

Abstract
Xia-Gibbs syndrome (XGS: OMIM # 615829) results from de novo truncating mutations within the AT-Hook DNA Binding Motif Containing 1 gene (AHDC1). To further define the phenotypic and molecular spectrum of this disorder, we established an XGS Registry and recruited patients from a worldwide pool of approximately 60 probands. Additional de novo truncating mutations were observed among 25 individuals, extending both the known number of mutation sites and the range of positions within the coding region that were sensitive to alteration. Detailed phenotypic examination of 20 of these patients via clinical records review and data collection from additional surveys showed a wider age range than previously described. Data from developmental milestones showed evidence for delayed speech and that males were more severely affected. Neuroimaging from six available patients showed an associated thinning of the corpus callosum and posterior fossa cysts. An increased risk of both scoliosis and seizures relative to the population burden was also observed. Data from a modified autism screening tool revealed that XGS shares significant overlap with autism spectrum disorders. These details of the phenotypic heterogeneity of XGS implicate specific genotype/phenotype correlations and suggest potential clinical management guidelines.
Keywords: AHDC1, de novo mutation, intellectual disability
1 |. INTRODUCTION
Xia-Gibbs syndrome (XGS: OMIM #615829) is a newly described disorder characterized by developmental delay, hypotonia, speech delay, sleep apnea, and seizures (Xia et al., 2014). Patients with XGS have de novo truncating mutations within a critical region of the AT-Hook DNA Binding Motif Containing 1 gene (AHDC1). Probands present with features that are associated with several different syndromes and are observed in many undiagnosed patients who likely harbor other genetic disorders, making DNA testing and the establishment of molecular diagnosis the primary diagnostic tool for XGS. Indeed, the original identification of the disorder depended on the identification of molecular variation in AHDC1, rather than recognition of a distinctive set of shared clinical features. This initial report of XGS focused on four probands and their relatives who were clinically assessed after identification of AHDC1 truncating alleles, revealing a shared set of phenotypic features (Xia et al., 2014). Subsequently, an additional seven patients were described with features consistent with the original four that were reported (Yang et al., 2015). These published cases each have truncating AHDC1 mutations, but display clinical heterogeneity, including the variable occurrence of seizures, the extent of anomalies detectable by brain magnetic resonance imaging (MRI), differences in the overall levels of cognitive function, the age of language development, and the possible presence of aggressive or unruly behavior in some cases (Xia et al., 2014; Yang et al., 2015). Whether this reflects allelic heterogeneity and if specific genotype/phenotype correlations can be discerned, remains unknown.
Approximately 60 probands worldwide have now been identified through additional publication (Bosch et al., 2016; Garcia-Acero & Acosta, 2017; Miller et al., 2017; Park, Kim, Jang, & Jang, 2017; Quintero-Rivera et al., 2015), self-referral, physician contact, and social media. XGS is, therefore, an example of a disorder where the diagnostic impact of whole-exome sequencing, when combined with social media, has allowed families to rapidly build networks, establish support groups, and participate as stakeholders in research (Enns et al., 2014).
To more systematically evaluate the consequences of de novo truncating mutations in this gene, and to provide a resource for the future study of XGS and AHDC1-related disorders, we established an XGS Registry to gather genotype and phenotype data from multiple families. The detailed evaluation of the molecular and clinical spectrum of 20 cases provides a basis for both improved forecast of prognoses and support for research studies.
2 |. METHODS
2.1 |. Identification of AHDC1 families
Sixty families were identified worldwide via self-reporting, physician referral, and social media.
2.2 |. Registry
A Registry was established utilizing RedCap (Research Electronic Data Capture), which is a secure, web-based application designed to support data capture for research studies (Harris et al., 2009). This application was initially housed in a health insurance portability and accountability act of 1996 (HIPAA) compliant environment hosted by Amazon Web Services and later migrated to a local, compliant host.
2.3 |. Ethics, consent, and permissions
The infrastructure for the Registry and outreach to identified probands and families was approved by the Baylor College of Medicine Institutional Review Board. Invitation emails were sent to parents and caretakers, and positive responders were provided informed consent for Registry participation. The Registry includes full contact information, detailed patient records from each participant’s clinical consultations, and parent responses to a questionnaire designed to capture clinical and developmental features of the affected child. A second institutional review board (IRB) approved Protocol was used to further consent individuals who were in the Registry, for whom there were additional requests for research participation.
2.4 |. Clinical ascertainment
Parents of probands consented to participation in the registry, together with their affected children. Participants join the registry by completing an initial survey and by providing their contact information and a genetic testing report documenting a pathogenic variant in the AHDC1 gene. A further clinical survey was then administered (complete survey details are in the Supporting Information). In addition to the clinical survey, 14 probands and their families attended a clinical and family conference held in April of 2017, in Houston, Texas. At that conference, clinical observations from the survey data were clarified by the family and physicians. Additional medical records and MRI images were also obtained for some of the subjects. Available MRI images were independently reviewed by a pediatric neuroradiologist (JVH).
2.5 |. Facial feature analysis
Facial feature analyses were performed using the Facial Dysmorphology Novel Analysis software (FDNA, Inc., Boston, MA) and facial images provided by families. A mask depicting the composite, characteristic appearance of XGS was created.
2.6 |. Data analysis
Statistical analyses were performed using R. The Fisher exact test was used to compare the language ability between male and female patients. A binomial test was used to compare the prevalence of scoliosis in the patients with the frequency in the general population.
3 |. RESULTS
3.1 |. Overview of the patients, gene, and AHDC1 mutations
Twenty individuals with de novo truncating AHDC1 mutations had detailed clinical assessments and were included for the phenotypic comparison described in this study. These individuals represent a subset of the 60 families described worldwide as of November 2017, for whom our review of the available records of mutation data was consistent with a molecular XGS diagnosis in 34 families. Of these 34 families, 25 joined the registry and all but five families provided sufficient clinical data for inclusion.
