total Case | Relapse Case (N) | Relapse Case (%) | Median Disease Free (month) | Total Case | Relapse Case (N) | Relapse Case (%) | Median Disease Free (month) | |||
| GMPPA | 0.061 | 468 | 165 | 35.26% | 49.01 | 3 | 3 | 100.00% | 28.09 | 0.055 | 399 | 166 | 41.60% | 33.15 | 2 | 2 | 100.00% | 6.87 |
| DRD3 | 0.075 | 466 | 168 | 36.05% | 46.68 | 5 | 0 | 0.00% | NA | 0.049 | 396 | 168 | 42.42% | 30.98 | 5 | 0 | 0.00% | NA |
| DNAJC2 | 0.0000000067 | 469 | 166 | 35.39% | 47.77 | 2 | 2 | 100.00% | 0 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| SETX | 0.113 | 462 | 168 | 36.36% | 46.68 | 9 | 0 | 0.00% | NA | 0.062 | 392 | 168 | 42.86% | 30.98 | 9 | 0 | 0.00% | NA |
| MMRN2 | 0.000042 | 468 | 165 | 35.26% | 47.77 | 3 | 3 | 100.00% | 11.27 | 0.000043 | 399 | 166 | 41.60% | 33.15 | 2 | 2 | 100.00% | 4.57 |
| ZNF560 | 0.032 | 462 | 168 | 36.36% | 45.3 | 9 | 0 | 0.00% | NA | 0.034 | 392 | 167 | 42.60% | 30.39 | 9 | 1 | 11.11% | NA |
To evaluate the association of mutations in multiple genes and survival, mutations in 19 genes and those in genes associated with survival were analyzed (Fig 2 and S2 Table). Mutations in these 19 genes were not associated with overall survival or disease-free survival. Mutations in DNAJC2 or MMRN2 were negatively associated with survival. They significantly decreased the median survival time to 9.95 months and the median disease-free survival time to 4.57 months. However, mutations in DRD3 or ZNF560 were positively associated with survival. They significantly increased both survival time and disease-free survival time. Furthermore, mutations in DNAJC2, GMPPA, or MMRN2 significantly decreased median survival time to 11.27 months and the median disease-free survival time to 6.87 months (Fig 3). Mutations in DRD3, SETX, or ZNF560 significantly increased both survival time and disease-free survival time. Patients with LUAD who had mutations in DNAJC2, GMPPA, or MMRN2 exhibited significantly shorter survival and earlier recurrence than those without mutations. However, patients with mutations in DRD3, SETX, or ZNF560 exhibited longer survival and later recurrence than those without these mutations.
Fig 2. Survival analysis for mutations in 19 genes and survival-related genes.

Survival and disease-free survival time of patients with or without specific gene mutations were analyzed using Kaplan-Meier curves. These 19 gene mutations, two gene mutations negatively associated with survival (DNAJC2 or MMRN2), and two gene mutations positively associated with survival (DRD3 or ZNF560) were analyzed. Overall survivals is shown in A-C while disease-free survival is shown in D-F. Red lines indicate mutation positive while blue lines indicate mutation negative. P-values were obtained from Log rank test.
Fig 3. Comparative survival analysis for mutations in survival-related genes.

Survival and disease-free survival time of patients with or without specific gene mutations were analyzed using Kaplan-Meier curves. Mutations in three genes (DNAJC2, GMPPA, or MMRN2) negatively associated with survival and mutations in three genes (DRD3, SETX, or ZNF560) positively associated with survival were analyzed. Overall survival is shown in A and B while disease-free survival is shown in C and D. Red lines indicate mutation positive while blue lines indicate mutation negative. P-values were obtained from Log rank test.
The frequency of mutations in six gene associated with survival was further analyzed in other lung cancer types such as lung squamous cell carcinoma, small cell lung cancer, and another two data sets of LUAD (Table 3 and S3 Fig). Mutations in these six genes were found not only in another two LUAD data sets (n = 21 and n = 4), but also in lung squamous cell carcinoma (n = 28), small cell lung cancer (n = 12), and Pan-lung cancer (n = 146).
