

Abstract
A nicotine-degrading enzyme termed NicA2 was altered (NicA2-J1) through fusion of an albumin binding domain to increase its half-life. Examination of NicA2-J1 in vivo demonstrated a complete blockade of brain nicotine access, which in turn blunted nicotine’s psychoactive effects. These data further support development of pharmacokinetic nicotine cessation therapeutics.
Smoking is the leading cause of preventable illness in the world today with tobacco use killing 6 million people per year.1 Effective cessation aids are essential to assist in reducing the prevalence of cigarette smoking and related illness.2 Among the pharmacological treatments that are approved for nicotine cessation, Varenicline has shown the most success, however, such treatment only increases the odds of abstinence at 6 months approximately 3-fold compared to placebo.4
Due to a low success rate with “pharmacodynamic” treatment strategies, the past decade has seen what has been termed a “pharmacokinetic” approach emerge for treating drug addiction. Of these, immunopharmacotherapy is a methodology wherein vaccines are crafted to stimulate the production of antibodies specific to the drug of interest.5 Many drugs of abuse have been targeted including, cocaine, methamphetamine, heroin and nicotine.6 The overall premise of immunopharmacotherapy is that the composite size of antibody-drug union is too large to cross the blood-brain barrier, reducing the concentration/rate of drug entering the brain, ultimately blunting the reinforcing and addictive effects of the drug. Unfortunately, vaccine strategies for nicotine addiction that have been tested in clinical trials have failed to achieve their primary end-point of increased cessation rates compared to placebo.7 However, the results from clinical trials revealed that individuals who possessed higher levels of antibodies were associated with significantly higher abstinence rates compared to placebo.6b Complementing this idea of diminished nicotine delivered to the brain, multiple clinical studies have shown that very low nicotine content cigarettes (VLNC) lead to reduced nicotine intake, increased continuous and 7-day point prevalence abstinence rates, and increase in time to relapse.8 Thus, a pharmacokinetic approach for treating nicotine addiction could still have a significant effect on human health, but only if sufficient pharmacokinetic capacity can be generated to substantially reduce free drug concentration.
In contrast to simple antibody-mediated sequestering of nicotine entering the brain, an alternative strategy to reducing nicotine’s brain concentration would be through its degradation. By decreasing nicotine’s peripheral circulation through nicotine catabolism an effective concentration would not be reached or maintained in the brain. Toward this effort, we previously reported the characterization of NicA2,3 a nicotine degrading enzyme isolated from P. putida S16,9. In our inaugural study we revealed that this enzyme exhibits favorable kinetic parameters with a KM of 43.5 nM and kcat of (6.64 ±0.17) × 10−3 s−1. In addition we reported an impressive stability of this enzyme with a half-life of >30 days in buffer and 3 days in serum, while products formed from NicA2’s enzymatic degradation of nicotine were found to be non-toxic.3
Although, NicA2’s in vitro metrics were quite favorable, the enzyme would still need to show success with in vivo assessment to be a considered as a viable nicotine cessation aid. With these thoughts in mind as a first challenge we viewed half-life in circulation as a point of focus, as without an increase in NicA2’s in vivo stability behavioral assessment studies would be limited. With a molecular weight of 50 kDa, NicA2 is susceptible to kidney filtration, which has an upper threshold of 60 kDa. One approach to increasing a protein’s stability in vivo is through fusion to another protein with an extended half-life in serum.10 Albumin, (~67 kDa) is the most abundant protein in plasma, present at 35 – 50 mg/mL, with a half-life of 19 days in humans.10a Albumin helps maintain plasma pH, contributes to colloidal blood pressure, functions as a carrier for many metabolites and fatty acids, and serves as a major drug transport protein in plasma.10a While an albumin fusion was considered the most promising approach, simple addition of this fusion protein to either the N or C-terminal end of NicA2 could greatly alter its molecular architecture, and enzymatic activity. Fusion-structural ambiguity was reduced when the three-dimensional structure of WT NicA2 holo enzyme was disclosed by Tararina et al.11 Interestingly the first 52 amino acids (N-terminus) in WT-NicA2 were not observed in its crystal structure.11 It was hypothesized that this structural sequence within NicA2 is intrinsically disordered and may not be critical to the enzyme’s properties. With these structural reservations now adequately addressed the albumin binding domain (ABD)035 created by Per-Åke et al., which possesses fM binding to human serum albumin (HSA) was engaged as NicA2’s fusion partner.12
The first 50 amino acids from the N-terminus of NicA2 were deleted to give Δ50-NicA2. Residues 51Gly and 52Gly were left as native linkers for fusion with ABD035 to afford ABD035-Δ50-NicA2, termed NicA2-J1. The catalytic properties of the enzyme variant were examined by liquid chromatography-mass spectrometry (LC-MS) (Table 1).3 Compared with NicA2, NicA2-J1 exhibited a similar kcat and KM, indicating that the 50 amino acid deletion and fusion at the N-terminus had no effect on the enzyme’s kinetics. Additional binding kinetic parameters were determined with a series of serum albumins (human, rat and mouse) by surface plasmon resonance (SPR) with a Biacore 3000 system (GE healthcare) (Table 2). NicA2-J1 demonstrated high affinity to human and rat albumin, both with KDs in the pM range.