The 20 described individuals include 6 previously published and 14 novel cases. Medical records were sought and successfully obtained for 18 of the probands, while two others were determined to have already provided sufficient clinical information. MRI images and reports were collected and independently reviewed by a pediatric neuroradiologist, for six cases.
We directly confirmed the presence of the AHDC1 mutations by sanger DNA sequencing in 13 individuals, where samples were available to be tested. The confidence in the accuracy of the mutation data from the remaining seven cases was based upon the availability of clinical diagnostic reports from major providers. Figure 1 shows the position of all 25 mutations from XGS patients in the Registry. In five of the cases described here, we also had data from whole-exome sequencing and we considered other gene candidates as contributing to disease, apart from AHDC1. We were able to exclude all such candidates based on phenotype mismatch, frequency, or lack of a second variant contributing to an established recessive mode of inheritance (Supporting Information Table S1). A de novo NF1 variant reported in one individual was also excluded as contributing to the clinical profile, due to the phenotype mismatch.
FIGURE 1.
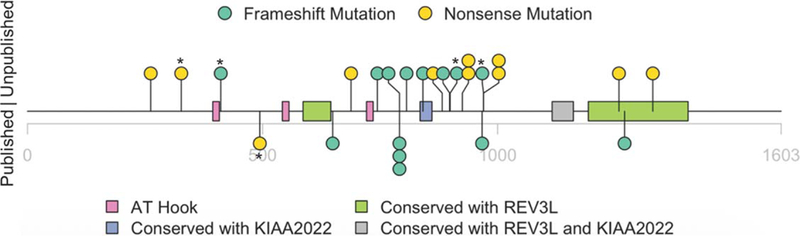
Schematic representation of mutations identified in 25 XGS patients that have participated in the XGS Registry. Mutations in five individuals who are not clinically assessed in detail, in this study, are indicated by an asterisk. Previously reported variants are indicated below the line, newly reported variants above. The x-axis shows amino acid positions in the encoded protein, pink rectangles indicate ATHook domains, green rectangles indicate REV3L homology domains, purple rectangle indicates KIAA2022 homology domains and gray rectangle indicates REV3L and KIAA2022 homology domains. Green circles indicate frameshift variants, yellow indicates nonsense variants (see Supplementary Material for further description of homologies and conservation)
In addition to the more severely affected individuals with truncating mutations who are included in the Registry, one less severe case has been putatively associated with a de novo heterozygous missense mutation in AHDC1. This patient is diagnosed with autism spectrum disorder, without other reported features of XGS. Confirmation of missense mutations leading to mild phenotypes would be of considerable interest for delineating gene function and disease pathology. At this time, however, it is impossible to eliminate mutations in other loci as the cause of this patient’s disorder. A search for variants contributing to this individual’s phenotype is underway, and so these data are not included in the present report.
3.2 |. Mutation position
The previously reported cases represented nine distinct mutations that spanned the amino acid positions in the AHDC1 gene product from codon 375 to 1,270 (Figure 1). In the present report, we now describe an additional 14 cases, including 11 novel AHDC1 truncating mutations, extending this range from codon 262 to 1,330 and doubling the number of known pathogenic mutation sites of AHDC1. There are three recurring mutations in our cohort, including the previously reported p. Cys791Trpfs*57 (c.2373_2374delTG), which is now known to be present in 4/20 (20%) of these unrelated patients. Two additional newly observed mutations, p.Arg925* (c.2773C> T) and p.Gln970* (c.2908C> T), each occurred in two patients.
The mutations in the 20 probands all are predicted to lead to premature chain termination (PCT) during translation and include stopgain changes (9 sites) or frameshifts leading to downstream truncation events (11 Sites). The truncation peptide sequences predicted by conceptual translation to arise as a result of the frameshift mutations did not show high homology to other proteins. Two different mutations gave rise to similar truncation peptides and, with one exception, all truncation products terminate within the boundaries of other nonsense sites.
Database searches revealed functional domains within AHDC1, that are shared with REV3L (DNA polymerase zeta catalytic subunit) (Gan, Wittschieben, Wittschieben, & Wood, 2008) and/or KIAA2022 (Van Maldergem et al., 2013). The cross-species alignments further revealed striking conservation across multiple domains with both REV3L and KIAA2022 (Figure 1; Supporting Information Figure S1).
3.3 |. Patient phenotype
The current ascertainment of the clinical spectrum associated with XGS is described in Table 1. Ascertainment for the reported cases includes data from initial clinical visits, family survey, and subsequent consultancy (see Supporting Information Table S2 for details). The 20 cases include 11 males and 9 females, with a median age of 8 years old. Two of the participants were more than 18 years old.
TABLE 1.
