

Abstract
A protocol is presented for generating human induced pluripotent stem cells (hiPSCs) that express endogenous proteins fused to in-frame N- or C-terminal fluorescent tags. The prokaryotic CRISPR/Cas9 system (clustered regularly interspaced short palindromic repeats/CRISPR-associated 9) may be used to introduce large exogenous sequences into genomic loci via homology directed repair (HDR). To achieve the desired knock-in, this protocol employs the ribonucleoprotein (RNP)-based approach where wild type Streptococcus pyogenes Cas9 protein, synthetic 2-part guide RNA (gRNA), and a donor template plasmid are delivered to the cells via electroporation. Putatively edited cells expressing the fluorescently tagged proteins are enriched by fluorescence activated cell sorting (FACS). Clonal lines are then generated and can be analyzed for precise editing outcomes. By introducing the fluorescent tag at the genomic locus of the gene of interest, the resulting subcellular localization and dynamics of the fusion protein can be studied under endogenous regulatory control, a key improvement over conventional overexpression systems. The use of hiPSCs as a model system for gene tagging provides the opportunity to study the tagged proteins in diploid, nontransformed cells. Since hiPSCs can be differentiated into multiple cell types, this approach provides the opportunity to create and study tagged proteins in a variety of isogenic cellular contexts.
Keywords: Genetics, Issue 138, Stem Cells, hiPSC, Molecular Biology, CRISPR, Cas9, Genome Engineering, Gene Knock-in, Gene Tagging, Fluorescent Protein
Introduction
The use of genome-editing strategies, especially CRISPR/Cas9, to study cellular processes is becoming increasingly accessible and valuable1,2,3,4,5,6,7. One of the many applications of CRISPR/Cas9 is the introduction (via homology directed repair (HDR)) of large exogenous sequences such as GFP into specific genomic loci that then serve as reporters for the activity of a gene or protein product8. This technique can be used to join a fluorescent protein sequence to an endogenous open reading frame where the resulting endogenously regulated fusion protein can be used to visualize the subcellular localization and dynamics of the protein of interest5,6,9,10,11. While endogenously tagged proteins offer many benefits compared to overexpression systems, inserting large sequences into the human genome is an inefficient process typically demanding a selection or enrichment strategy to obtain a population of cells that can be easily studied5,12.
This protocol describes the insertion of a DNA sequence encoding a fluorescent protein (FP) into a desired genomic locus. The protocol includes design and delivery of the donor template plasmid, and the ribonucleoprotein (RNP) complex (wild type S. pyogenes Cas9 protein combined with synthetic CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA)). Also described is the enrichment of putatively edited cells via fluorescence activated cell sorting (FACS) and the clonal cell line generation process. To date, this method has been used to generate hiPSC lines with either monoallelic or (rarely) bi-allelic green fluorescent protein (GFP) tags labeling twenty-five proteins representing major cellular structures. The resulting edited cells from these efforts have been confirmed to have the expected genetic insertion, express a correctly localizing fusion protein, and maintain pluripotency and a stable karyotype12 (and unpublished data). This method has also been used to generate multiple other single and dual (two different proteins tagged in the same cell) edited populations of hiPSCs (unpublished data).
Human iPSCs derived from a healthy donor were chosen for these genome-editing efforts because, unlike many conventional cell lines, they are diploid, karyotypically stable, non-transformed, and proliferative. These properties provide an attractive model for studying fundamental cell biology and disease modeling. Furthermore, the differentiation potential of hiPSCs provides the opportunity to study multiple developmental stages in parallel across various lineages and cell types using isogenic cells including organoids, tissues and "disease in a dish" models13,14,15. While this protocol was developed for hiPSCs (WTC line), it may be informative for the development of protocols using other mammalian cell lines.
Protocol
1. In Silico Design of crRNA and Donor Template Plasmid for FP Knock-in
Obtain the annotated reference sequence from NCBI16 or the UCSC Genome Browser17 (e.g., GenBank format) of the gene of interest and import it into a bioinformatics software of choice. If the host genome sequence is known to contain variants relative to the reference, include those now by adjusting the reference sequence in the bioinformatics software (see Discussion).