Table 1.
Michaelis-Menten parameters of NicA2 WT and NicA2-J1
| NicA2 variants | KM [nM] | kcat [10−3·s−1] | kcat/KM [s−1·M−1] |
|---|---|---|---|
| WT-NicA23 | 43.5±4.7 | 6.64±0.17 | 1.53×105 |
| NicA2-J1 | 53.3±8.6 | 6.16±0.26 | 1.16×105 |
Table 2.
Binding kinetics for NicA2-J1 and albumin interaction.
| Albumin | ka (M−1·s−1) | kd (s−1) | KD (nM) | t1/2 (h)* |
|---|---|---|---|---|
| HSA (human) | 1.66 × 104 | 5.75 × 10−6 | 0.347 | 33.33 |
| RSA (rat) | 2.26 × 104 | 9.56 × 10−6 | 0.423 | 20.05 |
| MSA (mouse) | 2.82 × 104 | 9.45 × 10−5 | 3.36 | 2.03 |
Complex half-life: the time required for 50% of the NicA2-J1-albumin complex from being dissociated.
To confirm improved serum stability of NicA2-J1, the in vivo half-life of this fusion protein compared to NicA2 was evaluated in rodents. Rats (n=3) were dosed intraperitoneally (IP) with NicA2-J1 or NicA2 (10 mg/kg) and serum was collected at 1, 4, 16, 24, 48 and 72 h. The serum was analyzed by enzyme linked immunosorbent assay (ELISA) and NicA2/NicA2-J1 concentrations were plotted against time to generate the pharmacokinetic curves (Fig. 1), with the parameters shown in Table 3. Both NicA2 and NicA2-J1 serum concentrations peaked at ~16 h after injection. Moreover, the area under the curve (AUC) at 72 h of NicA2-J1 was 12044 µg/mL·h compared to 4115 µg/mL·h of NicA2. After 72 h, NicA2-J1 remained at 100 µg/mL while NicA2 was fully eliminated, suggesting that albumin binding occurred, which significantly slowed clearance of NicA2-J1. Remarkably, the half-life of the NicA2 variant was extended to over 5 days.
Fig.1.