Table 3. Comparative analysis of mutation frequencies of lung adenocarcinoma-associated six genes in six data sets of different lung cancer cohorts.
| Data set \ Gene name | GMPPA | DRD3 | DNAJC2 | SETX | MMRN2 | ZNF560 |
|---|---|---|---|---|---|---|
| Lung Adenocarcinoma TCGA (n = 471) | 3 (0.64%) | 5 (1.06%) | 2 (0.42%) | 9 (1.91%) | 3 (0.64%) | 9 (1.91%) |
| Pan-Lung Cancer Nat Genet 2016 (n = 954) | 10 (1.05%) | 33 (3.46%) | 12 (1.26%) | 44 (4.61%) | 10 (1.05%) | 37 (3.88%) |
| Lung Squamous Cell Carcinoma TCGA (n = 498) | 2 (0.40%) | 5 (1.00%) | 0 (0.00%) | 10 (2.01%) | 0 (0.00%) | 11 (2.21%) |
| Small Cell Lung Cancer U Cologne, Nature 2015 (n = 101) | 2 (1.98%) | 3 (2.97%) | 1 (0.99%) | 5 (4.95%) | 1 (0.99%) | 0 (0.00%) |
| Lung Adenocarcinoma Broad, Cell 2012 (n = 135) | 1 (0.74%) | 7 (5.19%) | 0 (0.00%) | 4 (2.96%) | 4 (2.96%) | 5 (3.70%) |
| Lung Adenocarcinoma MSKCC 2015 (n = 34) | 0 (0.00%) | 0 (0.00%) | 0 (0.00%) | 1 (2.94%) | 0 (0.00%) | 3 (8.82%) |
| Sum | 18 (0.82%) | 53 (2.42%) | 15 (0.68%) | 73 (3.33%) | 18 (0.82%) | 65 (2.96%) |
Associations of mutations in six genes with survival were analyzed using three data sets of lung cancer cohorts with survival information (Table 4). Mutations in GMPPA, DNAJC2, and MMRN2 were significantly associated with patient mortality in lung adenocarcinoma of Pan-Lung Cancer cohort. Mutations in DNAJC2 and MMRN2 were significantly associated with shortened overall survival (median survival of 10 and 21.9 months, respectively) (S4 Fig). Mutations in DRD3 and SETX were significantly associated with patient survival, and mutations in SETX extended overall survival. These gene mutations were not associated with mortality or overall survival in other types of lung cancer. These results suggested that mutations in DNAJC2, GMPPA, MMRN2, DRD3, and SETX could be significantly associated with survival in patients with LUAD. They might be considered as biomarkers for predicting survival or recurrence in patients with LUAD.
Table 4. Comparative analysis of six-gene mutation frequency and survival in three data sets of lung cancer cohorts.