Locate the desired FP insertion site. For C-terminal tags, the sequence for the FP tag will be introduced between the last base of the last codon and the first base of the stop codon. For N-terminal tagging, the sequence for the FP tag will typically be introduced between the last base of the start codon and the first base of the next codon. In some cases, such as when the start codon is a single codon exon, or where a signal sequence exists near the protein terminus, the desired FP insertion site may be situated in a more 3ʹ position, so long as it continues to be in frame.
Use 50 bp on each side of the desired insertion site as the input sequence for any publicly available crRNA design tool. Once 2-4 crRNA targets are identified near the insertion site, annotate the crRNA binding sites and protospacer-adjacent motif (PAM) sequences (NGG) in the bioinformatics software. These crRNAs will be used to induce double stranded breaks (see Discussion for more guidance on crRNA design). NOTE: Custom crRNA sequences can be submitted for synthesis with a commercial vendor (recommended), or the sequence can be used as a starting point to design a cloning or in vitro synthesis strategy, which is beyond the scope of this protocol (see Discussion).
To initiate the donor template plasmid, use 1 kb of sequence upstream of the desired insertion site as the 5ʹ homology arm (this should include the start codon for N-terminal insertions), and use 1 kb of sequence downstream of the desired insertion site as the 3ʹ homology arm (this should include the stop codon for C-terminal insertions). Bases between the two homology arms are typically not omitted. Including cell-line specific variants in the homology arms will preserve these genetic variants in the resulting edited cells.
Between the two homology arms, insert the sequence for the FP (or other knock-in sequence) and the linker sequence (see Discussion for more guidance on linkers). For N-terminal tags, the linker sequence should be directly 3ʹ of the FP; for C-terminal tags, the linker sequence should be directly 5ʹ of the FP.
Disrupt crRNA binding sites in the donor template plasmid to prevent Cas9 cutting of donor sequence (see Discussion for considerations when altering crRNA binding sites). If possible, disruption of the PAM to a sequence other than NGG or NAG is preferred. Alternatively, introducing point mutations to three bases in the seed region of the crRNA (10 bases proximal to the PAM) is predicted to sufficiently disrupt crRNA binding. Some crRNA binding sites are disrupted by introduction of the FP sequence in the donor template plasmid; ensure that no PAM, or intact binding region still exists in these cases. NOTE: In silico donor template plasmid can be submitted for gene synthesis by a commercial vendor, or it can be used as a starting point to design a cloning strategy, which is beyond the scope of this protocol. A simple backbone such as pUC19 or pUC57 is sufficient.
2. Ribonucleoprotein (RNP) Transfection for CRISPR/Cas9 Mediated Knock-in in hiPSCs
NOTE: In this protocol, the term 'gRNA' describes synthetic crRNA and tracrRNA properly re-suspended, quantified, and pre-complexed per manufacturer's instructions (see Table of Materials). Supplement all media with 1% Penicillin Streptomycin. General culturing guidelines of the WTC hiPSC line are described in more detail at the Allen Cell Explorer18,19. WTC hiPSCs are used in this protocol, but with proper transfection optimization, electroporation of RNP and donor template plasmid may be successfully adapted to other cell types.
Prepare 10 µM working stocks of gRNA and wild type S. pyogenes Cas9 protein2,20; keep on ice. Prepare 1 µg/µL working stock of donor template plasmid; keep at room temperature (RT). Use pH 8.0 TE buffer for all dilutions.
Prepare a matrix-coated 6-well tissue culture plate with 5 mL of fresh growth media supplemented with 10 µM ROCK inhibitor (Ri) per well. Keep plate with media in the incubator at 37 °C and 5% CO2 until ready to plate cells after the transfection procedure (maximum 2 h). NOTE: All matrix-coated plates used in this protocol are made by adding a volume of ice-cold Matrigel diluted 1:30 in cold DMEM/F12 media according to the Allen Institute for Cell Science's protocol for culturing the WTC hiPSC line19.
- Using a gentle single-cell dissociation reagent as recommended in the Table of Materials, passage hiPSCs into single-cell suspension and count cells using an automated cell counter, or hemocytometer. NOTE: A detailed protocol for the WTC hiPSC line used here can be found at the Allen Cell Explorer19. Briefly, wash cells once with RT DPBS and treat with dissociation reagent for 3-5 minutes. Then triturate cells into single-cell suspension by gentle pipetting and pellet by centrifugation. Resuspend the live cell pellet in growth media supplemented with 10 µM Ri.
