

Abstract
Bicyclic aziridinium ions were generated by the removal of an appropriate leaving group through internal nucleophilic attack by nitrogen atom in the aziridine ring. The utility of bicyclic aziridinium ions, specifically 1-azoniabicyclo[3.1.0]hexane and 1-azoniabicyclo[4.1.0]heptane tosylate highlighted in the aziridine ring openings by the nucleophile with the release of the ring strain to yield the corresponding ring-expanded azaheterocycles such as pyrrolidine, piperidine and azepane with diverse substituents on the ring in regio- and stereospecific manner. Herein, we report a simple and convenient method for the preparation of the stable 1-azabicyclo[4.1.0]heptane tosylate followed by selective ring opening via a nucleophilic attack either at the bridge or at the bridgehead carbon to yield piperidine and azepane rings, respectively. This synthetic strategy allowed us to prepare biologically active natural products containing piperidine and azepane motif including sedamine, allosedamine, fagomine and balanol in highly efficient manner.
Keywords: Chemistry, Issue 138, Bicyclic aziridinium ions, ring-expansion, stereospecific, azaheterocycles, piperidines, azepane
Introduction
Among three membered cyclic compounds, aziridine has similar ring strain energy as cyclopropane and oxirane to afford various nitrogen-containing cyclic and acyclic compounds via ring opening1,2,3. However, the characteristics and reactivity of aziridine depend on the substituent of ring nitrogen. Aziridine with an electron-withdrawing group at the ring nitrogen4, is called "activated aziridine", which is activated to react with the incoming nucleophile without any additional activating reagent. On the other hand, "non-activated aziridine" with electron-donating substituent at nitrogen is quite stable and inert to the nucleophiles, unless it is activated as an aziridinium ion as Ia (Figure 1a)5,6,7. The ring opening of a non-activated aziridine depends on various factors such as the substituents at C2 and C3 carbon of aziridine, the electrophile to activate the aziridine ring and the incoming nucleophile. The isolation and characterization of an aziridinium ion is not possible due to its high reactivity towards ring-opening reaction by nucleophiles, but its formation and characteristics were observed spectroscopically with a non-nucleophilic counter anion5,8,9,10. The regio- and stereoselective ring-opening reaction of aziridinium ion by a suitable nucleophile yields nitrogen-containing acyclic valuable molecules (Pi and Pii)5,6,7,8,9,10.
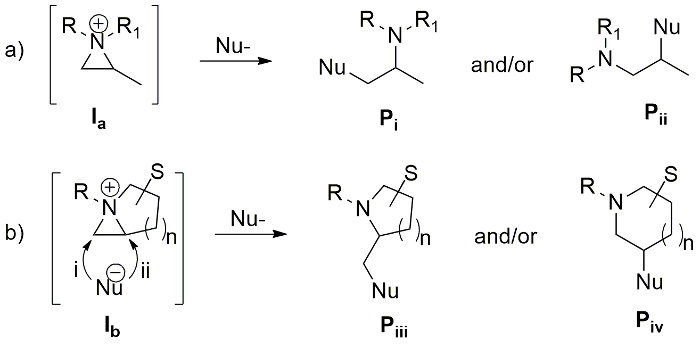
Similarly, a bicyclic aziridinium ion (Ib) is possibly generated via the removal of the leaving group by the nucleophilic attack of ring nitrogen of aziridine in intramolecular fashion (Figure 1b). Then, this intermediate undergoes ring-expansion with the incoming nucleophile via the release of ring strain. The formation and stability of bicyclic aziridinium ion are dependent on many factors such as the substituents, the size of the ring, and solvent medium9. The regio- and stereoselectivity of the aziridine ring-expansion is a pivotal aspect of its synthetic utility, which depends on the nature of the substituents in the starting substrate and the characteristics of applied nucleophile.
In our early study, we succeeded to prepare 1-azoniabicyclo [3.1.0]hexane tosylate Ib (n = 1) whose subsequent ring-expansion resulted in the formation of a pyrrolidine and a piperidine (Piii and Piv, n = 1, Figure 1)8. As a part of our continuing study on the bicyclic aziridinium ion chemistry, we describes herein the formation of 1-azoniabicyclo [4.1.0]heptane tosylate Ib (n = 2) as a representative example. This was prepared from 2-(4-toluensulfonyloxybutyl)aziridine and its ring-expansion was trigged by a nucleophile to afford valuable piperidine and azepane (Pi and Pii, n = 2, Figure 1) with diverse substituents around the ring11. The ring-expansion of enantiopure aziridine 4-[(R)-1-(R)-1-phenylethyl)aziridin-2-yl]butan-1-ol (1) resulted in the asymmetric synthesis of substituted azaheterocycles which are applicable to build biologically active molecules with piperidine and azepane skeleton. This synthetic protocol has been applied for various compounds ranging from simple 2-cyanomethylpiperidine 5f, 2-acetyloxymethylpiperidine 5h and 3-hydroxyazepane 6j to more complex molecules including natural products such as fagomine (9), febrifugine analogue (12) and balanol (15) in optically pure forms11.
Protocol
1. Synthesis of (6R)-1-[(R)-1-Phenylethyl)-1-Azoniabicyclo[4.1.0]Heptane Tosylate (4)
- Synthesis of 4-(R)-[1-(R)-1-phenylethyl)aziridin-2-yl]butyl 4-methylbenzenesulfonate (2)
- Add 100 mg of 4-[(R)-1-(R)-1-phenylethyl)aziridin-2-yl]butan-1-ol (1)12 (0.46 mmol, 1.0 equiv), 140 µL of triethylamine (Et3N, 1.0 mmol, 2.2 equiv), and a magnetic stir-bar into an oven-dried 25 mL two-neck round-bottom flask under nitrogen (N2) atmosphere.
