

Abstract
The embryonic chicken is commonly used as a reliable model organism for vertebrate development. Its accessibility and short incubation period makes it ideal for experimentation. Currently, the study of these developmental pathways in the chicken embryo is conducted by applying inhibitors and drugs at localized sites and at low concentrations using a variety of methods. In vitro tissue culturing is a technique that enables the study of tissues separated from the host organism, while simultaneously bypassing many of the physical limitations present when working with whole embryos, such as the susceptibility of embryos to high doses of potentially lethal chemicals. Here, we present an organotypic culturing protocol for culturing the embryonic chicken half head in vitro, which presents new opportunities for the examination of developmental processes beyond the currently established methods.
Keywords: Developmental Biology, Issue 138, Culture, developmental biology, tissue, embryo, microscopy, manipulations, development
Introduction
The embryonic chicken (Gallus gallus) is an excellent model organism commonly used in the field of biology. Its incubation period is roughly 21 days and many eggs can be incubated simultaneously, making experimentation quick and efficient. Perhaps most importantly, the embryo is also easily manipulated, enabling the extensive study of key developmental processes and of the genes and proteins that drive these processes.
The embryonic chicken eye is a complex organ that develops via the interaction of a number of different tissues similar to many other body systems. This method enables the study of the development of these tissues, particularly at advanced stages of development. For example, the multi-layered retina may be of particular interest to those studying the development of the nervous system. Alternative methods that enable the study of other eye tissues such as the cornea, the vitreal body, the lens, the sclera, and the eyelids are of benefit to researchers. The chicken embryonic eye also contains a series of flat bones, the scleral ossicles, which can be used as a model for the study of intramembranous bone induction and ossification in vertebrates1.
Currently, there are a number of methods used to study embryonic development. Microinjections of inhibitory antibodies or other inhibitory molecules2,3, surgically implanted microbeads soaked in inhibitor4, and electroporation5 are all methods that can be used to downregulate genes or proteins of interest in an embryo. Similar methods are used to upregulate proteins. These methods are not without their limitations. For example, when using chemicals to alter the embryonic development, the lethal effects on the embryo must be evaluated, and this limits the use of the aforementioned methods to localized sites of application at doses low enough to ensure the survivability of the embryo.
In vitro tissue culturing has been used in a wide range of organisms to study development and can be used to bypass some of the aforementioned limitations. For example, the femora6, feather buds7,8, and limbs9 of the chicken have all been studied using tissue culturing methods, as have the testes of the mouse10 and the roots and stems of plants11. These methods grant scientists a high degree of control over the tissue development, such as the ability to fluctuate the temperature and alter the nutrient availability. The isolation of the tissue from the whole embryo also makes it far less susceptible to the lethal effects of chemicals, thus enabling manipulation studies on a global scale at higher concentrations. Another notable advantage of in vitro culturing is the preservation of the tissue's cellular environment; the arrangement of tissues remains relatively unchanged, making it possible to study the interactions between different tissue types9. Thus, in vitro culturing opens doors to additional experimental approaches not available in in vivo or in ovo models.
Currently, studying the development of the embryonic chicken eye using chemicals is particularly challenging. A number of extraembryonic membranes cover the embryo, making it difficult to apply microbeads or chemicals; the embryo is also very active within the egg as it gets older, further complicating an already difficult method. This protocol enables easy access to the eye and its surrounding tissues, eliminating these barriers, while also providing new opportunities to examine the developmental processes within the eye. This protocol was established to study the induction of the scleral ossicles within the embryonic eye.
Protocol
NOTE: For embryo stages, utilize the Hamburger and Hamilton12 (HH) staging table.
1. Embryo Incubation
Incubate fertilized chicken eggs in a sterile, temperature-controlled incubator at 37 °C ± 1 °C and ~40% humidity.
Turn the eggs 1x per day and allow them to incubate to HH34 (8 days post-fertilization).