- Prepare an aliquot of 1.84 x 106 cells for each experimental condition to be transfected in separate 1.5 mL tubes. NOTE: All volumes are calculated for a 4.5 µL total reaction volume in a 100 µL electroporation volume, times 2.3 reactions. This accounts for duplicate transfection and excess for pipetting error.
Prepare ribonucleoprotein (RNP) complex tubes for each experimental condition by adding 2.88 µL of 10 µM gRNA and 2.88 µL of 10 µM Cas9 to a 1.5 mL tube. Incubate at RT for a minimum of 10 min (maximum 1 h).
Pellet one cell aliquot (prepared in step 2.3.1) at 211 x g for 3 min at RT. Aspirate supernatant and resuspend cell pellet in 220 µL of manufacturer's electroporation buffer.
Add 220 µL of resuspended cells from step 2.5 into the 1.5 mL RNP complex tube prepared in step 2.4.
Add 4.60 µL of 1 µg/µL donor template plasmid to the 1.5 mL tube prepared in step 2.4.
Use the nucleofection tip and pipette to mix the tube contents 2-3 times, then transfer 100 µL of suspension to the prepared electroporation device. Avoid introducing any bubbles in the tip. Apply 1300 V for 1 pulse of 30 ms.
Gently transfer the suspension into the prepared 6-well plate from step 2.2 with a swirling motion. Disperse cells by gently moving the plate side-to-side and front-to-back.
- Using a new nucleofection tip, repeat steps 2.8-2.9 with the remaining 100 µL of suspension and transfer into a second well of the prepared 6-well plate.
- Repeat steps 2.4 through 2.10 for each gRNA and donor template plasmid combination, including non-targeting gRNA, donor template plasmid only, and buffer only controls. Take care to change pipette and nucleofection tips to avoid cross-contamination.
Incubate transfected cells at 37 °C and 5% CO2. Change the media to regular growth media (no Ri) at 24 h, and continue feeding hiPSCs every 24 h for 72-96 h, monitoring confluency. When hiPSCs reach 60-80% confluence, proceed to step 3. NOTE: Heavy cell death (>70%, estimated) is normal 24-48 h after transfection.
3. FACS-Enrichment of Putatively Edited hiPSCs
Note: When sorting stem cells, adapt instrument settings to promote cell survival as suggested in the Discussion. Briefly, use the largest nozzle possible (130 µm), a low flow-rate (≤ 24 µL/min), preservative-free sheath fluid (such as saline, see Table of Materials), and low sample pressure (10 psi).
Prior to beginning a FACS experiment, change media to growth media supplemented with 10 µM Ri and incubate cells at 37 °C and 5% CO2 for 2-4 h to promote survival after FACS.
Using a gentle single-cell dissociation reagent as recommended in the Table of Materials, passage hiPSCs into single-cell suspension in growth media supplemented with 10 µM Ri19.
Filter hiPSC suspension through 35 µm mesh filter into polystyrene round-bottomed tubes.
Sort cells using forward scatter and side scatter (including height vs. width) to exclude debris and doublets. Use live, buffer only control cells to set the FP-positive gate, such that <0.1% of buffer only cells fall within the gate.
Sort the entire population of FP-positive cells into a 1.5-15 mL polypropylene tube containing 0.5-2 mL of RT growth media supplemented with 10 µM Ri. NOTE: Polypropylene reduces the potential for cell adhesion to the plastic.
Centrifuge collected cells at 211 x g for 3 min at RT.
Carefully aspirate supernatant and resuspend cell pellet in 200 µL of growth media supplemented with 10 µM Ri. Transfer up to 3,000 sorted cells to a single well of a fresh matrix-coated 96-well plate19. NOTE: With appropriate instrument setup, cells can also be sorted (in bulk) directly into a single well of a matrix-coated 96-well tissue culture plate containing 200 µL of growth media supplemented with 10 µM Ri at a recommended density of 1,000-3,000 cells per well for hiPSC.
- Incubate sorted cells at 37 °C and 5% CO2. Change the media to growth media supplemented with 5 µM Ri at 24 h. At 48 h begin feeding cells regular growth media (no Ri) every 24 h for 72-96 h, monitoring confluency. Survival after FACS is estimated to be greater than 50% if a minimum of 500 cells are seeded in one well of a 96-well plate.