- Add anhydrous dichloromethane (CH2Cl2, 5 mL) to the reaction flask using airtight syringe.
- Cool the reaction mixture at 0 °C using ice bath and stir the mixture for 5 min.
- Add p-toluenesulfonic anhydride (164 mg, 0.50 mmol, 1.10 equiv) to the reaction mixture and stir for another 45 min.
- Warm the reaction mixture to room temperature (RT) and stir for 30 min.
- Monitor the reaction using a thin layer chromatography (TLC) by using hexanes:ethyl acetate (EtOAc) (1:1 v/v, retention factor Rf = 0.55) as an eluent.
- After the complete consumption of starting alcohol 1, quench the reaction mixture with water (5 mL). Extract the heterogeneous mixture with CH2Cl2 (3 x 15 mL), dry the combined organic layer over anhydrous sodium sulphate (Na2SO4) for 10 min and concentrate in vacuo using a rotary evaporator.
- Purify the crude product with flash column chromatography eluting with hexanes:EtOAc (2:1 to 1:2 v/v) to afford 165 mg (0.44 mmol, 96% yield) of 4-(R)-[1-(R)-1-phenylethyl)aziridin-2-yl]butyl 4-methylbenzenesulfonate (Rf = 0.55, hexanes:EtOAc (1:1 v/v)) as a viscous liquid.
- Synthesis of (6R)-1-[(R)-1-phenylethyl)-1-azoniabicyclo[4.1.0]heptane tosylate (4)
- Transfer 5 mg of freshly prepared 2 in a NMR tube and add acetonitrile-d3 (CD3CN, 300 µL).
- Keep the above solution at RT for 24 h to attain the complete conversion to (6R)-1-[(R)-1-phenylethyl)-1-azoniabicyclo[4.1.0]heptane tosylate 4.
- Monitor the conversion of 2 to 4 by nuclear magnetic resonance (NMR) spectrum in CD3CN at different time points (10 min, 1, 5, 7 and 24 h).
2. General Procedure for the Expansion of 1-Azoniabicyclo[4.1.0]Heptane Tosylate
Use steps 1.1.1 to 1.1.7 for the preparation of corresponding tosylate 2.
Add the above crude tosylate, and a magnetic stir-bar into an oven-dried 25 mL round-bottom flask.
Add anhydrous CH3CN (4.0 mL) to the reaction flask using airtight syringe.
Add the corresponding nucleophile (1.37 mmol, 3.0 equiv) to the reaction mixture and stir for 8-15 h.
Quench the reaction mixture with water (5 mL), extract with CH2Cl2 (3 x 15 mL), dry the combined organic layer over anhydrous Na2SO4 and concentrate in vacuo using a rotary evaporator.
Purify the crude product with column chromatography by using hexanes:EtOAc (19:1 to 7:3 v/v) to afford pure ring expanded products. NOTE: All compounds were synthesized using above procedure except 6j (entry j, Table 1). Caution: Dried CH3CN is prepared by distilling it from calcium hydride (CaH2) under N2 atmosphere. Sodium cyanide is toxic among the most poisonous substances known and it is used under chemical fume hood with proper safety protection. Quenching of NaCN was performed with potassium permanganate (KMnO4) solution.
3. Synthesis of (S)-1-[(R)-1-Phenylethyl]azepan-3-ol (6j)
Transfer 4-(R)-[1-(R)-1-Phenylethyl)aziridin-2-yl]butyl 4-methylbenzenesulfonate (100 mg, 0.27 mmol, 1.0 equiv), and a magnetic stir-bar into an oven-dried 25 mL round-bottom flask fitted with reflux condenser.
Add 1,4-dioxane (4 mL) to the reaction flask using syringe.
Add 0.4 mL of 2.0 M sodium hydroxide solution (NaOH, 0.80 mmol, 3.0 equiv) to the reaction mixture and heat to reflux for 2.0 h.
Quench the reaction mixture with water (4 mL), extract with CH2Cl2 (4 x 15 mL), dry the combined organic layer over anhydrous Na2SO4 and concentrate in vacuo using a rotary evaporator.
Purify the crude product with column chromatography eluting with hexanes:EtOAc (2:1 to 1:1 (v/v)) (Rf = 0.60, hexanes:EtOAc (1:1 (v/v)) to afford 46 mg of (S)-1-[(R)-1-Phenylethyl]azepan-3-ol (0.21 mmol, 79% yield).
4. Synthesis of [(2R,3R,4R)-3,4-Bis(Benzyloxy)-1-(S)-1-Phenylethyl)Piperidin-2-Yl]Methyl Acetate (8):
Transfer (3R,4R)-3,4-bis(benzyloxy)-4-[(R)-1-(S)-1-phenylethyl)aziridin-2-yl]butan-1-ol ( 7) (220 mg, 0.51 mmol, 1.0 equiv), 180 µL of Et3N (1.27 mmol, 2.5 equiv), and a magnetic stir-bar into an oven-dried 25 mL two-neck round-bottom flask under N2 atmosphere.
Add anhydrous CH2Cl2 (4 mL) to the reaction flask using airtight syringe.