Detailed patient phenotypes
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | Summary |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Referencea | #1 (Patient 1) | #2 (Patient 5) | #2 (Patient 1) | #1 (Patient 3) | #1 (Patient 2) | #2 (Patient 6) | |||||||||||||||
| Mutation | |||||||||||||||||||||
| Nucleotide change | c.2373_2374delTG | c.3809delA | c.2644C>T | c.2520delT | c.2773C>T | c.2229delG | c.l945delG | c.2062C>T | c.2908C>T | c.2373_2374delTG | c.2691delA | c.2908C>T | c.784C>T | c.3989C>A | c.2415delG | c.2898delC | c.2773C>T | c.2373_2374delTG | c.2373_2374delTG | c.3773C>G | |
| Protein change | p.Cys791Trpfs*57 | p.Glnl270Argfs*75 | p.Gln882* | p.Arg841Alafs*91 | p.Ser744Profs*188 | p.Arg925* | p.Ala649Profs*83 | p.Arg688* | p.Gln970* | p.Cys791Trpfs*57 | p.Va!898Trpfs*34 p.Gln970* | p.Gln262* | p.Serl330' | p.Leu806Trpfs*126 | p.Tyr967Thrfs*175 | p.Arg925* | p.Cys791Trpfs*57 | p.Cys791Trpfs*57 | p.Serl258* | ||
| Age | 5 years | 12 years | 10 years | 9 years | 13 years | 3 years | 4 years | 20 years | 6 years | 12 years | 4 years | 11 years | 6 years | 17 years | 6 years | 8 years | 6 years | 8 years | 21 years | 2 years | 8 years (2~21 years) |
| Gender | F | M | M | M | M | M | M | F | F | M | F | F | F | M | F | F | F | M | M | M | Female 9 (45%) |
| Ethnicity | White | White/Asian | White | White | White | White | White | White | White | White | White | White | White | White | White | Asian | White | White | White | White | White 18 (90%) |
| Growth | |||||||||||||||||||||
| Height | 0.05 | 0.01 | 0.05 | 0.15 | 0.03 | <0.01 | <0.01 | >0.99 | (0.05, 0.15) | 0.05 | <0.01 | NA | 0.05 | <0.01 | (0.25,0.5) | <0.01 | (0.15,0.25) | 0.15 | <0.01 | 0.01 | |
| Scoliosis | N | N | Y | N | Y | N | N | Y | N | N | N | N | N | N | N | N | N | Y | N | 4 (20%) | |
| Language | |||||||||||||||||||||
| M-CHAT score | 12 | 6 | 9 | 13 | 10 | 5 | 9 | 10 | 1 | 2 | 0 | 9 | 1 | 14 | 0 | 4 | 8 | 4 | 10 | 13 | 8.5 (0~14) |
| Prior autism diagnosis | Y | N | Y | N | N | N | Y | Y | N | N | N | N | Y | N | N | N | N | N | N | 5 (25%) | |
| Current languageb | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 3 | 3 | 1 | 3 | 3 | 0 | 3 | 1 | 1 | 3 | 2 | 0 | 1 (0—3) |
| Age at first word | 4 years | NA | NA | NA | NA | NA | NA | 5 years | 2.5 years | 12 months | 2.5 years | 2 years | 4 years | NA | Around 3 years | UKN | Around 2.5–3 years 2 years | 5 years | NA | 2.75 years (1~5 years) |
|
| Age, two words together | NA | NA | NA | NA | NA | NA | NA | 6 years | 3.5 years | 24 months | 3.5 years | 3 years | 5 years | NA | 3 years | 4 years | NA | 2 years | 7 years | NA | 3.5 years (2~7 years) |
| Age following command | 2 years | 8 years | 8 years | NA | 4 years | 23 months | 3 years 9 months | 4 years | 3–4 years | 18 months | 2 years | 6 years | 5 years | 5 years | 2 years | 6 years | 3 years | 4 years | 3 years | NA | 3.875 (1.5~8 years) |
| Mobility | |||||||||||||||||||||
| Hypotonia diagnosis | Y | Y | Y | Y | Y | Y | Y | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | N | 18 (90%) |
| Independent walking | Y | Y | Y | N | Y | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | N | N | 16 (80%) |
| Age independent walking | 3 years | 6 years | 3 years | NA | 3 years | NA | 3 years 5 months | 22 months | 2.5 years | 18 months | 2.5 years | 2 years | 3 years | 19 months | 2 years | 2 years | 2 years 4 months 4 years | NA | NA | 2.5 years (1.5~6 years) |
|
| Sleep/airway | |||||||||||||||||||||
| Sleep apnea | Y | N | N | N | Y | Y | Y | N | N | Y | N | N | N | N | N | Y | N | Y | Y | Y | 9 (45%) |
| Breathing support | Y | N | N | N | N | N | N | N | N | Y | N | N | N | N | N | N | N | N | Y | N | 3 (15%) |
| Neuro | |||||||||||||||||||||
| MRI | Abnormal | Abnormal | Abnormal | Abnormal | Normal | Normal | Abnormal | Abnormal | Normal | Abnormal | Abnormal | Normal | Normal | Normal | Normal | Normal | Abnormal | Abnormal | Abnormal | Abnormal | 1 12 (60%) |
| EEG | Normal | NA | Normal | Normal | NA | Normal | NA | Normal | Normal | Abnormal | Normal | Normal | Normal | NA | Normal | Abnormal | NA | Abnormal | Abnormal | Normal | 4 (20%) |
| Seizure | N | N | Y | N | N | N | N | Y | N | Y | N | N | N | N | Y | N | N | Y | Y | N | 6 (30%) |
| Age at first seizure | 9 years | 9 months | 3 years | 2 years | 5 years | 12 years | 4 years (9 months~12 years) |
||||||||||||||
| Ataxia | Y | Y | N | Y | N | N | Y | Y | Y | Y | Y | Y | N | N | Y | N | Y | Y | Y | N | 13(65%) |
| Vision | |||||||||||||||||||||
| Glasses/contacts | Y | No | N | Y | N | N | Y | Y | N | N | N | N | Y | N | N | N | Y | N | N | N | 6 (30%) |
| Vision | N.asc. | N.asc. | N.asc. | Mild myopia | N.asc. | N.asc. | Slight astig. | N.asc. | N.asc. | Prismatic lenses | NA | Sunglasses 40/70 | 20/20 | 100% | NA | 6/9.5 | NA | NA | |||
| Strabismus | Y | N | Y | N | N | Y | N | N | Y | N | Y | N | Y | N | N | N | Y | N | Y | N | 8 (40%) |
| Dysmorphic features | |||||||||||||||||||||
| Small ear lobes | N | NA | Y | N | N | N | N | N | N | Y | NA | NA | N | NA | Y | N | N | N | N | NA | |
| Upturned earlobes | N | NA | N | Y | N | N | Y | N | N | N | NA | NA | Y | NA | N | Y | N | N | N | NA | |
| Low-set ears | N | NA | N | Y | N | N | N | Y | N | N | NA | NA | Y | NA | N | N | N | N | N | NA | |
| Protuberant ears | N | NA | Y | N | N | N | N | N | Y | Y | NA | NA | N | NA | N | Y | N | N | N | NA | |
| Deep-set eyes | N | NA | N | N | Y | Y | N | Y | N | Y | NA | NA | N | NA | N | N | N | N | N | NA | |
| Upslanting palpebral fissures | N | NA | N | Y | N | N | N | N | Y | N | NA | NA | Y | NA | N | N | N | N | N | NA | |
| Downslanting palpebral fissure | N | NA | N | N | Y | N | Y | N | N | N | NA | NA | N | NA | N | N | N | N | N | NA | |
| Mild ptosis | N | NA | N | Y | N | Y | N | N | N | N | NA | NA | Y | NA | N | Y | N | N | Y | NA | |
| Esotropia | N | NA | N | Y | N | Y | N | N | Y | N | NA | NA | Y | NA | N | N | N | N | N | NA | |
| Hypertelorism | N | NA | Y | N | N | N | Y | Y | Y | N | NA | NA | Y | NA | N | N | Y | N | Y | NA | |
| Flat nasal bridge | Y | NA | Y | Y | N | Y | Y | N | Y | N | NA | NA | N | NA | N | N | N | N | Y | NA | |
| Micrognathia | N | NA | N | N | N | Y | N | N | N | N | NA | NA | N | NA | N | N | N | N | Y | NA | |
| Thin upper lip | Y | NA | Y | Y | Y | Y | Y | Y | Y | Y | NA | NA | Y | NA | Y | Y | Y | N | Y | NA | |
| Broad forehead | Y | NA | N | Y | N | Y | Y | Y | Y | N | NA | NA | Y | NA | Y | N | Y | N | Y | NA | |
Height was shown in percentage (WHO child growth standards 2–5 years http://www.who.int/childgrowth/standards/height_for_age/en/ and WHO growth references for 5–19 years http://www.who.int/growthref/en/, percentage for two adult patients are assessed as 19 year olds).