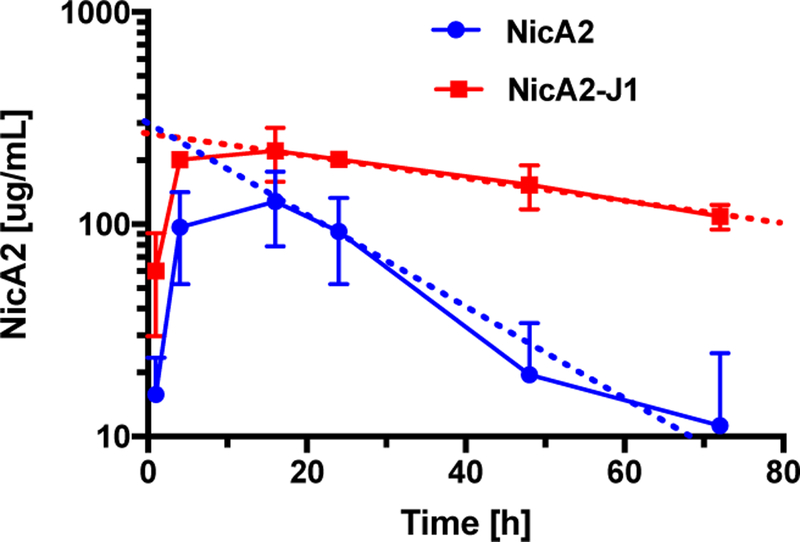
Pharmacokinetics of NicA2 and NicA2-J1 in rat serum.
Table 3.
Pharmacokinetic parameters of NicA2 and NicA2-J1.
| PK parameters | NicA2 | NicA2-J1 |
|---|---|---|
| t1/2 (h) | 25 | 128 |
| Cmax (µg/mL) | 127.9 | 221.5 |
| 72 h AUC (µg/mL·h) | 4115 | 12044 |
| Total AUC (µg/mL·h) | 4223 | 32627 |
t1/2: half-life; Cmax: maximum concentration. Total AUC was estimated by extending the elimination phase to X-intercepts.
With a greatly improved AUC and half-life, we next looked to examine the efficacy of NicA2-J1 in vivo, specifically its ability to prevent the development of nicotine dependence in rats. Nicotine dependence is characterized by the emergence of nicotine abstinence syndrome after the cessation of chronic nicotine exposure. Such an abstinence syndrome has been characterized in both humans and rats and is associated with both somatic and motivational components.13 In rats, the somatic signs of nicotine withdrawal include abdominal constrictions, facial fasciculation, ptosis, and hyperalgesia.14 The motivational components include hyperalgesia and irritability-like behavior. To test the effect of NicA2-J1 on the development of nicotine dependence,15 irritability (bottle-brush test),16 hyperalgesia (von Frey test),17 and somatic signs of withdrawal18 after 24–48 h of abstinence were measured in dependent rats (n = 8 for each group, equal number of males and females), with two additional groups receiving saline as controls. Rats were exposed to nicotine (3.16 mg/kg/day) or saline-containing osmotic minipumps for 7 days and were treated every other day with NicA2-J1 (10 mg/kg) or vehicle (phosphate buffered saline (PBS), pH 7.4). Behaviors were measured 24–48 h after removal of the minipumps during spontaneous withdrawal and withdrawal signs were scored.19 Rats were placed inside a transparent cylinder (50 cm high x 30 cm diameter) and their behavior was observed for 30 min. The number of wet dog shakes, front paw tremors, teeth chattering, genital licks, and abdominal contractions were counted. A global withdrawal score was calculated for each animal. When tested 24 h into nicotine withdrawal, there was a significant effect of treatment on the withdrawal scores, F(3;28)=14.06; P<0.001. The Newman Keuls post hoc test showed that the nicotine exposed rats (Nico+PBS) had significantly higher numbers of somatic signs (P< 0.001) (41.4±5.9) compared with both saline+PBS and saline+ NicA2-J1-exposed rats (9.9±1.4 and 12.6 ± 2.1). The animals exposed to nicotine with NicA2-J1 treatment showed significantly less somatic signs (P< 0.001) compared to the animals without NicA2-J1 treatment (16.6±4.3), demonstrating the efficacy of NicA2-J1 in preventing the somatic signs of nicotine withdrawal (Fig.2A).
Fig. 2.