Cool the reaction mixture at 0 °C and stir for 5 min.
Add p-toluenesulfonic anhydride (200 mg, 0.61 mmol, 1.20 equiv) to the reaction mixture and stir for another 45 min.
Warm the reaction mixture to RT and stir for 30 min.
Monitor the progress of reaction by TLC using hexanes:EtOAc (7:3 v/v, Rf = 0.60) as an eluent.
After the complete consumption of alcohol 7, quench the reaction mixture with water (5 mL), extract with CH2Cl2 (3 x 15 mL), dry the combined organic layer over anhydrous Na2SO4 and concentrate in vacuo using a rotary evaporator.
Transfer the above crude tosylate, and a magnetic stir-bar into an oven-dried 25 mL round-bottom flask.
Add anhydrous CH3CN (4 mL) to the reaction flask using airtight syringe.
Add 125 mg of sodium acetate (NaOAc, 1.53 mmol, 3.0 equiv) to the reaction mixture and stir for 12 h.
Quench the reaction mixture with water (5 mL), extract with CH2Cl2 (3 x 15 mL), dry the combined organic layer over anhydrous Na2SO4 and concentrate in vacuo using a rotary evaporator.
Purify the crude product with column chromatography by using hexanes: EtOAc (9:1 to 4:1 v/v) to afford pure compound 8 (200 mg, 83% yield).
5. Synthesis of (3R,4R)-3-Azido-1-[(R)-1-Phenylethyl]azepan-4-Ol (14):
Transfer (R)-1-[(S)-1-(R)-1-phenylethyl)aziridin-2-yl]butane-1,4-diol (13) (235 mg, 1.0 mmol, 1.0 equiv), 0.35 mL of Et3N (2.50 mmol, 2.5 equiv), and a magnetic stir-bar into an oven-dried 25 mL two-neck round-bottom flask under nitrogen atmosphere.
Add anhydrous CH2Cl2 (8 mL, 0.12 M) to the reaction flask using airtight syringe.
Cool the reaction mixture at 0 °C and stir for 5 min.
Add p-toluenesulfonic anhydride (359 mg, 1.10 mmol, 1.10 equiv) to the reaction mixture and stir for another 45 min.
Monitor the progress of reaction by TLC using hexanes: EtOAc (1:1 v/v, Rf = 0.40) as an eluent.
After the complete consumption of starting alcohol 13, quench the reaction mixture with water (10 mL), extract with CH2Cl2 (3 x 15 mL), dry the combined organic layer over anhydrous Na2SO4 and concentrate in vacuo using a rotary evaporator.
Add the above crude tosylate, and a magnetic stir-bar into an oven-dried 25 mL round-bottom flask.
Add anhydrous CH3CN (8 mL, 0.12 M) to the reaction flask using airtight syringe.
Add 214 mg of NaOAc (3.30 mmol, 3.30 equiv) to the reaction mixture and stir for 12 h.
Quench the reaction mixture with water (10 mL), extract with CH2Cl2 (15 mL x 3 times), dry the combined organic layer over anhydrous Na2SO4 and concentrate in vacuo using a rotary evaporator.
Purify the crude product with column chromatography by using hexanes: EtOAc (9:1 to 4:1 v/v) to afford pure compound 14 (133 mg, 51% yield).
6. Characterization of All Products
Characterize all the new compounds by 1H, 13C NMR spectroscopy and high-resolution mass spectrometry (HRMS).
Representative Results
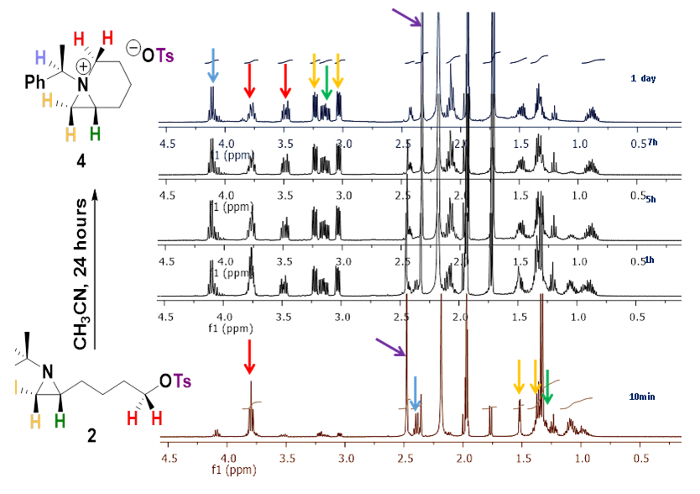
The reaction of 4-[(R)-1-(R)-1-phenylethyl)aziridin-2-yl]butan-1-ol (1)12 with p-toluenesulfonic anhydride and triethylamine in CH2Cl2 at room temperature for 1.0 h yielded the corresponding 2-(4-tosyloxybutyl)aziridine 2 in 96% yield11. 1H NMR (400 MHz) spectrum of compound 2 in CD3CN at different time intervals shows the conversion of 2-(4-tosyloxybutyl)aziridine 2 to 1-azabicyclo[4.1.0]heptane tosylate 4. Among all down field shifted peaks, a distinctive quartet peak at 2.34 ppm (J = 6.5 Hz) for tertiary proton of phenylethyl group was shifted to at 4.11 ppm (J = 6.9 Hz) by the formation of the quaternary ammonium ion (Figure 2).