- When hiPSCs reach 60-80% confluence and show mature morphology (smooth, well-packed colony centers), passage into a larger format plate such as a 24-well plate, then from a 24-well plate into a 6-well plate.
- When the hiPSCs in a 6-well plate reach 60-80% confluence and show mature morphology, expand to a 100 mm plate, re-plate for imaging, cryopreserve, or seed at clone picking density (step 4)19.
4. Generating Putatively Edited Clonal hiPSC Lines
Using a gentle single-cell dissociation reagent as recommended in the Table of Materials, passage hiPSCs into single-cell suspension and determine the number of cells per mL19.
Seed 10,000 cells of the edited population of hiPSCs onto a fresh matrix-coated 100 mm tissue culture dish using growth media supplemented with 10 µM Ri19. Change the media to growth media without Ri 24 h after seeding, and feed the hiPSCs with fresh growth media every 24 h for 5-7 days.
When hiPSCs have formed colonies that are visible macroscopically (approximately 500 µm) they are large enough to be isolated. Prepare a matrix-coated 96-well plate by aspirating excess matrix and adding 100 µL of growth media supplemented with 10 µM Ri per well19.
- On a dissecting microscope use a P-200 pipette, or similar, to gently scrape and aspirate individual colonies from the plate surface. Transfer volume (~20-100 µL) containing the colony to a single well of the 96-well plate prepared in step 4.3.
- After all colonies have been transferred, incubate the plate in a tissue culture incubator at 37˚C and 5% CO2. Change the media to regular growth media (no Ri) at 24 h, and continue feeding cells every 24 h for 72-96 h until colonies have approximately tripled in size (approximately 1500 µm). NOTE: Picking 24-96 colonies per crRNA used in the transfection is recommended. Survival of isolated clones is typically greater than 95%.
- Using a gentle single-cell dissociation reagent as recommended in the Table of Materials, passage hiPSC clones into a new matrix-coated 96-well plate as follows19.
- Using an 8-channel aspirator, remove and discard media from the first column of the 96-well plate.
- Using a P-200 multichannel pipette, add ~200 µL of DPBS to the first column of the 96-well plate to wash the cells. Using an 8-channel aspirator, remove and discard DPBS wash from the first column of the 96-well plate.
- Using a P-200 multichannel pipette, add 40 µL of dissociation reagent to the first column of the 96-well plate.
- Repeat steps 4.5.1-4.5.3 for up to a total of six columns of the 96-well plate, changing tips to be sure to not cross-contaminate wells. Place the plate in the 37 °C incubator for 3-5 minutes from the time the dissociation reagent was added to the first column (step 4.5.3). NOTE: It is recommended to only passage a maximum of six columns (48 wells) at a time due to the time it takes to perform these steps. When performing this protocol for the first time, start with only passaging one or two columns at a time. Limiting the number of columns passaged at one time ensures that cells are not left in the dissociation reagent for too long, which could be harmful to hiPSCs.
- When the cells in the first column of the plate have begun to lift off the plate bottom, use a P-200 multichannel pipette to add 160 µL of DPBS to the first column of the 96-well plate and gently triturate the cells at the "12:00", "3:00", "6:00", and "9:00" positions of each well. Transfer the entire volume of cell suspension (200 µL) to a V-bottom 96-well plate.
- Repeat step 4.5.5 for the remaining columns of cells that have dissociation reagent in them; change tips as to not cross-contaminate wells.
- Spin the V-bottom plate in a centrifuge at 385 x g for 3 min at RT.
- Using a P-200 multichannel pipette, gently remove the supernatant and resuspend cells in 200 µL of fresh growth media supplemented with 10 µM Ri per well. Repeat for all wells, changing tips as to not cross-contaminate.
- Transfer all of the cell suspension to a fresh matrix-coated 96-well plate19. Incubate the plate in a tissue culture incubator at 37 °C and 5% CO2. Change the media to regular growth media (no Ri) at 24 h, and continue feeding cells every 24 h for 72-96 h, until the majority of clones reach 60-80% confluence. NOTE: This passage helps to spread out the cells and allow for more growth over the entire area of the 96-well plate.