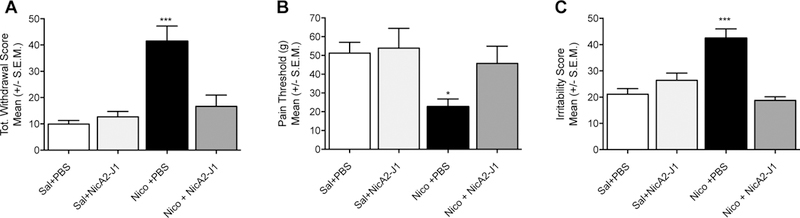
Effect of NicA2-J1 during nicotine withdrawal in rats. NicA2-J1 blocks total withdrawal score (A), hyperalgesia (B) and irritability like behaviour (C) during withdrawal. (Sal: saline; Nico: nicotine)
The hindpaw withdrawal threshold was determined by using von Frey filaments, ranging from 3.63 to 125.89 g. Testing began after 10 min of habituation to the testing environment. The series of von Frey hairs was applied from below the wire mesh to the central region of the plantar surface of the left hindpaw in ascending order, beginning with the lowest filament (3.63 g). The filament was applied until buckling of the hair occurred, and it was maintained for approximately 2 s. A withdrawal response was considered valid only if the hindpaw was completely removed from the platform. If withdrawal did not occur during three applications of a particular filament, then the next larger filament in the series was applied in a similar manner. Once the threshold was determined for the left hindpaw, the same testing procedure was repeated for the right hindpaw after 5 min. The one-way ANOVA revealed that 7-day exposure to nicotine significantly increased pain sensitivity F(3;28)=3.312; P<0.05 during nicotine withdrawal. Rats that were exposed to nicotine exhibited a decrease in mechanical thresholds during spontaneous withdrawal (22.7 ± 4.1) compared to the saline+PBS (51.3 ± 5.8); saline+ NicA2-J1 (53.9 ± 10.5); and Nico+NicA2-J1 (45.8 ± 9.1) (Fig. 2B).
The bottle-brush test was used to test irritability-like behavior during nicotine withdrawal (48h) based on the methods previously described by Kimbrough et al. 2017.16 Testing consisted of 10 trials per rat in plastic cages (10.5 in x 19 in x 8 in; Ancare, Bellmore, NY) with fresh bedding. Three observers blind to the treatment scored the behaviors and the average of the aggressive responses (smelling, biting, boxing, following, exploring the target) and defensive responses (escaping, burying, jumping, climbing, grooming and vocalization) were calculated by averaging the observers’ sums. The data are expressed as the sum of aggressive and defensive scores that corresponds to the total irritability score. When tested 48h into nicotine withdrawal, there was a significant effect of treatment on irritability responses, F(3;28)=17.03; P<0.001 (Fig. 2C). The Newman Keuls post hoc test showed that the nicotine exposed rats (Nico+PBS) had significantly higher numbers of irritability-like responses (P<0.001) (42.4±3.6) compared with both saline+PBS and saline+ NicA2-J1-exposed rats (21.1±2.1 and 26.4±2.8 respectively). The animals exposed to nicotine and treated with the NicA2-J1 showed irritability-like responses similar to the saline exposed groups (18.8±1.3). In summary, NicA2-J1 completely prevented the development of irritability-like behavior, hyperalgesia, and somatic signs of withdrawal in animals exposed to chronic nicotine, strongly supporting our hypothesis that NicA2 variants may prevent the development of addiction-like behaviors.
As an additional means to illustrate the correlation between the behavior changes observed nicotine dependent rats and enzyme administration, we analyzed nicotine blood and brain levels in a similar experiment. Rats (n=4) were exposed to nicotine (3.16 mg/kg/day) containing osmotic minipumps for 7 days and were treated every other day with NicA2-J1 (10 mg/kg) or vehicle (PBS, pH 7.4). Blood was collected after 1 and 5 days and brains were collected after 7 days of first dosing of NicA2-J1. Nicotine was extracted from the blood and tissues to be analyzed by LC-MS. Remarkably, there was no nicotine detected in the blood or brains in the treated group, while the control group exhibited expected concentrations of nicotine in both blood and brains (Fig. 3). These results clarify at a molecular level why the Nico+ NicA2-J1 group exhibited the same behavior as the groups receiving saline. With complete elimination of nicotine in the blood, it can no longer reach the brain to trigger the neuroadaptations leading to nicotine dependence. To put this in perspective at the clinical level nicotine vaccines were only able to reduce brain nicotine concentrations 30% – 64%, which has been a suspected cause of the vaccines lack of efficacy.6b
Fig. 3.