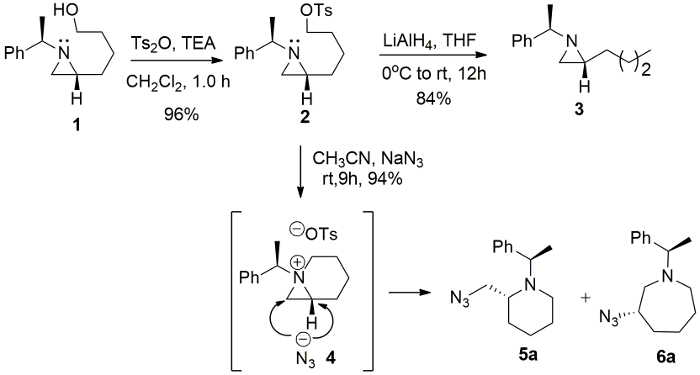
The intermediacy of 1-azabicyclo[4.1.0]heptane tosylate 4 from 2-(4-tosyloxybutyl)aziridine 2 was further confirmed by the formation of azidated products, 2-azidomethylpiperidine 5a and 3-azidoazepane 6a, with sodium azide (NaN3) in CH3CN, while a simple reduction product 2-butylaziridine 3 was obtained from the reaction of the same starting substrate with lithium aluminium hydride (LiAlH4) in tetrahydrofuran (THF) (Figure 3).
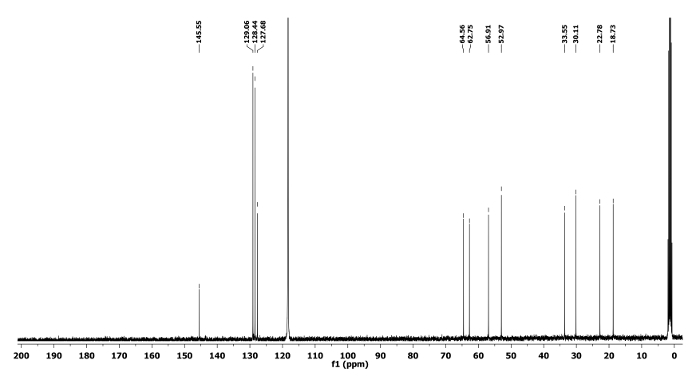
The 1H and 13C NMR spectra of the ring expanded product 5a and 6a are depicted in Figure 5, Figure 6, Figure 7 and Figure 8, respectively. A distinguish quartet peak at 4.07 ppm with a coupling constant of 6.7 Hz in 1H NMR spectrum of compound 5a corresponds to the C-H proton of phenylethyl group present at nitrogen. Similar quartet at 3.81 ppm with a coupling constant of 6.8 Hz was observed for compound 6a. Similar NMR spectral changes were observed for all other compounds (Table 1). The detailed characterization data of 5a, 6a, 5f, 5h and 6j are reported as follows:
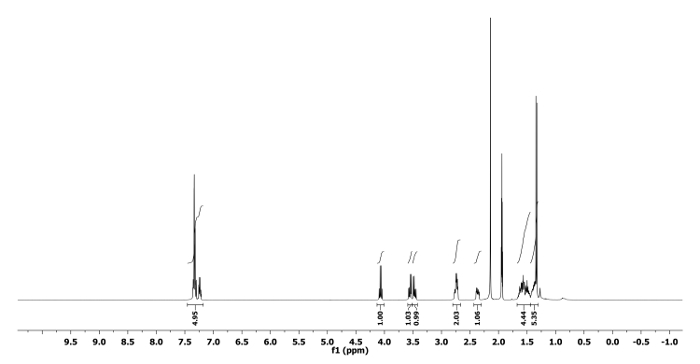
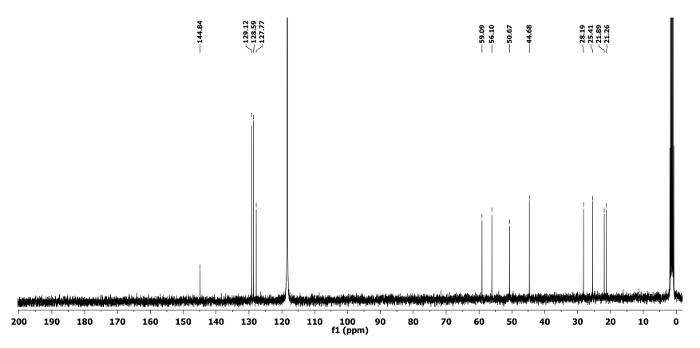
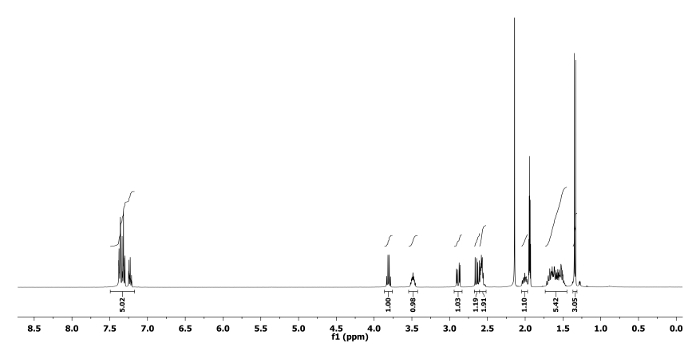
Compound 5a: Rf = 0.50 (hexanes/EtOAc 9:1 v/v). [α]20D = +64.3 (c = 0.6, CHCl3). 1H NMR (400 MHz, CD3CN) δ 7.43-7.17 (m, 5H), 4.07 (q, J = 6.7 Hz, 1H), 3.55 (dd, J = 12.7, 5.1 Hz, 1H), 3.47 (dd, J = 12.7, 6.8 Hz, 1H), 2.82-2.66 (m, 2H), 2.44-2.30 (m, 1H), 1.68-1.44 (m, 4H), 1.44-1.31 (m, 2H), 1.34 (d, J = 6.7 Hz, 3H). 13C NMR (CD3CN, 101 MHz): δ 144.8, 129.1, 128.5, 127.7, 59.0, 56.1, 50.6, 44.6, 28.1, 25.4, 21.8, 21.2. HRMS-MALDI (m/z): calcd. for C14H21N4 [M+H]+ 245.1760; found 245.1766.