References: 1, Xia et al. (2014); 2, Yang et al. (2015).
Language: 0. No words 1. < 50 words; 2. no sentence but >50 words; 3. full sentence > 200 words.
3.4 |. Language and cognitive ability
3.4.1 |. Overall cognitive ability
A diagnosis of autism, or autism spectrum disorder, had previously been reported in 5 of the 20 patients described here (25%). Methods for assessing autism spectrum disorder can be heterogeneous, hence we used the modified checklist for autism in toddlers (M-CHAT) as a standardized method to assess cognitive ability in the individuals in the cohort (Robins et al., 2014). This tool is widely used as a preliminary screen for children to identify increased autism risk, for which a formal, full neuropsychological evaluation would be recommended. In general practice, those who have a score indicating medium or high risk of autism will receive further testing. The 20 patients had a median M-CHAT score of 8 (0~4). Of these, 5 (25%) had an M-CHAT score less than 2, which is considered low risk for autism, 4 (20%) had an M-CHAT score between 3~7, at medium risk for autism, and 11 (55%) had an M-CHAT score between 8~20 and were at high risk for autism. All five individuals who had been diagnosed with autism, or autism spectrum disorder prior to this study, had an M-CHAT score higher than 8 (Figure 2a).
FIGURE 2.
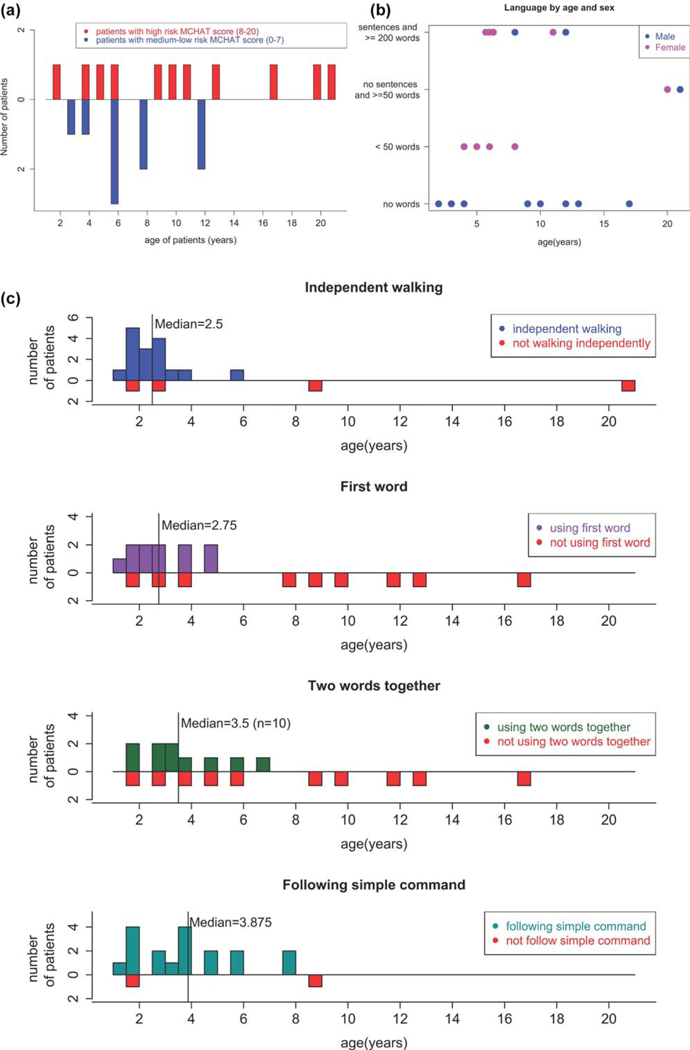
(a) M-CHAT score of the 20 patients. Patients with M-CHAT scores 8~20 are considered high risk and those who scored 0~7 are considered to have medium-low risk. (b) Language capability distributed by age and sex. (c) Age at onset distribution of patients who achieve different milestones (from up to bottom): independent walking, using first word, using two words together and following simple commands. For patients who have not reached any of the milestones, their current ages are shown in red in each panel
In addition to M-CHAT, the patients were assessed on their language skills and on the capability to follow simple commands (Figure 2b,c). A total of 18 (90%) of 20 patients were reported as able to follow simple commands, with a median age of 3.9 (1.~58) years (Figure 2c).