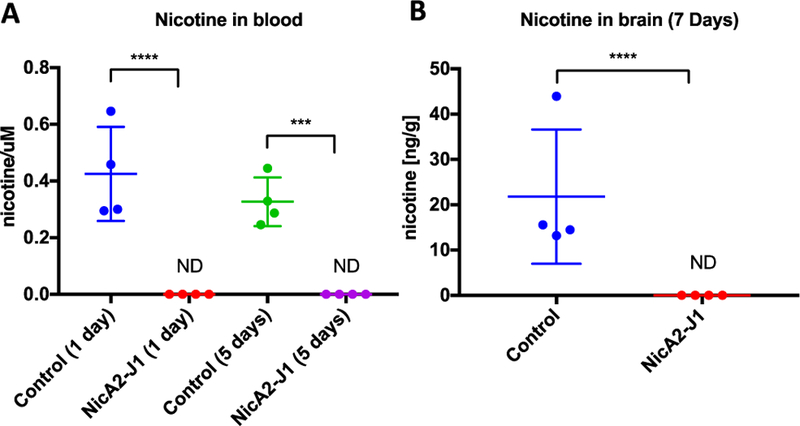
Nicotine concentrations in rat blood (A) after 1 or 5 days and brains (B) after 7 days of NicA2-J1 administration. (ND: not detected)
In conclusion, we have disclosed a proof of concept that a nicotine-degrading enzyme can impact nicotine’s behavioral effects. The sequence of events demonstrating NicA2’s robust impact on nicotine dependence were initiated by improving the enzymes in vivo stability by fusion of an albumin binding domain (ABD035) to the N- terminus of a 50 a.a. truncated NicA2 (Δ50-NicA2). The catalytic properties of the new construct were characterized by LC-MS and presented equivalent catalytic activity as the WT. NicA2-J1 was further evaluated for therapeutic value in pharmacokinetic and in vivo efficacy studies. NicA2-J1 bound tightly to albumin granting a much longer in vivo half-life than the WT. Serum nicotine distribution and behavioral testing revealed that the enzyme completely eliminates blood nicotine content, thereby halting the drug’s psychoactive effects. This research is still a work in progress, however, with these results, we believe the objective of creating an enzyme for nicotine cessation therapy is firmly established.
Supplementary Material
Acknowledgments
We acknowledge funding from National Institute on Drug Abuse (DA041839 to K. Janda and DA036691 to O. George). P.O.M (IPNA-CSIC) was supported by a Marie Curie IOF from the European Union’s Seventh Framework Program FP7/2007–2013 under REA Grant Agreement No. 623155. We thank Dr. Cody Wenthur for assistance in pharmacokinetic analysis. This is manuscript # 29619 from The Scripps Research Institute.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.World Health Organization., WHO Report on the Global Epidemic, 2013: Enforcing Bans on Tobacco Advertising, Promotion, Sponsorship, WHO Press, Geneva,Switzerland, 2013. [Google Scholar]
- 2.Biener L, Reimer RL, Wakefield M, Szczypka G, Rigotti NA and Connolly G, Am. J. Prev. Med, 2006, 30, 217–224. [DOI] [PubMed] [Google Scholar]
- 3.Xue S, Schlosburg JE and Janda KD, J. Am. Chem. Soc, 2015, 137, 10136–10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDonough M, Aust Prescr, 2015, 38, 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janda KD and Treweek JB, Nat. Rev. Immunol, 2011, 12, 67–72. [DOI] [PubMed] [Google Scholar]
- 6.(a) Shen XY, Orson FM and Kosten TR, Clin. Pharmacol. Ther, 2012, 91, 60–70; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Goniewicz ML and Delijewski M, Hum. Vaccin. Immunother, 2013, 9, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Hoogsteder PHJ, Kotz D, van Spiegel PI, Viechtbauer W and van Schayck OCP, Addiction, 2014, 109, 1252–1259; [DOI] [PubMed] [Google Scholar]; (b) Wolters A, de Wert G, van Schayck OC and Horstman K, Addiction, 2014, 109, 1268–1273. [DOI] [PubMed] [Google Scholar]