Compound 6a: Rf = 0.70 (hexanes/EtOAc 9:1 v/v).[α]20D = -3.2 (c = 0.6, CHCl3). 1H NMR (400 MHz, CD3CN) δ 7.45-7.18 (m, 5H), 3.81 (q, J = 6.8 Hz, 1H), 3.49 (tt, J = 8.0, 5.1 Hz, 1H), 2.89 (dd, J = 13.9, 4.4 Hz, 1H), 2.64 (dd, J = 13.9, 7.7 Hz, 1H), 2.60-2.52 (m, 2H), 2.05-1.97 (m, 1H), 1.72-1.45 (m, 5H), 1.34 (d, J = 6.8 Hz, 3H). 13C NMR (CD3CN, 101 MHz): δ 145.5, 129.0, 128.4, 127.6, 64.5, 62.7, 56.9, 52.9, 33.5, 30.1, 22.7, 18.7. HRMS-MALDI (m/z): calcd. for C14H21N4 [M+H]+ 245.1760; found 245.1764.
2-[(R)-1-(R)-1-Phenylethyl)piperidin-2-yl]acetonitrile (5f): Rf = 0.85 (hexanes/EtOAc 19:1 v/v). [α]20D = +35.3 (c = 0.65, CHCl3). 1H NMR (400 MHz, CD3CN) δ 7.42-7.20 (m, 5H), 3.89 (q, J = 6.7 Hz, 1H), 2.85-2.75 (m, 2H), 2.68-2.54 (m, 2H), 2.45-2.37 (m, 1H), 1.71-1.44 (m, 5H), 1.42-1.31 (m, 1H), 1.34 (d, J = 6.7 Hz, 3H). 13C NMR (CD3CN, 101 MHz): δ 144.1, 129.2, 128.5, 127.9, 120.1, 59.3, 54.0, 44.3, 30.6, 26.1, 21.5, 21.2, 17.8. HRMS-MALDI (m/z): calcd. for C15H21N2 [M+H]+ 229.1699; found 229.1694.
[(R)-1-(R)-1-Phenylethyl)piperidin-2-yl)]methyl acetate (5h): Rf = 0.50 (hexanes/EtOAc 7:3 v/v). [α]20D = +75.8 (c = 0.55, CHCl3). 1H NMR (400 MHz, CD3CN) δ 7.41-7.26 (m, 4H), 7.25-7.17 (m, 1H), 4.29 (dd, J = 11.3, 4.8 Hz, 1H), 4.16 (dd, J = 11.3, 6.7 Hz, 1H), 4.05 (q, J = 6.8 Hz, 1H), 2.81-2.68 (m, 2H), 2.40 (ddd, J = 12.1, 6.2, 3.4 Hz, 1H), 1.97 (s, 3H), 1.65-1.41 (m, 5H), 1.39-1.29 (m, 1H), 1.32 (d, J = 6.8 Hz, 3H). 13C NMR (CD3CN, 101 MHz): δ 171.5, 145.2, 129.0, 128.5, 127.6, 63.8, 59.4, 55.7, 45.3, 28.8, 26.3, 22.1, 21.1, 21.0. HRMS-MALDI (m/z): calcd. for C16H24NO2 [M+H]+ 262.1801; found 262.1800.
(S)-1-[(R)-1-Phenylethyl]azepan-3-ol (6j): Rf = 0.40 (hexanes/EtOAc 1:1 v/v). [α]20D = -11.1 (c = 0.55, CHCl3). 1H NMR (400 MHz, CD3CN) δ 7.41-7.27 (m, 4H), 7.27-7.19 (m, 1H), 3.80 (q, J = 6.8 Hz, 1H), 3.67-3.56 (m, 1H), 2.71 (dd, J = 13.5, 3.1 Hz, 1H), 2.67-2.53 (m, 3H), 1.72-1.43 (m, 6H), 1.35 (d, J = 6.8 Hz, 3H). 13C NMR (CD3CN, 101 MHz): δ 145.4, 129.0, 128.6, 127.7, 70.2, 64.4, 57.1, 53.1, 37.9, 29.7, 21.7, 17.6. HRMS-MALDI (m/z): calcd. for C14H22NO [M+H]+ 220.1695; found 220.1693.
The aziridine-ring-expansion has been observed with several other nucleophiles and the results are summarised in Table 1. Among all nucleophiles, cyanide, thiocyanide and acetate preferred the selective formation of piperidine rings (Table 1, entry f, g, h), while azide, hydroxide and amine nucleophiles yielded azepane rings (Table 1, entry a, j, k).