3.4.2 |. Timing of speech
Eight out of 20 patients (40%) were reported to have no speech, four (20%) were reported to use less than 50 words, two (10%) were reported to have a vocabulary of more than 50 words but do not speak in full sentences, and six (30%) patients were reported to use complete sentences with a vocabulary of more than 200 words (Figure 2b). Among the 12 patients who use at least one word other than “mama” or “dada,” the median age for using one word is 2.75 (1~5) years, while the median age for using two words together is 3.5 (2~6) years (Figure 2c). These data are consistent with a relationship between gender and speech as male patients are significantly more likely to be nonverbal than female patients (p< .01). One of the male patients is 2 years old, which is younger than the median age at which first words were spoken, however, the ρ value is still <.01 when he is removed from this analysis. It is noteworthy that all the patients reported with no speech are male (Figure 2b).
3.4.3 |. Correlation of M-CHAT and speech
Multilinear regression showed that a linear relationship exists between the M-CHAT score, the square root of age and language capability ratings (0= nonverbal; 1= using less than 50 words; 2= not using sentence but using more than 50 words; 3=using sentences with a vocabulary of more than 200 words). The equation is:
M-CHAT score = 1:99*sqrt (age)– 2:27
*language capability rating (adjusted R2= 0:42)
This indicates that the M-CHAT score increases with age, but the rate of increase slows down with the gain of age. It also suggested for patients of the same age, for every point increase in language capability rating, the M-CHAT score decreases by approximately 2 points.
3.5 |. Neurology
3.5.1 |. Abnormal MRI
An abnormal MRI was reported in 12 out of 20 (60%) patients who have had MRI performed. MRI images for re-review were available for six patients. We found that 6/6 had thinning of the corpus callosum (Figure 3, compare b–f to “control” individuals a and d). In addition, several subjects were found to have a cyst in the posterior fossa (Subject 4, Figure 3b, Subject 3 Figure 3e, Subject 7, Figure 3h).
FIGURE 3.
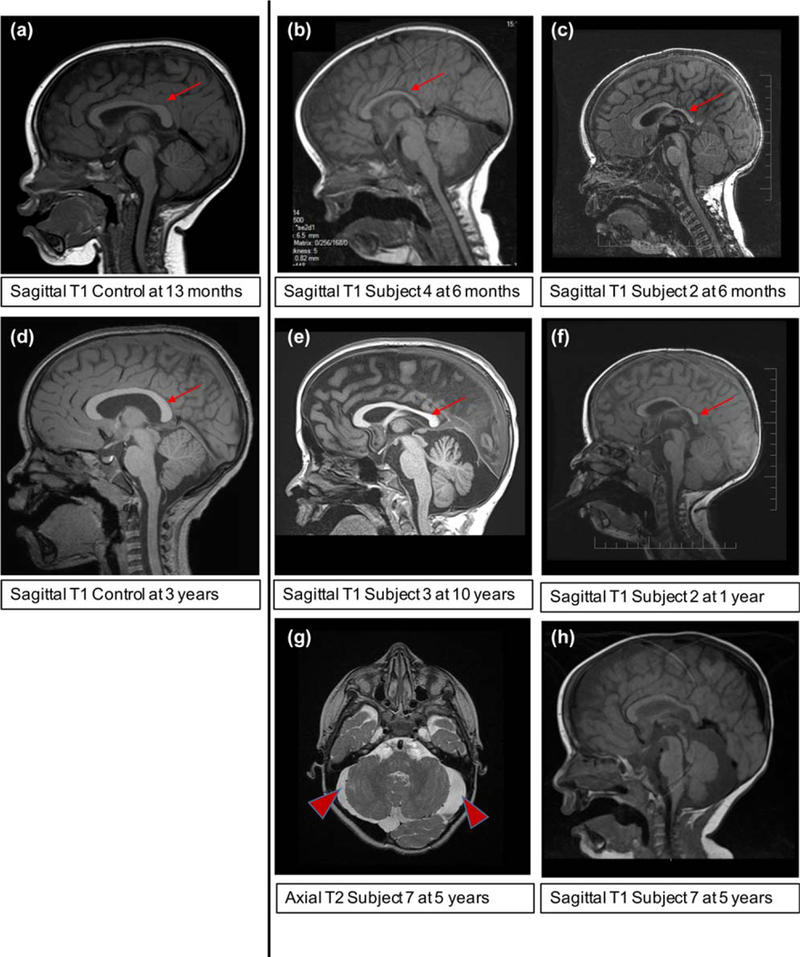
Brain MRI features of the subjects, unless otherwise indicated a Sagittal T1 image is shown. (a) A control subject (a clinical MRI for history of minor head trauma) in a subject at age 13 months showing a typical corpus callosum (red arrow). (b) Subject 4 at age 6 months, shows thinning of the corpus callosum posteriorly (red arrow) in addition there is a large extra-axial fluid space posterior to the cerebellum. (c) Subject 2 at age 6 months, although the young age limits the assessment, the corpus callosum appears thin (red arrow). (d) A control subject (a clinical MRI for history of seizure) in a subject at age 3 years showing a typical corpus callosum (red arrow). (e) Subject 3 at age 10 years, shows thinning of the corpus callosum (red arrow), particularly in the posterior portion, there is also a large extra-axial fluid space posterior to the cerebellum. (f) Subject 2 (also shown in c) now at age 1 year, a later study confirms the suspicion of the 6-month study suggesting thinning of the corpus callosum. (g) Subject 7 at age 5 years, a T2 axial image shows the extra-axial fluid space around the cerebellum is larger than normal and dysmorphic in appearance. (h) Subject 7 at age 5 years, a T1 sagittal image shows thinning of the corpus callosum and a large extra-axial fluid space posterior to the cerebellum
3.5.2 |. Seizures
Seizures were reported in 6 out of 20 (30%) patients. The median onset age of seizure is 4 years old (9 months~12 years old). An abnormal EEG was reported in three out of these six (50%) patients.