| Chromosome | 2 | 3 | 7 | 9 | 10 | 19 | ||
|---|---|---|---|---|---|---|---|---|
| Mutated gene | GMPPA | DRD3 | DNAJC2 | SETX | MMRN2 | ZNF560 | ||
| Pan-Lung Cancer Nat Genet 2016 (n = 954) | Mutation | Total | 10 (1.05%) | 33 (3.46%) | 12 (1.26%) | 44 (4.61%) | 10 (1.05%) | 37 (3.88%) |
| Living | 5 | 25 | 6 | 37 | 3 | 28 | ||
| Deceased | 5 | 8 | 6 | 7 | 7 | 9 | ||
| Fisher’s exact | 0.158 | 0.697 | 0.111 | 0.061 | 0.0075* | 0.711 | ||
| Overall Survival | P-value | 0.12 | 0.757 | 0.0066** | 0.063 | 0.00014*** | 0.89 | |
| Median Months | 28.1 | 31.51 | 14.1 | 63.59 | 15 | 39.1 | ||
| Pan-Lung Cancer Nat Genet 2016; Lung Adenocarcinoma (n = 481) | Mutation | Total | 6 (1.25%) | 14 (2.91%) | 5 (1.04%) | 22 (4.57%) | 8 (1.66%) | 18 (3.74%) |
| Living | 2 | 14 | 1 | 21 | 3 | 16 | ||
| Deceased | 4 | 0 | 4 | 1 | 5 | 2 | ||
| Fisher’s exact | 0.034* | 0.027* | 0.014* | 0.023* | 0.024* | 0.27 | ||
| Overall Survival | P-value | 0.074 | 0.16 | 0.00014*** | 0.027* | 0.0025** | 0.65 | |
| Median Months | 28.1 | NA | 14.1 | NA | 21.9 | NA | ||
| Lung Squamous Cell Carcinoma TCGA (n = 498) | Mutation | Total | 2 (0.40%) | 5 (1.00%) | 0 | 10 (2.01%) | 0 | 11 (2.21%) |
| Living | 1 | 2 | NA | 6 | NA | 5 | ||
| Deceased | 1 | 3 | NA | 4 | NA | 6 | ||
| Fisher’s exact | 1 | 0.657 | NA | 1 | NA | 0.545 | ||
| Overall Survival | P-value | 0.33 | 0.107 | NA | 0.919 | NA | 0.747 | |
| Median Months | 1.71 | 9.82 | NA | 63.5 | NA | 30.09 | ||
| Small Cell Lung Cancer U Cologne, Nature 2015 (n = 101) | Mutation | Total | 2 (1.96%) | 3 (2.94%) | 1 (0.98%) | 5 (4.90%) | 1 (0.98%) | 0 |
| Living | 1 | 1 | 1 | 3 | 1 | NA | ||
| Deceased | 1 | 2 | 0 | 2 | 0 | NA | ||
| Fisher’s exact | 1 | 1 | 0.333 | 0.33 | 0.333 | NA | ||
| Overall Survival | P-value | 0.722 | 0.812 | 0.266 | 0.159 | 0.351 | NA | |
| Median Months | 3 | 27 | NA | 72 | NA | NA | ||
Fisher’s exact P-value: Log rank test, NA: not available,
* indicates p<0.05,
** indicates p<0.01,
*** indicates p<0.001
Discussion
Lung cancer is the second most common cancer. It has a high mortality rate. The discovery of biomarkers that can predict overall survival of lung cancer patients is essential for treatment of patients. Identification of survival-specific gene mutations is important not only for understanding genetic disparities associated with survival, but also for predicting the survival of LUAD patients. These gene mutations can be significant biomarkers for LUAD. In this study, TCGA LUAD data set was used to derive gene mutations by machine learning. Patients with LUAD were divided into surviving and non-surviving groups and machine learning was performed with four feature selection methods to identify gene mutations associated with survival of patients with LUAD from mutations in about 40,000 genes [16,19–24]. The most frequently observed mutations determined by machine learning were in SETX and ZNF560 genes. Mutational incidence of SETX, ZNF560, GMPPA, and MMRN2 was significant. Mutations in MMRN2 and DNAJC2 were significantly and negatively associated with patient survival while those in ZNF560 and DRD3 were positively associated with patient survival. Mutations in genes determined by machine learning seem to influence survival in LUAD.
Because of the relatively small number of mutations in LUAD cohort, mutations in six genes were applied to other data sets of lung cancer cohorts to analyze mutation frequencies and association with survivals such as Lung Adenocarcinoma (Broad and MSKCC) cohorts, Lung Squamous Cell Carcinoma TCGA cohort, Pan-Lung Cancer cohort, and Small Cell Lung Cancer cohort. The average frequency of mutations in six genes was 0.81% ~ 3.30% and their associations with survival were similar between LUAD cohort and Pan-Lung Cancer cohort. Data set of Pan-Lung Cancer cohort was composed of LUAD and lung squamous cell carcinoma. In LUAD of Pan-Lung Cancer cohort, mutations in DNAJC2, GMPPA, MMRN2, DRD3, and SETX were significantly associated with survival status, and those in DNAJC2, MMRN2, and SETX were significantly associated with overall survival. This result supports that mutations in these six genes can predict the survival of patients with LUAD and overall survival time. They could be considered as biomarkers of LUAD.