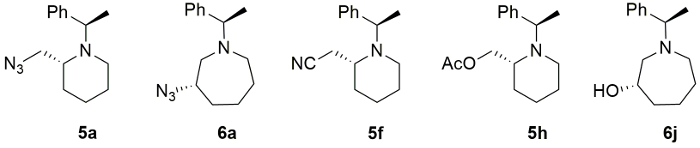
The structures and their stereochemistry of major compounds 5f, 5h and 6j originatd from (2R)-2-(4-hydroxybutyl)-1-[(1R)-(1-phenylethyl)]aziridine were further confirmed by their conversion to known compounds13,14,15 and we found out that the nucleophilic ring-expansion proceeds in completely stereospecific manner. Representative examples include 2-azidomethyl- (5a), 3-azidoazopane (6a), 2-cyanomethyl-(5f), 2-acetyloxymethylpiperidine (5h) and 3-hydroxyazepane (6j) in their optically pure froms (Figure 4).
The presented protocol was applicable to synthesize natural azasugar D-Fagomine (9) and its 3-epimer16. The reaction of suitably functionalized aziridinyl alcohol 7 underwent aziridine ring-expansion under similar reaction condition to afford protected fagomine 8. The removal of all protecting group in compound 8 produces D-fagomine (9) in 94% yields (Figure 9)11.
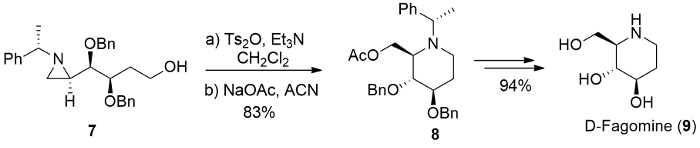
In the same manner, benzyl protected alcohol 10 was treated with p-toluenesulfonic anhydride and Et3N, followed by nucleophilic ring opening with sodium cyanide (NaCN), resulting in the ring expanded cyanomethylpiperidine 11 in 90% yield as a single isomer, which can be used as synthetic precursor for febrifugine analogue (Figure 10)17.
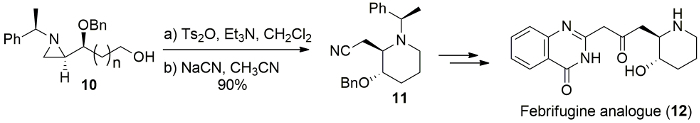
The ring-expansion toward azepane also succeeded via azoniabicycle prepared from alcohol 13 followed by the reaction with NaN3 to yield the desired product 14, which was used for azepane core of balanol (15) by one-pot benzyl deprotecton and azide reduction under catalytic hydrogenation (Figure 11)11.
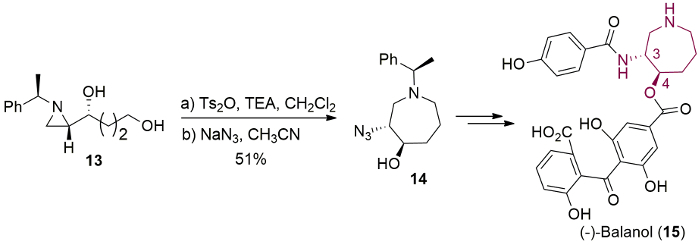
Figure 1. Ring opening of aziridinium ion. Possible ring opening products: (a) Pi and Pii from monocyclic aziridinium ion (Ia); (b) Piii and Piv from bicyclic aziridinium ions (Ib). Please click here to view a larger version of this figure.
Figure 2. Generation of bicyclic aziridinium ion.1H NMR (400 MHz) spectral changes by the conversion of 2-(4-tosyloxybutyl)aziridine 2 to 1-azoniabicyclo[4.1.0]heptane tosylate 4 in CD3CN in different time intervals as 10 min, 1, 5, 7 and 24 h11. Please click here to view a larger version of this figure.
Figure 3. Ring expansion of aziridine. The formation of 1-azoniabicyclo-[4.1.0]heptane tosylate 4 and its ring-expansion reaction with NaN3. Please click here to view a larger version of this figure.
Figure 4. Piperidines and azepanes products. Representative ring expansion products (5a, 6a, 5f, 5h and 6j) Please click here to view a larger version of this figure.
Figure 5. 1H NMR spectrum of 5a. Chemical shifts and relative integrations of characteristic protons are shown11. Please click here to view a larger version of this figure.
Figure 6. 13C NMR spectrum of 5a. Chemical shifts of characteristic carbons are shown11. Please click here to view a larger version of this figure.
Figure 7. 1H NMR spectrum of 6a. Chemical shifts and relative integrations of characteristic protons are shown11. Please click here to view a larger version of this figure.
Figure 8. 13C NMR spectrum of 6a. Chemical shifts of characteristic carbons are shown11. Please click here to view a larger version of this figure.
Figure 9. Synthesis of fagomine. Synthesis of fagomine (9) from the ring-expansion of (3R,4R)-3,4-bis(benzyloxy)-4-[(R)-1-(S)-1-phenylethyl)aziridin-2-yl]butan-1-ol (7). Please click here to view a larger version of this figure.
Figure 10. Synthesis of febrifugine. Synthesis of febrifugine analogue (12) from the ring-expansion of (S)-4-(benzyloxy)-4-[(R)-1-(R)-1-phenylethyl)aziridin-2-yl]butan-1-ol (10). Please click here to view a larger version of this figure.