2. Preparation and Sterilization of the Materials
For 12 embryos, autoclave 2 L of distilled water, 1 L of 0.85% chick saline, 12 glass pipettes, 1 box of paper tissue, 24 2.5 cm x 2.5 cm squares of semi-porous filter paper, and 24 2.5 cm x 2.5 cm squares of steel wire mesh with the edges curved downwards. To sterilize the materials, autoclave them at 121 °C for at least 15 min. NOTE: The autoclaving of the materials can be done in advance.
Sterilize the incubator with 70% ethanol by wiping the sides, shelves, and door. Place 2 plastic containers containing autoclaved distilled water inside the incubator to maintain humidity. Prewarm the culture incubator to 37 °C ± 1 °C and ~40% humidity. Ensure the incubator is completely dark; if the door is transparent, cover it with tinfoil.
Prewarm 100 mL of nutrient medium and 1 mL of penicillin by placing them in the culture incubator.
Cover the workspace with a paper towel and sterilize the bench by spraying it with 70% ethanol. Sterilize 2 pairs of forceps, dissection scissors, a razor blade, and a plastic spoon with 70% ethanol in a similar manner.
Place 12 sterile 100 mm Petri dishes and 24 sterile 35 mm Petri dishes on the bench for use. Ensure the dishes remain in a sterile bag until use.
3. Embryo Preparation
NOTE: From this point onwards, wear a protective dust face mask to avoid contaminating the cultures. A bacterial or fungal infection will ruin the culture and there is a risk of it spreading quickly to all cultures in the incubator.
Crack open the egg and transfer the embryo to a 100 mm Petri dish containing 0.85% chick saline. Stage the embryo according to the anatomical features described in the Hamburger and Hamilton staging guidelines12 and confirm the embryo is at HH34.
Cut the neck and then bisect the head of the embryo at the midline using a sterilized razor blade. Using sterile forceps, remove the brain. Leave the beak intact.
4. Culture Setup
Place 2 of the 35 mm Petri dishes inside one 100 mm Petri dish.
Place a steel wire mesh inside each 35 mm Petri dish.
Using forceps, transfer 1 of the bisected heads to a piece of 2.5 cm x 2.5 cm semi-porous filter paper with the eye facing up. Ensure the tissue is secure and does not slip off by tilting it slightly.
Using forceps, carefully place the eye tissue and the filter paper on top of the steel wire mesh in 1 of the 35 mm Petri dishes, creating a raised stage with the eye tissue on top.
Repeat steps 4.3 and 4.4 with the other half head.
Using a sterile glass pipette, carefully add nutrient medium directly into each 35 mm Petri dish until it reaches the level of the filter paper. Do not submerge the half head in the nutrient medium.
Add 50 µL of 10,000 U/mL penicillin-streptomycin to the nutrient medium in each Petri dish.
Fold a piece of tissue paper into a small square and place it inside the 100 mm Petri dish. Moisten it with autoclaved distilled water.
Place the culture dish into the sterile, dark incubator at 37 °C ± 1 °C and ~40% humidity for up to 4 days.
Repeat steps 3 and 4 for each embryo. NOTE: The number of embryos needed will depend on the specific experiment; here, we describe the protocol for 12 embryos.
5. Culture Maintenance
Once per day, top up the plastic containers in the incubator with fresh, autoclaved distilled water.
Daily check all cultures for bacterial or fungal infections. Cultures that have disintegrated or in which the medium has changed color are infected. Discard all cultures that are infected.
6. Fixation
Following culturing, remove all dishes from the incubator.
Using a pair of forceps, gently remove the eye from the filter paper, taking care not to tear the tissue.
Fix the eye tissue in 4% paraformaldehyde in 1x phosphate-buffered saline overnight at 4 °C or in 10% neutral-buffered formalin overnight at room temperature.
Store the tissue at 4 °C in 1x phosphate-buffered saline.
Representative Results
Using this method, an embryonic chicken eye can be cultured from day 8 of its development (HH34) in vitro for 4 days. Four days of in ovo development corresponds to HH38.