- 8.(a) Hatsukami DK, Donny EC, Koopmeiners JS and Benowitz NL, Cancer Epidemiol. Biomarkers Prev, 2015, 24, 472–476; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hatsukami DK, Hertsgaard LA, Vogel RI, Jensen JA, Murphy SE, Hecht SS, Carmella SG, al’Absi M, Joseph AM and Allen SS, Cancer Epidemiol Biomarkers Prev, 2013, 22, 1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Tang H, Wang L, Wang W, Yu H, Zhang K, Yao Y and Xu P, PLoS genetics, 2013, 9, e1003923; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yu H, Tang H, Wang L, Yao Y, Wu G and Xu P, J. Bacteriol, 2011, 193, 5541–5542; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yu H, Tang H and Xu P, Sci Rep, 2014, 4, 5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Dennis MS, Zhang M, Meng YG, Kadkhodayan M, Kirchhofer D, Combs D and Damico LA, Journal of Biological Chemistry, 2002, 277, 35035–35043; [DOI] [PubMed] [Google Scholar]; (b) Duttaroy A, Kanakaraj P, Osborn BL, Schneider H, Pickeral OK, Chen C, Zhang GY, Kaithamana S, Singh M, Schulingkamp R, Crossan D, Bock J, Kaufman TE, Reavey P, Carey-Barber M, Krishnan SR, Garcia A, Murphy K, Siskind JK, McLean MA, Cheng S, Ruben S, Birse CE and Blondel O, Diabetes, 2005, 54, 251–258. [DOI] [PubMed] [Google Scholar]
- 11.Tararina MA, Janda KD and Allen KN, Biochemistry, 2016, 55, 6595–6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jonsson A, Dogan J, Herne N, Abrahmsen L and Nygren PA, Protein Eng Des Sel, 2008, 21, 515–527. [DOI] [PubMed] [Google Scholar]
- 13.(a) Zhao-Shea R, DeGroot SR, Liu L, Vallaster M, Pang X, Su Q, Gao G, Rando OJ, Martin GE, George O, Gardner PD and Tapper AR, Nat Commun, 2015, 6, 6770; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Grieder TE, Herman MA, Contet C, Tan LA, Vargas-Perez H, Cohen A, Chwalek M, Maal-Bared G, Freiling J, Schlosburg JE, Clarke L, Crawford E, Koebel P, Repunte-Canonigo V, Sanna PP, Tapper AR, Roberto M, Kieffer BL, Sawchenko PE, Koob GF, van der Kooy D and George O, Nat. Neurosci, 2014, 17, 1751–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Skjei KL and Markou A, Psychopharmacology (Berl.), 2003, 168, 280–292; [DOI] [PubMed] [Google Scholar]; (b) Hamouda AK, Jackson A, Bagdas D and Damaj MI, Nicotine & tobacco research : official journal of the Society for Research on Nicotine and Tobacco, 2017, DOI: 10.1093/ntr/ntx183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen A and George O, Front Psychiatry, 2013, 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimbrough A, de Guglielmo G, Kononoff J, Kallupi M, Zorrilla EP and George O, Alcohol. Clin. Exp. Res, 2017, 41, 1886–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Pitcher GM, Ritchie J and Henry JL, J. Neurosci. Methods, 1999, 87, 185–193; [DOI] [PubMed] [Google Scholar]; (b) Cohen A, Treweek J, Edwards S, Leao RM, Schulteis G, Koob GF and George O, Addict. Biol, 2015, 20, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Watkins SS, Stinus L, Koob GF and Markou A, J. Pharmacol. Exp. Ther, 2000, 292, 1053–1064; [PubMed] [Google Scholar]; (b) Kallupi M and George O, Curr. Protoc. Neurosci, 2017, 80, 8 41 41–48 41 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malin DH, Lake JR, Newlin-Maultsby P, Roberts LK, Lanier JG, Carter VA, Cunningham JS and Wilson OB, Pharmacol. Biochem. Behav, 1992, 43, 779–784. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.