Figure 11. Synthesis of azepane natural product. Synthesis of (-)-balanol (15) from the ring-expansion of (R)-1-[(S)-1-(R)-1-phenylethyl)aziridin-2-yl]butane-1,4-diol (13) as a key step. Please click here to view a larger version of this figure.
| ||||||
| Entry | Substrate | Nucleophile | Solventa,b | Time (h) | Ratio (5/6)c | Yieldd (%) |
| a | Aziridine (1) | NaN3 | CH3CN | 9 | 41/59 | 94 |
| b | Aziridine (1) | NaN3 | 1,4-dioxane | 2.0 | 40/60 | 84 |
| c | Aziridine (1) | CsF | CH3CN | 8.0 | 46/54 | 79 |
| d | Aziridine (1) | n-Bu4NBr | CH3CN | 12.0 | Complex | - |
| e | Aziridine (1) | NaI | CH3CN | 12.0 | Complex | - |
| f | Aziridine (1) | NaCN | CH3CN | 8.0 | 92/08 | 92 |
| g | Aziridine (1) | NaSCN | CH3CN | 11.0 | 91/09 | 93 |
| h | Aziridine (1) | NaOAc | CH3CN | 12.0 | 60/40 | 90 |
| i | Aziridine (1) | NaOMe | CH3CN | 11.0 | 58/42 | 93 |
| j | Aziridine (1) | NaOH | 1,4-dioxane | 2.0 | 15/85 | 93 |
| k | Aziridine (1) | BnNH2 | CH3CN | 14.0 | 35/65 | 87 |
| l | Aziridine (1) | Phenol | CH3CN | 15.0 | 47/53 | 56 |
| m | Aziridine (1) | NaOBz | CH3CN | 13.0 | 48/52 | 76 |
| n | Aziridine (1a) | NaN3 | CH3CN | 9.5 | 40/60 | 91e |
| [a]CH2Cl2 was used as solvents for the preparation of 2-(4-tosyloxybutyl)aziridine. [b]Tosylation was performed at 0 to 25 °C. | ||||||
| [c]Determined by 1H NMR. [d]Combined yield of 5 and 6. [e]Inseparable mixture of 5n and 6n from isomeric aziridine 1a. | ||||||
| [f] All reactions were performed at 25 °C except the entries b and j (110 °C). [g]Concentration is 0.1 M |
Table 1. Selected examples of aziridine ring expansion. The formation and ring-expansion of 1-azoniabicyclo[4.1.0]heptane tosylate 4 with various nucleophiles11.
Discussion
Piperidine and azepane are two most abundant azaheterocycles in many life-saving drugs and antibiotics including various biologically active natural products16. In order to access both enantiopure piperidine (5) and azepane (6) with diverse substituents, we develped an efficient synthetic method through the formation of 1-azoniabicyclo[4.1.0]heptane tosylate from entiopure 2-(4-hydroxybutyl)aziridne followed by regiospecific nucleophilic attack either at the bridge or at the bridgehead carbon. Azoniabicyclic intermediate was easily prepared by dissolving corresponding 2-(4-tosyloxybutyl)aziridine in acetonitrile. The criticality of present protocol is how efficiently we can prepare the starting aziridine with proper substitution at desired position in pure isomeric forms. Using this protocol, we were able to prepare 2-cyano- (5f), 2-acetoxymethylpiperidine (5h) and 3-hydroxyazepane (6j) as representative examples in optically pure forms.
The analytical data for all reported compounds were found to be in good agreement with the literature values, which implies that nucleophilic ring openings from azoniabicycle (4) are stereospecific with the retention (path i) and inversion (path ii) of the configurations.
After developing a method for the conversion of aziridine to piperidine and azepane ring system, the main challenge was to utilize the present protocol for the synthesis of few natural products. In this way, we have selected fagomine as a representative example of piperidine natural product and balanol as a azepane natural product. We found that the synthesis of natural fagomine requirs the E-configuration of starting vinylic aziridine, which was difficult to synthesize by Wittig reaction using unstable 3-carbon ylide, but we were able to synthesize the target precursor using slightly modified procedure with better selectivity11.
This ring-expansion protocol via 1-azoniabicyclo[4.1.0]heptane was further utilized for the synthesis of natural products bearing piperidine and azepane. Based on the observed regioselectivity shown by acetate and nitrile nucleophiles towards 2-acetoxy and 2-cyanomethylmethylpiperidine ring, we prepared 1-azoniabicyclo[4.1.0]heptanes from properly decorated alcohols 7 and 10 whose subsequent ring-expansion gave substituted piperidine 8 and 11 with all desired configurations, respectively. Global deprotection of ring-expanded product 8 and 11 afforded D-fagomine (9) and febrifugine analogue (12) in excellant yields. Similarly, we prepared another starting aziridinyl alcohol 13, which underwent the ring-expansion under similar reaction condition with hydroxide nucleophile to give 3-azido-4-hydroxyazepane 14 and was converted to the azepane core of balanol (15). Especially, this synthetic method for 3-hydroxyazepane core presented in the structure of balanol is more efficent than two previously known methods15.
The present strategy for the ring-expansion of aziridine via the formation of azoniabicyclic intermediate is applicable for the synthesis of various chiral piperidine and azepane ring system in a highly selective manner. In the future, it can be adapted to synthesize various complex natural and unnatural products in highly efficient manner.
In summary, we have prepared 1-azabicyclo[4.1.0]heptane tosylate from 2-(4-hydroxybutyl)aziridine whose subsequent regio- and stereospecfic ring-expansion by nucleophiles produced substituted piperidines and azepanes. This operationally simple ring-expansion strategy enabled us to develop the facile, stereoselective, and divergent synthesis of piperidines and azepanes with various substituents including natural products such as fagomine, febrifugine analogue and balanol in highly efficient manner.