This culturing method supports the development of feather buds surrounding the eye and on the eyelids (Figure 1B). These feather buds are not present in ovo at HH34 prior to the culturing (Figure 1A), indicating that feather bud development is initiated during the culturing period, as it is in ovo13 (Figure 1C). The eyelids and nictitating membrane grow to cover the eye during the culturing (Figure 1B). The eyelids appear more mature after the in vitro culturing than at the start of the culturing at HH34 (Figure 1A) but are less developed than at HH38 (Figure 1C). This data shows that the key aspects of embryonic development are supported, albeit with some small delay.
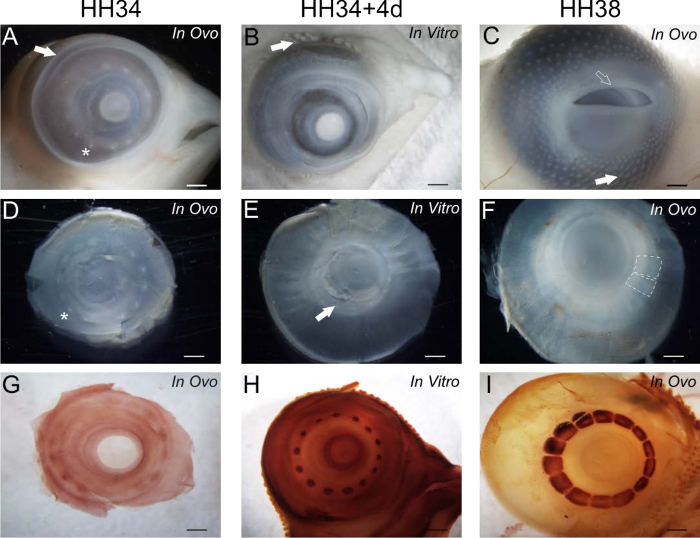
After culturing, we dissected the anterior portion of the eye to show the conjunctival papillae (a transient ring of 13 - 14 epithelial protrusions), which act as yet another landmark indicating that the developmental events are supported in culture. Typically, these papillae are fully formed by HH34 (Figure 1D), and degenerate over time as they induce skeletogenic condensations in the underlying mesenchyme; they disappear by HH381 (Figure 1F). Our data shows that this degeneration occurs in vitro (Figure 1E) and is preceded by a flattening of the papillae following the in ovo pattern of development. Unstained condensations, however, are not visible in the in vitro samples, unlike at HH38 (Figure 1F). We used alkaline phosphatase staining, which detects early osteoblast activity to further assess the induction of these condensations14; this process occurs alongside the degeneration of the conjunctival papillae in ovo. The induction of the skeletogenic condensations, which begins at HH34 (Figure 1G), does proceed in vitro as evidenced by the alkaline phosphatase staining (Figure 1H). These condensations, however, are not as large as they are at HH3814 (Figure 1I), again indicating that there is a small delay in the development. We estimate that the 4 days of culturing represent approximately 2 days of embryonic development; hence a delay of about 50%. Collectively, these results show that the induction and development of the intramembranous bones, the scleral condensations, is supported in vitro and that the developmental processes such as eyelid growth and feather bud formation proceed in vitro.
Figure 1. Morphological characteristics of in vitro cultured eyes, HH34 eyes, and HH38 eyes. A - F are unstained and G - I are stained for the alkaline phosphatase (AP) enzyme to distinguish active osteoblasts within the scleral condensations. A, D, and G show eyes at the start of the culturing period at HH34. B, E, and H show eyes after 4 days of in vitro culturing. C, F, and I show eyes after 4 days of in ovo culturing, at HH38. All eyes are viewed laterally. (A) The conjunctival papillae are visible in a ring surrounding the cornea. An asterisk indicates papilla #12, the first one to form. The arrow indicates the edge of the eyelid in the nasal region of the eye. (B) The conjunctival papillae have flattened and begun to degenerate. The arrow indicates a growing feather bud. (C) The conjunctival papillae have completely degenerated and are covered by eyelids and the nictitating membrane. The open arrow indicates the edge of the eyelid in the nasal region of the eye. The solid arrow indicates a growing feather bud. (D) A dissected anterior portion of the HH34 eye. The asterisk indicates papilla #12. (E) A dissected anterior portion of an HH34+4 in vitro eye showing flattened papillae. The papillae in the temporal region (solid arrow) have begun to degenerate; these are the oldest papillae in the ring. (F) A dissected anterior portion of an HH38 eye with the eyelids and nictitating membranes removed, showing that all papillae have degenerated. The dashed lines outline the position of two adjacent unstained condensations.(G) A HH34 eye stained for AP showing very small skeletogenic condensations indicative of the start of induction. (H) A HH34+4 eye stained for AP showing enlarged, growing skeletogenic condensations. (I) A HH38 eye stained for AP showing large skeletogenic condensations. All scale bars are 1 mm. Please click here to view a larger version of this figure.