Mutations in MMRN2 and DNAJC2 were observed to be important for predicting the survival and prognosis negatively. MMRN2 encodes a multimerin2 which is an elastin microfibril interface-located (EMILIN)-like protein, extracellular matrix glycoprotein [25]. MMRN2 acts as a modified growth factor β antagonist. It can interfere with VEGF-A/VEGFR2 pathway in endothelial cells [25]. Recent studies have demonstrated that CLEC14A-MMRN2 binding has potential for future anti-angiogenic therapy because it plays a role in inhibiting angiogenesis during tumor growth [26]. The DNAJC2 gene encodes a phosphorylated protein with a J-domain and a Myb DNA-binding domain. Its protein is observed in both nucleus and cytoplasm [27]. DNAJC2 protein can form a heterodimeric complex with the ribosome to acts as a molecular protector for the initial polypeptide chain when exiting the ribosome [27]. DNAJC2 protein has been identified as a leukemia-associated antigen. Its expression is increased in those with leukemic seizures [28]. In addition, chromosomal abnormalities involving the DNAJC2 gene are associated with primary head and neck squamous cell tumors [29]. These studies have revealed molecular mechanisms that MMRN2 and DNAJC2 either cause or exacerbate cancers. However, further studies are needed to determine the role of MMRN2 and DNAJC2 in LUAD.
Mutations in ZNF560 and SETX were observed to be important for predicting the survival and prognosis positively. The ZNF560 gene has been reported in colorectal cancer studies [30]. Left-sided colon cancer (LSCC) and right-sided colon cancer (RSCC) differ in their genetic susceptibilities to neoplastic transformation. Patients with LSCC had low mortality and improved overall 5-year survival rate than patients with RSCC [30]. ZNF560 was down-regulated in LSCC compared to that in RSCC. It may be useful for predicting a positive prognosis. SETX is a RNA/DNA helicase that splices RNA, regulates gene expression, terminates transcription, and stabilizes telomere and genome [31]. Mutations in SETX are linked to neurodegenerative disorders, ataxia oculomotor apraxia type 2, and amyotrophic lateral sclerosis type 4 [31]. Although the role of SETX in cancer has not been known, its expression level is relatively lower than other genes. Since there is little research on the role of ZNF560 and SETX in cancers, more researches are needed to understand their roles in LUAD.
Since this study focused on the yield and selection of gene mutation rather than deducing an efficient algorithm through machine learning, a dependent algorithm was used. In this case, the weighted results of the independent algorithm could not be obtained. Utilizing 100% trained data is closer to probability statistics than machine learning. Of 19 gene mutations, six gene mutations were significantly associated with survival in LUAD, showing a relatively high rate (about 32%). Further study is needed to determine the differences between using dependent and independent algorithms in machine learning methods for analyzing medical information of solid tumors.
It is important to apply the optimal feature selection method that classifies human cancer genetic mutations according to specific factors among various feature selection methods. Previously reported feature selection methods in medical studies have used Weka that can implement information gain, correlation, and ranker algorithms, and ensemble learning methods [32–34]. However, in order to classify gene mutations using dependent algorithm, selection methods for prediction of economic demand as well as the above feature selection method were applied to feature selection in this study. Of combination algorithms used, the combination with the highest classification prediction rate was information gain-Naïve bayes combination. It can be adopted to analyze RNA sequence or other medical information in LUAD.
In summary, machine learning was conducted to obtain information necessary to select mutations in genes associated with survival of patients with LUAD. We identified specific mutational markers associated with survival of patients with LUAD. Mutations in DNAJC2, GMPPA, and MMRN2 can be used as biomarkers of negative prognosis for patient’s overall survival and disease-free survival while mutations in DRD3, SETX, and ZNF560 can be used as biomarkers of positive prognosis. This study also suggested a predictive classification model of LUAD based on mutation expression.