3.5.3 |. Timing of walking
Independent walking is reported in 16 out of 20 patients (80%), with median beginning walking age of 2.5 (1.5~6) years. All the patients who are not able to walk independently are male. However, there is no significant difference between the ability to walk independently between male and female patients (ρ =.09). Hypotonia is reported in 18 out of 20 patients (90%).
3.6 |. Other significant features
3.6.1 |. Scoliosis
Scoliosis was reported in 4 of 20 (20%) patients (ages 10~21 years), and three had received surgery for the condition. Compared to a general population prevalence of idiopathic scoliosis of 0.47–5.2% with an age of onset of 10–15 years, the risk of scoliosis in XGS individuals is significantly increased (ρ=018, compared to 5.2% prevalence) (Konieczny, Senyurt, & Krauspe, 2013; Mirtz, Thompson, Greene, Wyatt, & Akagi, 2005). No sex bias was apparent.
3.6.2 |. Airway/Sleep
Sleep apnea was reported in 9 out of 20 patients (45%), three of these nine patients (33.3%) use respiratory support during sleep.
3.6.3 |. Vision
Strabismus was reported in 8 out of 20 patients (40%).
3.6.4 |. Dysmorphic features
Facial features (Figure 4a) consistently revealed dysmorphic features such as broad forehead, hypertelorism, flat nasal bridge, and thin upper lip (Table 1). These features are also shown in the composite mask depicting the characteristic appearance of XGS patients (Figure 4b). To date, no patient has been clinically diagnosed based on a recognizable pattern of facial dysmorphisms, or a facial gestalt.
FIGURE 4.

(a) Frontal face images of some of the patients (accession number in this study are shown at top left corner) (b) A computer generated masking depicting the characteristic features of Xia-Gibbs syndrome. This image was generated using the FDNA software. Overlapping features include a broad forehead, hypertelorism, flat nasal bridge, and thin upper lip
3.6.5 |. Aggressive behaviors
Aggressive behavior in at least one patient was suggested by Yang et al. (2015). We were unable to ascertain any history of overtly aggressive behavior in the reported cases, although several parents described a history of self-injury.
3.7 |. Genotype and phenotype association
To explore the correlation between the specific site of mutation and phenotype, we compared three sets of patients who share the same mutations. Patients 1, 10, 18, and 19 share mutation p. Cys791Trpfs*57, Patients 6 and 17 share mutation p.Arg925* and Patients 9 and 12 share mutation p.Gln970*. For the mutation p. Cys791Trpfs*57, all four patients are verbal and reported to have sleep apnea and abnormal brain MRI. Seizures were reported in three of the four patients. Strabismus and a high risk M-CHAT score were reported in two of the four patients. It is challenging to compare the two patients with mutation p.Arg925* as one of them (Patient #6) is only 3 years old, and is yet to reach some developmental milestones. Patients 9 and 12 were each reported to be verbal, using sentences with a vocabulary of more than 200 words. They were both reported to have normal MRI and EEG. They achieved developmental milestones, such as using words and walking, at similar ages. Strabismus was reported in Patient 9 but not in Patient 12.
To further explore correlations between the mutations and phenotypes, multiple phenotypes were plotted against the mutation sites (data not shown). No significant association was observed.
4 |. DISCUSSION
The ability to aggregate data from the observation and care of individuals with XGS is a key step for clinical management, family counseling, and molecular research into the condition. A concerted effort to identify patients worldwide has identified approximately 60 families, of whom 20 are described in detail here, including 14 not previously reported. The ascertainment combined records from initial clinical visits and subsequent clinical and family-reported findings.
Previously known truncating mutations in AHDC1 leading to XGS were observed to cluster around the middle of the coding region (from p.Gly375Argfs*3 to p.Gln1270Argfs*75), and this report expands these positional boundaries (from p.Gln262* to p.Ser1330*, Figure 1). Importantly, the number of different known variants increased, revealing the sensitivity of the protein to truncating events throughout much of its length. All mutations discussed here are de novo in the proband. The mutations each are truncating and lead to a shorter predicted protein.
The AHDC1 gene contains a single coding exon and database searches yielded some insights into the possible protein function. Domains are shared with KIAA2022 and REV3L. KIAA2022 is a target for truncating mutations that cause intellectual disability (Van Maldergem et al., 2013). REV3L is the catalytic subunit of polymerase zeta and is implicated in genome stability and DNA translesion repair (Gan et al., 2008) and it is therefore conceivable that AHDC1 mutations may interfere with DNA repair. Previous work has shown the interaction of AHDC1 with Tax Interaction Protein 1 which was proposed to occur in vivo under DNA damaging conditions (Shalaby, Hampson, Oliver, & Hampson, 2012), supporting this model.
The preponderance of de novo truncating pathogenic AHDC1 alleles could indicate that missense mutations do not cause disease. Alternatively, the absence of pathogenic missense variants may simply reflect the early period of discovery and reduced ascertainment due to a potentially milder phenotype. A similar, illustrative example is BainbridgeRopers Syndrome (BRS: OMIM #615485), where severe phenotypes resulting from de novo truncating mutations in four cases led to the initial disease gene identification of ASXL3 (Bainbridge et al., 2013) and follow up discovery of additional truncating mutations (Balasubramanian et al., 2017). Recently, however, compound missense heterozygous alleles in ASXL3 have been suggested to cause a mild form of BRS in one patient (Giri et al., 2017) and by analogy, future observation of missense mutations in AHDC1 leading to pathogenicity cannot be discounted.