Discussion
This protocol makes use of established tissue culturing techniques to achieve the growth of a chicken eye from embryonic day 8 (HH34) in vitro for 4 days. This grid-culturing method was originally described by Trowell15. We optimized a protocol from Pinto and Hall's 1991 study16 utilizing a semi-porous membrane with the grid to study inductive signals between separated tissue layers of the embryonic chicken eye15. Using this method, Roach cultured 14-day old chicken femur6. This method has also proven effective in culturing mouse tissues6,17. The use of the filter paper is critical to the success of the protocol as it helps the tissue to take up nutrients from the medium effectively without saturating or submersing the tissue.
Nutrient medium at a proper pH is critical for optimal growth. The medium should be at or near the physiological pH, and the use of phenol red in the medium as an indicator provides an advantage in that pH changes can be easily detected. Serum-free media can be used, and changing the media daily is not necessary and does not affect the results. The protocol was developed using a commercially available medium (see Table of Materials), which contains high levels of amino acids and glucose; no further supplementation of the media is needed. Most occurrences of poor tissue growth using this protocol occurred when the nutrient media was either at an improper pH or contaminated.
The eye can survive and grow for up to 4 days in vitro, starting at embryonic day 8, when the protocol is performed under optimal conditions. One limitation of this protocol is that the survivability is impacted beyond 4 days of culturing. Thus, the developmental processes that span a longer timeframe will require modifications to the protocol. Another limitation of the protocol is the absence of an active blood flow and vascularization. Blood vessels are important for many developmental processes, not just for the delivery of nutrients to the tissue9, which is achieved in vitro via capillary action through the filter paper on which the tissue rests.
This protocol can be used to study the development of entire organs or large pieces of tissue in vitro. Vertebrate organs are highly complex, and their development is regulated by many distinct spatiotemporal patterns. The study of these developmental pathways relies on the ability to manipulate the signals that regulate them. Current methods limit this manipulation to relatively small, localized areas, and to low enough concentrations to avoid embryonic lethality. For example, the ex ovo method of culturing the entire chicken embryo in culture18 or the method to window eggs19 to conduct manipulations are limited in that either the entire embryo must be treated, which can cause lethality, or only a small localized area is treated. The advantage of the protocol presented here is that the effect of these signals can be studied on entire organs or large amounts of tissue, without affecting the embryonic viability. This protocol does not require any supplementation with carbon dioxide as in earlier protocols15. The delay in development that we observed is an advantage in that rapid developmental processes can be studied.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to thank Gregory Haller (Mount Saint Vincent University) for his preliminary work in the development of the protocol. The authors would also like to thank Nicholas Jones (Mount Saint Vincent University) for his technical expertise and assistance with the filming and production of the audio/visual portion of the manuscript. Daniel Andrews was supported by funding from MSVU and the Natural Sciences and Engineering Research Council of Canada (NSERC) via an Undergraduate Student Research Award. Tamara A. Franz-Odendaal is supported by an NSERC Discovery Grant.