Supporting information
The general machine learning method is Method I and our machine learning method is Method II. FS indicates feature selection.
(JPG)
{kind=link}
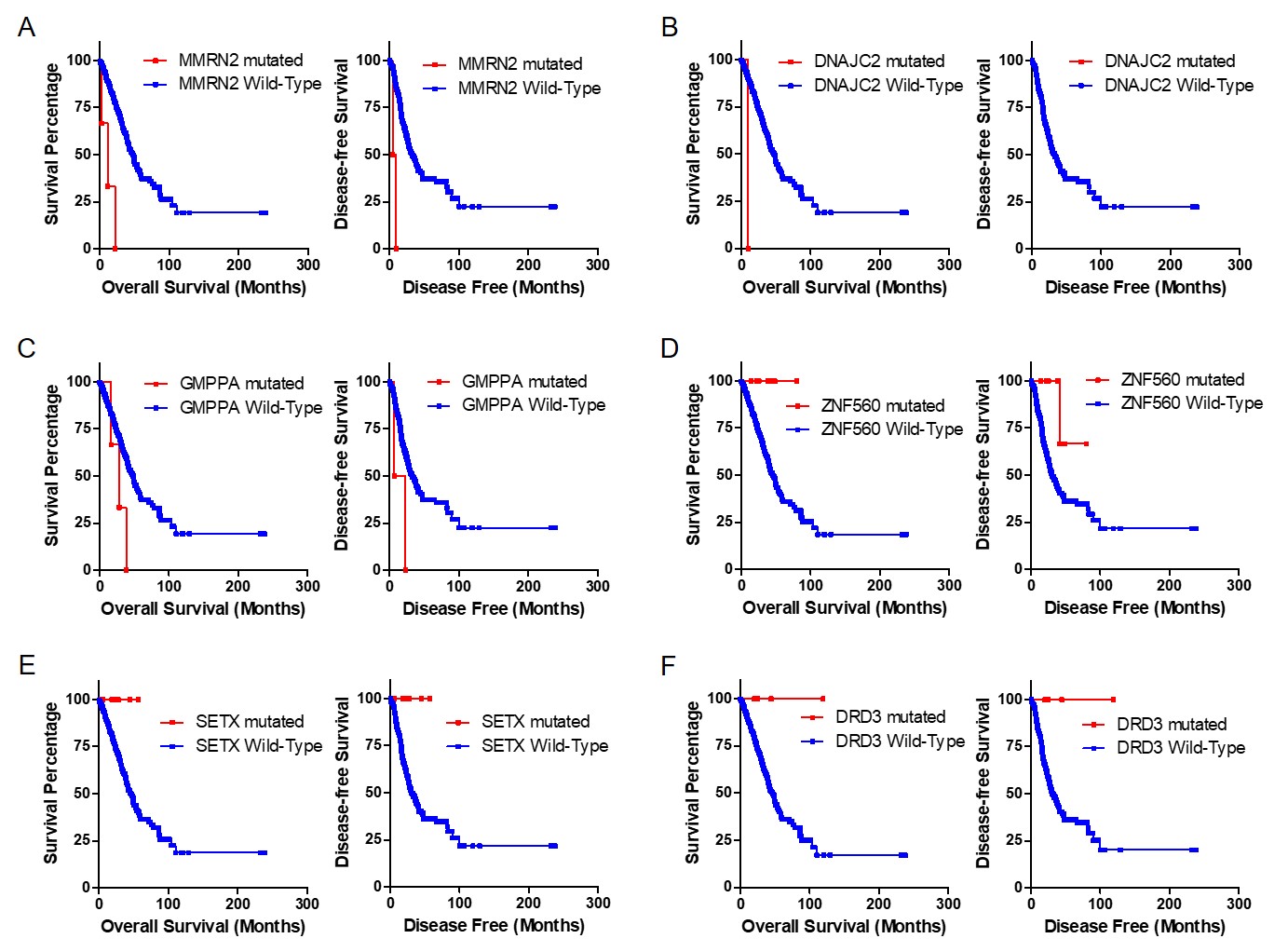
Survival and disease-free survival time of patients with or without specific gene mutations were analyzed using Kaplan-Meier curves. Overall survival is shown in the left column while disease-free survival is shown in the right column. P-values were obtained from Log rank test.