- Observe clones and identify an appropriate split ratio for each individual clone in the 96-well plate (e.g., 1:10, or 1:8). Using a gentle single-cell dissociation reagent as suggested in the Table of Materials, passage hiPSC clones into a new matrix-coated 96-well plate (steps 4.5.1-4.5.8) transferring a ratio of the cell suspension appropriate for each clone. Incubate the plate in a tissue culture incubator at 37˚C and 5% CO2. Change the media to regular growth media (no Ri) at 24 h, and continue feeding cells every 24 h for 72-96 h, until the majority of clones reach 60-80% confluence, and show mature morphology. NOTE: Each clone may have a different split ratio because of slightly different growth rate or survival from the previous passage, so this passage helps to normalize the number of cells per well of each clone for the freezing step to follow. Due to the varying rates of survival and growth, some clones may overgrow or fail to grow during these passaging steps.
- Save the remainder of the cell suspension and pellet for gDNA isolation by centrifuging cells in a V-bottom 96-well plate at 385 x g for 3 min at RT. Remove supernatant and proceed to gDNA isolation using a 96-well kit, or store plate of pelleted cells at -20˚C for up to three weeks.
5. Cryopreservation of clonal cell lines in 96-well plate format
Using a single-cell dissociation reagent, passage hiPSC clones as previously described (steps 4.5.1-4.5.7). Aspirate the supernatant using a P-200 multichannel pipette and re-suspend in 60 µL of growth media supplemented with 10 µM Ri. Repeat for all wells; change tips as to not cross-contaminate.
Transfer 30 µL of cell suspension to a non-matrix coated 96-well tissue culture plate. Then quickly add 170 µL freezing buffer (see Table of Materials) to each well, without mixing. Repeat by transferring the remaining 30 µL of suspension into a sister plate and adding 170 µL freezing buffer. NOTE: This process is done in duplicate sister plates so that a back-up population of cells exists after thawing one of the individual plates. Putting cryopreserved cells into only every other column of a 96-well plate allows for faster thawing (step 5.6).
Wrap plate with parafilm and place in a RT Styrofoam box with lid. Place the whole box in a -80 °C freezer.
After 24 h plates can be transferred out of the Styrofoam box and stored at -80˚C for up to four weeks. NOTE: While the cells are temporarily stored at -80 °C, genetic quality control assays can be performed with the gDNA harvested from cells obtained in step 4.6.1 in order to identify the clones to thaw and propagate further, as discussed in previously published work12. Briefly, a copy number droplet digital PCR assay can be used to identify clones that contain one or two copies of GFP and no donor template plasmid backbone integration. A combination of end-point PCR assays and Sanger sequencing can then identify clones that contain a precise insert.
- To thaw, bring the entire plate to 37 °C in a tissue culture incubator, watching carefully for the ice pellets to melt. Wells at the edge of the plate tend to thaw first.
- When the ice pellet of desired clone melts, gently transfer entire 200 µL to a 15 mL conical tube containing 3 mL of RT growth media supplemented with 10 µM Ri and centrifuge at 211 x g for 3 min at RT.
- Aspirate supernatant and resuspend pelleted cells in 1 mL of RT growth media supplemented with 10 µM Ri. Transfer to a fresh matrix-coated 24-well plate and incubate at 37˚C and 5% CO2. Change the media to regular growth media (no Ri) at 24 h, and continue feeding cells every 24 h for 72-96 h until the clone reaches 60-80% confluency and has mature morphology19.
Representative Results
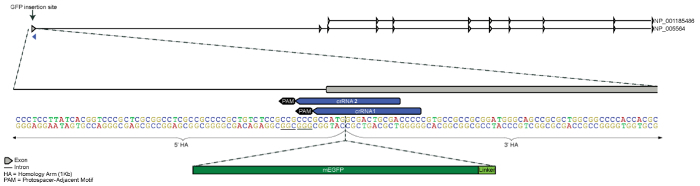
The goal of this experiment was to fuse mEGFP (monomeric enhanced GFP) to the nuclear lamin B1 protein by introducing the mEGFP sequence to the 5ʹ end of the LMNB1 gene (N-terminus of the protein). A linker (amino acid sequence SGLRSRAQAS) was chosen based on previous cDNA constructs from the Michael Davidson Fluorescent Protein Collection21. Because the crRNA binding region in the donor template plasmid for each candidate crRNA was disrupted after the in silico insertion of mEGFP and the linker sequence, no point mutations needed to be made to disrupt potential crRNA recognition and cleavage by Cas9 of the donor sequence (Figure 1). The donor sequence contained 1 kb homology arms flanking both ends of the mEGFP-linker sequence. The resulting 2,734 bp of DNA was cloned into a pUC57 backbone, sequence verified, and the resulting donor template plasmid was purified using an endotoxin-free maxi prep. The donor template plasmid and RNP complex were transfected, putatively edited cells were enriched, and the localization of the mEGFP-nuclear lamin B1 fusion protein was confirmed by fluorescence microscopy (Figure 2). Only results from the crRNA1 transfection are described here, although both crRNA sequences produced putatively edited populations12.