References
- Franz-Odendaal TA. Towards understanding the development of scleral ossicles in the chicken, Gallus gallus. Developmental Dynamics. 2008;237(11):3240–3251. doi: 10.1002/dvdy.21754. [DOI] [PubMed] [Google Scholar]
- Scholey JM. Functions of motor proteins in echinoderm embryos: an argument in support of antibody inhibition experiments. Cell Motility and the Cytoskeleton. 1998;39(4):257–260. doi: 10.1002/(SICI)1097-0169(1998)39:4<257::AID-CM1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Horakova D, et al. Effect of FGFR inhibitors on chicken limb development. Development, Growth & Differentiation. 2014;56(8):555–572. doi: 10.1111/dgd.12156. [DOI] [PubMed] [Google Scholar]
- Gerlach GF, Morales EE, Wingert RA. Microbead Implantation in the Zebrafish Embryo. Journal of Visualized Experiments. 2015. p. e52943. [DOI] [PMC free article] [PubMed]
- Luz-Madrigal A, Grajales-Esquivel E, Del Rio-Tsonis K. Electroporation of Embryonic Chick Eyes. Bio-protocol. 2015;5(12):e1498. doi: 10.21769/bioprotoc.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach HI. Long-term organ culture of embryonic chick femora: a system for investigating bone and cartilage formation at an intermediate level of organization. Journal of Bone and Mineral Research. 1990;5(1):85–100. doi: 10.1002/jbmr.5650050113. [DOI] [PubMed] [Google Scholar]
- Jung HS, et al. Local inhibitory action of BMPs and their relationships with activators in feather formation: implications for periodic patterning. Developmental Biology. 1998;196(1):11–23. doi: 10.1006/dbio.1998.8850. [DOI] [PubMed] [Google Scholar]
- McKinnell IW, Turmaine M, Patel K. Sonic Hedgehog functions by localizing the region of proliferation in early developing feather buds. Developmental Biology. 2004;272(1):76–88. doi: 10.1016/j.ydbio.2004.04.019. [DOI] [PubMed] [Google Scholar]
- Smith EL, Kanczler JM, Oreffo RO. A new take on an old story: chick limb organ culture for skeletal niche development and regenerative medicine evaluation. European Cells & Materials. 2013;26:91–106. doi: 10.22203/ecm.v026a07. [DOI] [PubMed] [Google Scholar]
- Sato T, et al. In Vitro Spermatogenesis in Explanted Adult Mouse Testis Tissues. PLoS One. 2015;10(6):e0130171. doi: 10.1371/journal.pone.0130171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa-Villarreal M, et al. Plant cell culture strategies for the production of natural products. BMB Reports. 2016;49(3):149–158. doi: 10.5483/BMBRep.2016.49.3.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. Journal of Morphology. 1951;88(1):49–92. [PubMed] [Google Scholar]
- Yu M, et al. The developmental biology of feather follicles. The International Journal of Developmental Biology. 2004;48(0):181–191. doi: 10.1387/ijdb.031776my. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabalee J, Hillier S, Franz-Odendaal TA. An investigation of cellular dynamics during the development of intramembranous bones: the scleral ossicles. Journal of Anatomy. 2013;223(4):311–320. doi: 10.1111/joa.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowell OA. The culture of mature organs in a synthetic medium. Experimental Cell Research. 1959;16(1):118–147. doi: 10.1016/0014-4827(59)90201-0. [DOI] [PubMed] [Google Scholar]
- Pinto CB, Hall BK. Toward an understanding of the epithelial requirement for osteogenesis in scleral mesenchyme of the embryonic chick. Journal of Experimental Zoology. 1991;259(1):92–108. doi: 10.1002/jez.1402590112. [DOI] [PubMed] [Google Scholar]
- Mbiene JP, MacCallum DK, Mistretta CM. Organ cultures of embryonic rat tongue support tongue and gustatory papilla morphogenesis in vitro without intact sensory ganglia. Journal of Comparative Neurology. 1997;377(3):324–340. doi: 10.1002/(sici)1096-9861(19970120)377:3<324::aid-cne2>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Cloney K, Franz-Odendaal TA. Optimized ex-ovo culturing of chick embryos to advanced stages of development. Journal of Visualized Experiments. 2015. p. e52129. [DOI] [PMC free article] [PubMed]
- Korn MJ, Cramer KS. Windowing chicken eggs for developmental studies. Journal of Visualized Experiments. 2007. p. e306. [DOI] [PMC free article] [PubMed]