(JPG)
{kind=link}
Mutations in six genes were demonstrated as frequency (percent).
(JPG)
{kind=link}
Overall survival time of patients with or without specific gene mutations were analyzed using Kaplan-Meier curves. P-values were obtained from Log rank test.
(JPG)
{kind=link}
(DOCX)
(DOCX)
Data Availability
All relevant data are within the manuscript and its Supporting Information file.
Funding Statement
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2017R1D1A1B03035616) and by the Bio & Medical Technology Development Program of NRF funded by the Ministry of Science & ICT (2016M3A9E8941667). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511: 543–550. 10.1038/nature13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Song NY, Zhu F, Wang Z, Willette-Brown J, Xi S, Sun Z, et al. IKKα inactivation promotes Kras-initiated lung adenocarcinoma development through disrupting major redox regulatory pathways. Proc Natl Acad Sci U S A. 2018;115: E812–E821. 10.1073/pnas.1717520115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen X, Duan Q, Xuan Y, Sun Y, Wu R. Possible pathways used to predict different stages of lung adenocarcinoma. Medicine (Baltimore). 2017;96:e6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen GK, Mellnick VM, Yim AK, Salter A, Ippolito JE. Synergy of Sex Differences in Visceral Fat Measured with CT and Tumor Metabolism Helps Predict Overall Survival in Patients with Renal Cell Carcinoma. Radiology. 2018;287:884–892. 10.1148/radiol.2018171504 [DOI] [PubMed] [Google Scholar]
- 5.Majeti BK, Lee JH, Simmons BH, Shojaei F. VEGF is an important mediator of tumor angiogenesis in malignant lesions in a genetically engineered mouse model of lung adenocarcinoma. BMC Cancer. 2013;13:213 10.1186/1471-2407-13-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Director’s Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma, Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med. 2008;14:822–827. 10.1038/nm.1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vural S, Wang X, Guda C. Classification of breast cancer patients using somatic mutation profiles and machine learning approaches. BMC Syst Biol. 2016;10 Suppl 3:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang S, Xu Y, Hui X, Yang F, Hu Y, Shao J, et al. Improvement in prediction of prostate cancer prognosis with somatic mutational signatures. J Cancer. 2017;8:3261–3267. 10.7150/jca.21261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shiao SPK, Grayson J, Lie A, Yu CH. Personalized Nutrition-Genes, Diet, and Related Interactive Parameters as Predictors of Cancer in Multiethnic Colorectal Cancer Families. Nutrients. 2018;10. pii: E795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hui X, Hu Y, Sun MA, Shu X, Han R, Ge Q, et al. EBT: a statistic test identifying moderate size of significant features with balanced power and precision for genome-wide rate comparisons. Bioinformatics. 2017;33:2631–2641. 10.1093/bioinformatics/btx294 [DOI] [PubMed] [Google Scholar]
- 11.Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016;48:607–616. 10.1038/ng.3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. 10.1038/nature11404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.George J, Lim JS, Jang SJ, Cun Y, Ozretić L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53. 10.1038/nature14664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. 10.1016/j.cell.2012.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Witten IH, Frank E. Data Mining: Practical machine learning tools and techniques 2005.