Disclosures
The authors declare no competing financial interests.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF-2012M3A7B4049645 and HUFS Research Fund (2018).
References
- Singh GS, D'hooghe M, De Kimpe N. Synthesis and Reactivity of C-Heteroatom-Substituted Aziridines. Chem. Rev. 2007;107:2080–2135. doi: 10.1021/cr0680033. [DOI] [PubMed] [Google Scholar]
- Yudin A. Aziridines and Epoxides in Organic Synthesis. Wiley-VCH Verlag GmbH & Co. KGaA; 2006. [Google Scholar]
- Lee WK, Ha HJ. Highlight of the Chemistry of Enantiomerically Pure Aziridine-2-carboxylates. Aldrichimica Acta. 2003;36:57–63. [Google Scholar]
- Ghorai MK, Bhattacharyya A, Das S, Chauhan N. Ring Expansions of Activated Aziridines and Azetidines. Top. Heterocycl. Chem. 2016;41:49–142. [Google Scholar]
- Kim Y, Ha H-J, Yun SY, Lee WK. The preparation of stable aziridinium ions and their ring-openings. Chem. Commun. 2008. pp. 4363–4365. [DOI] [PubMed]
- D’hooghe M, Van Speybroeck V, Waroquier M, De Kimpe N. Regio- and stereospecific ring opening of 1,1-dialkyl-2-(aryloxymethyl)aziridinium salts by bromide. Chem. Commun. 2006. pp. 1554–1556. [DOI] [PubMed]
- Stankovic S, D'hooghe M, Catak S, Eum H, Waroquier M, Van Speybroeck V, De Kimpe N, Ha HJ. Regioselectivity in the ring opening of non-activated aziridines. Chem. Soc. Rev. 2012;41:643–665. doi: 10.1039/c1cs15140a. [DOI] [PubMed] [Google Scholar]
- Ji MK, Hertsen D, Yoon DH, Eum H, Goossens H, Waroquier M, Van Speybroeck V, D'hooghe M, De Kimpe N, Ha HJ. Nucleophile-Dependent Regio- and Stereoselective Ring Opening of 1-Azoniabicyclo[3.1.0]hexane Tosylate. Chem. Asian J. 2014;9:1060–1067. doi: 10.1002/asia.201301551. [DOI] [PubMed] [Google Scholar]
- Mtro T-X, Duthion B, Gomez Pardo D, Cossy J. Rearrangement of β-amino alcohols viaaziridiniums: a review. Chem. Soc. Rev. 2010;39:89–102. doi: 10.1039/b806985a. [DOI] [PubMed] [Google Scholar]
- Dolfen J, Yadav NN, De Kimpe N, D'hooghe M, Ha HJ. Bicyclic Aziridinium Ions in Azaheterocyclic Chemistry-Preparation and Synthetic Application of 1-Azoniabicyclo[n.1.0]alkanes. Adv. Synth. Catal. 2016;358:3485–3511. [Google Scholar]
- Choi J, Yadav NN, Ha H-J. Preparation of a Stable Bicyclic Aziridinium Ion and Its Ring Expansion toward Piperidines and Azepanes. Asian J. Org. Chem. 2017;6:1292–1307. [Google Scholar]
- Yadav NN, Choi J, Ha H-J. One-pot multiple reactions: asymmetric synthesis of 2,6-cis-disubstituted piperidine alkaloids from chiral aziridine. Org. Biomol. Chem. 2016;14:6426–6434. doi: 10.1039/c6ob00806b. [DOI] [PubMed] [Google Scholar]
- Angoli M, Barilli A, Lesma G, Passarella D, Riva S, Silvani A, Danieli B. Remote Stereocenter Discrimination in the Enzymatic Resolution of Piperidine-2-ethanol. Short Enantioselective Synthesis of Sedamine and Allosedamine. J. Org. Chem. 2003;68:9525–9527. doi: 10.1021/jo035215g. [DOI] [PubMed] [Google Scholar]
- Shaikh TM, Sudalai A. Enantioselective Synthesis of (+)-α-Conhydrine and (-)-Sedamine by L-Proline-Catalysed α-Aminooxylation. Eur. J. Org. Chem. 2010. pp. 3437–3444.
- Miyabe H, Torieda M, Inoue K, Tajiri K, Kiguchi T, Naito T. Total Synthesis of (−)-Balanol. J. Org. Chem. 1998;63:4397–4407. [Google Scholar]
- Castillo JA, Calveras J, Casas J, Mitjans M, Vinardell MP, Parella T, Inoue T, Sprenger GA, Joglar J, Clapes P. Fructose-6-phosphate Aldolase in Organic Synthesis: Preparation of d-Fagomine, N-Alkylated Derivatives, and Preliminary Biological Assays. Org. Lett. 2006;8:6067–6070. doi: 10.1021/ol0625482. [DOI] [PubMed] [Google Scholar]
- Kikuchi H, Yamamoto K, Horoiwa S, Hirai S, Kasahara R, Hariguchi N, Matsumoto M, Oshima Y. Exploration of a New Type of Antimalarial Compounds Based on Febrifugine. J. Med. Chem. 2006;49:4698–4706. doi: 10.1021/jm0601809. [DOI] [PubMed] [Google Scholar]