The absence of well-characterized cases in this cohort with large deletions that either interrupt or overlap the gene may reflect a dominant negative mechanism of pathogenesis or the difficulties of precise ascertainment of the disorder. We note one published (Park et al., 2017) and twelve patients with copy number loss or gain covering regions of the genome ranging from 39kb to 19Mb at or near AHDC1, recorded in the DECIPHER database (Firth et al., 2009; https://decipher.sanger.ac.uk/search?q5AHDC1#consented-patients/results). One has thinning of the corpus callosum (Park et al, 2017). Seven of these share similar phenotypes with XGS patients such as developmental delay, hypotonia, and dysmorphic features, one has “ankyloglossia” and four have no phenotype information available. The interpretation of the phenotypes of these patients is confounded by the presence of genes near AHDC1 for which mutation can independently result in developmental disorders (Rocha, Vasques, Santos, & Paiva, 2016). These cases, therefore, warrant further investigation, including the precise mapping of the boundaries of the lesions.
Some AHDC1 mutation positions correlate with clinical observations and warrant follow up studies with much larger sample sizes to establish robust genotype-phenotype correlations. Patients with mutations occurring near the C-terminus of the protein (p.Ser1258*, p.Gln1270Argfs*75, and p.Ser1330*) are nonverbal with high M-CHAT scores, while the patient with the early stop-gain mutation p.Gln262* uses sentences and has an M-CHAT score of 1, which may suggest milder features. This may indicate that nonsense and frameshift variants closer to the Cterminus have a more severe clinical impact. Comparison of individuals with identical mutations suggests that there is heterogeneity among patients, even with the same alteration in AHDC1 gene. To further establish the correlation between phenotype and specific mutation site, larger numbers of patients will be needed to be analyzed.
The participating families indicated a wider range of patients’ age than previously recognized. The median age was 8 years—but two of the patients were adults (nineteen or older). As this is a newly described syndrome and younger children are more likely to seek genetic diagnosis, the true age distribution in the population is not known. This is a particularly important question for families as they plan ongoing care. The parents of older children reported challenging behavioral issues during puberty but declined to describe the patients as aggressive.
Approximately one-half of the patients either had a diagnosis of autism, autism spectrum disorder or were considered at high risk due to their M-CHAT assessment. The precise relationship between ASD and ID is controversial and complex (Matson & Shoemaker, 2009). In general, the younger children in this group exhibit hallmarks of ID, however, the relative cognitive abilities of older children reveal higher function and therefore are more likely to suggest ASD. Relatedly, ASD has an elevated frequency in males (Matson & Shoemaker, 2009), and the data here from the assessment of language capabilities suggest that male XGS patients are significantly more likely to be nonverbal than female patients. This feature warrants further investigation.
Previous reports on XGS have focused on somewhat different clinical features. In particular, clarification of the neurological features was more in-depth from this larger group of subjects. We observed that six patients (30%) developed seizures, while 13 (65%) had ataxia, in line with previous reports, further reinforcing the CNS involvement in XGS.
Upper airway abnormalities also appear to be a common feature in XGS. In the original report, three of four probands had obstructive sleep apnea, and the fourth had suspected tracheomalacia in infancy (Xia et al., 2014). Sleep apnea was present in 45% of the XGS cases described here, with many requiring continuous positive airway pressure at night. This suggests careful monitoring of airway function is essential and there should be a low threshold for referral to pulmonology.
A surprising finding from this detailed assessment was the frequency of scoliosis among XGS individuals. Scoliosis was not included as a characteristic phenotype in previous publications (only reported in Patient #2 in Yang et al., 2015). However, the usual age-at-onset of the feature is 10~15 years old (Konieczny et al., 2013) while the median age for the 11 patients reported here was 5 years (1.5~16) at the time of publication. Our study, therefore, suggests that patients are at significantly increased risk for developing scoliosis and would benefit from an orthopedic referral when appropriate.
The evaluation here suggests several features of XGS for which early diagnosis and regular surveillance can be of clinical benefit. All individuals should undergo detailed assessment by a physician who is familiar with the condition, with annual (more frequent during the first years of life) assessments of growth including height, weight, and head circumference to identify those at risk of failure to thrive or short stature necessitating intervention, and clinical evaluation for scoliosis. Neurodevelopmental delay is a ubiquitous feature of this condition, and regular childhood developmental assessments with early initiation of therapies will maximize developmental potential. Given the high frequency of structural brain anomalies, a baseline brain MRI should be considered, particularly if neurologic abnormalities such as hypotonia are present on exam. Neurologic and ophthalmologic involvement in some individuals may warrant consultation by the appropriate specialists. An evaluation for obstructive sleep apnea as well as an upper airway assessment should also be considered. Together, these practices can both provide better clinical diagnosis and suggest management strategies for symptoms that may be more severe in individual cases.
XGS has multiple features overlapping with autism, suggesting that XGS should be on the differential for unexplained intellectual disability and developmental delays. There are likely older children, adolescents, and adults previously diagnosed with an autism spectrum disorder or intellectual disability who have XGS and would benefit from a clinical genetics referral.
SUPPLEMENTARY MATERIALS:
Additional candidate variants in 5 patients,
Sources of data for clinical ascertainment
Conserved regions of AHDC1
ACKNOWLEDGMENTS
The authors wish to sincerely thank all the families with members who have AHDC1 variation for their contribution and participation. This work was supported in part by NHGRI awards to RAG (HG008898) and JRL (HG006542), and by a private donation. RAG was also supported as a visiting scholar of the Texas Institute for Advanced Studies (2016–2017 programs). JEP was supported by NHGRI K08 HG008986. CPP and LSP were funded by the UK Medical Research Council. All variant data have been submitted to ClinVar. Baylor College of Medicine receives revenue from genetic testing via co-ownership with Baylor Genetics Laboratories.