- 17.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ricketts CJ, Linehan WM. Gender Specific Mutation Incidence and Survival Associations in Clear Cell Renal Cell Carcinoma (CCRCC). PLoS One. 2015;10(10):e0140257 10.1371/journal.pone.0140257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN, et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell. 2018;173:338–354.e15. 10.1016/j.cell.2018.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu KH, Berry GJ, Rubin DL, Ré C, Altman RB, Snyder M. Association of Omics Features with Histopathology Patterns in Lung Adenocarcinoma. Cell Syst. 2017;5(6):620–627.e3. 10.1016/j.cels.2017.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bashiri A, Ghazisaeedi M, Safdari R, Shahmoradi L, Ehtesham H. Improving the Prediction of Survival in Cancer Patients by Using Machine Learning Techniques: Experience of Gene Expression Data: A Narrative Review. Iran J Public Health. 2017;46:165–172. [PMC free article] [PubMed] [Google Scholar]
- 23.Borisov N, Tkachev V, Suntsova M, Kovalchuk O, Zhavoronkov A, Muchnik I, et al. A method of gene expression data transfer from cell lines to cancer patients for machine-learning prediction of drug efficiency. Cell Cycle. 2018;17:486–491. 10.1080/15384101.2017.1417706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Way GP, Sanchez-Vega F, La K, Armenia J, Chatila WK, Luna A, et al. Machine Learning Detects Pan-cancer Ras Pathway Activation in The Cancer Genome Atlas. Cell Rep. 2018;23:172–180.e3. 10.1016/j.celrep.2018.03.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rao N, Lee YF, Ge R. Novel endogenous angiogenesis inhibitors and their therapeutic potential. Acta Pharmacol Sin. 2015;36:1177–1190. 10.1038/aps.2015.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noy PJ, Lodhia P, Khan K, Zhuang X, Ward DG, Verissimo AR, et al. Blocking CLEC14A-MMRN2 binding inhibits sprouting angiogenesis and tumour growth. Oncogene. 2015;34:5821–5831. 10.1038/onc.2015.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14:105–111. 10.1007/s12192-008-0068-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sahu SN, Lewis J, Patel I, Bozdag S, Lee JH, Sprando R, et al. Genomic analysis of stress response against arsenic in Caenorhabditis elegans. PLoS One. 2013;8:e66431 10.1371/journal.pone.0066431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Resto VA, Caballero OL, Buta MR, Westra WH, Wu L, Westendorf JM, et al. A putative oncogenic role for MPP11 in head and neck squamous cell cancer. Cancer Res. 2000;60:5529–5535. [PubMed] [Google Scholar]
- 30.Zhu H, Wu TC, Chen WQ, Zhou LJ, Wu Y, Zeng L, et al. Screening for differentially expressed genes between left-and right-sided colon carcinoma by microarray analysis. Oncol Lett. 2013; 6:353–358. 10.3892/ol.2013.1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Groh M, Albulescu LO, Cristini A, Gromak N. Senataxin: Genome Guardian at the Interface of Transcription and Neurodegeneration. J Mol Biol. 2017;429:3181–3195. 10.1016/j.jmb.2016.10.021 [DOI] [PubMed] [Google Scholar]
- 32.Assarzadeh Z, Naghsh-Nilchi AR. Chaotic particle swarm optimization with mutation for classification. J Med Signals Sens. 2015;5:12–20. [PMC free article] [PubMed] [Google Scholar]
- 33.Hemphill E, Lindsay J, Lee C, Măndoiu II, Nelson CE. Feature selection and classifier performance on diverse bio- logical datasets. BMC Bioinformatics. 2014;15 Suppl 13:S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kourou K, Exarchos TP, Exarchos KP, Karamouzis MV, Fotiadis DI. Machine learning applications in cancer prognosis and prediction. Comput Struct Biotechnol J. 2014;13:8–17. 10.1016/j.csbj.2014.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The general machine learning method is Method I and our machine learning method is Method II. FS indicates feature selection.
(JPG)
Survival and disease-free survival time of patients with or without specific gene mutations were analyzed using Kaplan-Meier curves. Overall survival is shown in the left column while disease-free survival is shown in the right column. P-values were obtained from Log rank test.
(JPG)
Mutations in six genes were demonstrated as frequency (percent).
(JPG)
Overall survival time of patients with or without specific gene mutations were analyzed using Kaplan-Meier curves. P-values were obtained from Log rank test.
(JPG)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information file.