Funding information
Texas A and M University, Grant/Award, Number: TIAS Scholar; National Human, Genome Research Institute, Grant/Award, Numbers: HG006542, HG008898, and K08, HG008986; Private Donation to Baylor, College of Medicine; UK Medical Research, Council
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
REFERENCES
- Bainbridge MN, Hu H, Muzny DM, Musante L, Lupski JR, Graham BH, … Ropers HH (2013). De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype withsimilarities to Bohring-Opitz syndrome. Genome Medicine, 5(2), 11 10.1186/gm415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian M, Willoughby J, Fry AE, Weber A, Firth HV, Deshpande C, … Tomkins S (2017). Delineating the phenotypic spectrum of Bainbridge-Ropers syndrome: 12 new patients with de novo, heterozygous, loss-of-function mutations in ASXL3 and review of published literature. Journal of Medical Genetics, 54(8), 537–543. 10.1136/jmedgenet-2016-104360 [DOI] [PubMed] [Google Scholar]
- Bosch DG, Boonstra FN, de Leeuw N, Pfundt R, Nillesen WM, de Ligt J, … de Vries BB (2016). Novel genetic causes for cerebral visual impairment. European Journal of Human Genetics, 24(5), 660–665. 10.1038/ejhg.2015.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enns GM, Shashi V, Bainbridge M, Gambello MJ, Zahir FR, Bast T, … Goldstein DB (2014). Mutations in NGLY1 cause an inherited disorder of the endoplasmic reticulum-associated degradation pathway. Genetics Medicine, 16(10), 751–758. 10.1038/gim.2014.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, … Carter NP (2009). DECIPHER: Database of chromosomal imbalance and phenotype in humans using Ensembl resources. American Journal of Human Genetics, 84(4), 524–533. 10.1016/j.ajhg.2009.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan GN, Wittschieben JP, Wittschieben BO, & Wood RD (2008). DNA polymerase zeta (pol zeta) in higher eukaryotes. Cell Research, 18(1), 174–183. 10.1038/cr.2007.117 [DOI] [PubMed] [Google Scholar]
- Garcia-Acero M, & Acosta J (2017). Whole-exome sequencing identifies a de novo AHDC1 mutation in a Colombian patient with XiaGibbs syndrome. Molecular Syndromology, 8(6), 308–312. 10.1159/000479357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri D, Rigden D, Didi M, Peak M, McNamara P, & Senniappan S (2017). Novel compound heterozygous ASXL3 mutation causing Bainbridge-ropers like syndrome and primary IGF1 deficiency. International Journal of Pediatric Endocrinology, 2017, 8 10.1186/s13633-017-0047-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, & Conde JG (2009). Research electronic data capture (REDCap): A metadatadriven methodology and workflow process for providing translational research informatics support. Journal of Biomedical Informatics, 42(2), 377–381. 10.1016/j.jbi.2008.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczny MR, Senyurt H, & Krauspe R (2013). Epidemiology of adolescent idiopathic scoliosis. Journal of Children’s Orthopaedics, 7(1), 3–9. 10.1007/s11832-012-0457-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson JL, & Shoemaker M (2009). Intellectual disability and its relationship to autism spectrum disorders. Research In Developmental Disabilities, 30(6), 1107–1114. 10.1016/j.ridd.2009.06.003 [DOI] [PubMed] [Google Scholar]
- Miller KA, Twigg SR, McGowan SJ, Phipps JM, Fenwick AL, Johnson D, … Wilkie AO (2017). Diagnostic value of exome and whole genome sequencing in craniosynostosis. Journal of Medical Genetics, 54(4), 260–268. 10.1136/jmedgenet-2016104215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirtz TA, Thompson MA, Greene L, Wyatt LA, & Akagi CG (2005). Adolescent idiopathic scoliosis screening for school, community, and clinical health promotion practice utilizing the PRECEDEPROCEED model. Chiropractic & Osteopathic, 13, 25 10.1186/1746-1340-13-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HY, Kim M, Jang W, & Jang DH (2017). Phenotype of a patient with a 1p36.11-p35.3 interstitial deletion encompassing the AHDC1. Annals of Laboratory Medicine, 37(6), 563–565. 10.3343/alm.2017.37.6.563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero-Rivera F, Xi QJ, Keppler-Noreuil KM, Lee JH, Higgins AW, Anchan RM, … Maas RL (2015). MATR3 disruption in human and mouse associated with bicuspid aortic valve, aortic coarctation and patent ductus arteriosus. Human Molecular Genetics, 24(8), 2375–2389. 10.1093/hmg/ddv004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robins DL, Casagrande K, Barton M, Chen CM, Dumont-Mathieu T, & Fein D (2014). Validation of the modified checklist for autism in toddlers, revised with follow-up (M-CHAT-R/F). Pediatrics, 133(1), 37–45. 10.1542/peds.2013-1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha CF, Vasques RB, Santos SR, & Paiva CL (2016). Minireview: Monosomy 1p36 syndrome – Reviewing the correlation between deletion sizes and phenotypes. Genetics and Molecular Research, 15(1). 10.4238/gmr.15017942 [DOI] [PubMed] [Google Scholar]
- Shalaby MA, Hampson L, Oliver A, & Hampson I (2012). Plexin D1: New potential biomarker for cervical cancer. Journal of Immunoassay and Immunochemistry, 33(3), 223–233. 10.1080/15321819.2011.634472 [DOI] [PubMed] [Google Scholar]
- Van Maldergem L, Hou Q, Kalscheuer VM, Rio M, Doco-Fenzy M, Medeira A, … Man HY (2013). Loss of function of KIAA2022 causes mild to severe intellectual disability with an autism spectrum disorder and impairs neurite outgrowth. Human Molecular Genetics, 22(16), 3306–3314. 10.1093/hmg/ddt187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia F, Bainbridge MN, Tan TY, Wangler MF, Scheuerle AE, Zackai EH, … Gibbs RA (2014). De novo truncating mutations in AHDC1 in individuals with syndromic expressive language delay, hypotonia, and sleep apnea. American Journal of Human Genetics, 94 (5), 784–789. 10.1016/j.ajhg.2014.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Douglas G, Monaghan KG, Retterer K, Cho MT, Escobar LF, … Chung WK (2015). De novo truncating variants in the AHDC1 gene encoding the AT-hook DNA-binding motif-containing protein 1 are associated with intellectual disability and developmental delay. Cold Spring Harbor Molecular Case Studies, 1(1), a000562 10.1101/mcs.a000562 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional candidate variants in 5 patients,
Sources of data for clinical ascertainment
Conserved regions of AHDC1