When compared to the negative control, which contained no gRNA, Cas9 protein, or donor template plasmid in the electroporation reaction (buffer only), the LMNB1 crRNA1 transfected cells contained 0.95% mEGFP-positive cells representing the putatively edited mEGFP-nuclear lamin B1 population (Figure 3a). This result was within the range of knock-in efficiencies observed across many genomic loci using this method, as previously reported12.
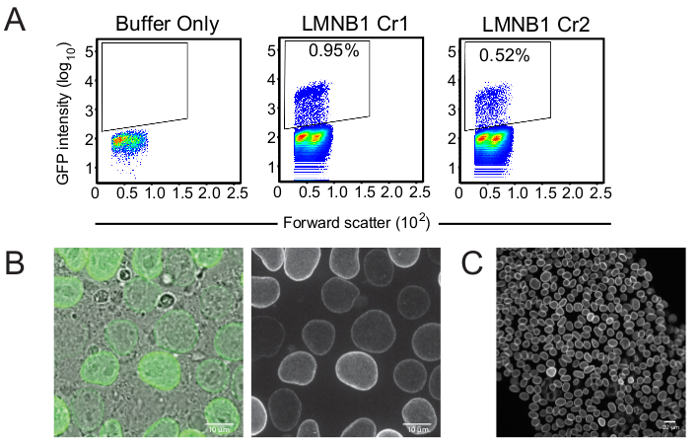
The mEGFP-positive cells were isolated by FACS and imaged by live microscopy to confirm expected localization of the mEGFP-nuclear lamin B1 fusion protein to the nuclear envelope. After FACS-enrichment, approximately 90% of sorted cells from the LMNB1 crRNA1 population were mEGFP-positive (as determined by microscopy), suggesting that some mEGFP-negative cells co-purified with the GFP-positive cells during the sorting procedure. This was an acceptable level of enrichment that allowed for picking of 96 clones that could then be genetically screened for successful editing. Generally, a cut-off for successful enrichment is 50% GFP positive.
The majority of the sorted cell population displayed fluorescence at the nuclear envelope (nuclear periphery) in nondividing cells and to an extended nuclear lamina within the cytoplasm during mitosis providing confidence in the correct genomic edit at the LMNB1 locus. The enriched population contained cells with either bright or dim signal. This difference in signal intensity may reflect a combination of correct and incorrect editing outcomes and highlights the utility of generating a genetically validated clonal line for further studies (see Discussion) (Figure 3b)12. After clonal line generation, genetically validated cells showed uniform GFP intensity in microscopy experiments (Figure 3c).
Figure 1. Design strategy for N-terminus GFP tagging of LMNB1 gene. The GFP tag was designed for N-terminal insertion 5ʹ of the first exon of LMNB1 located on chromosome 5. Both 5ʹ and 3ʹ homology arms are 1 kb each and meet between the start codon (ATG) and the second codon (homology arms only partially represented in figure). Two candidate crRNAs were designed to guide Cas9 to cleave as close to the intended insertion site as possible, while still being unique in the genome. Sequence for mEGFP and an amino acid linker were inserted just 3ʹ of the start codon (mEGFP and linker sequence not to scale). Please click here to view a larger version of this figure.
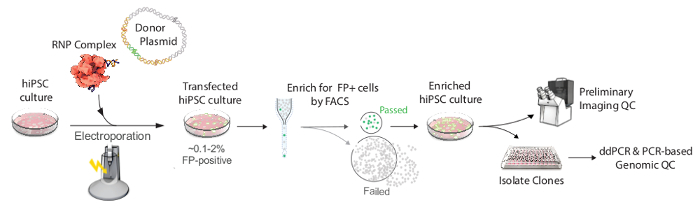
Figure 2. Workflow for producing endogenously tagged hiPSC clonal lines. Transfection components, including the Cas9/crRNA/tracrRNA RNP complex (shown as red Cas9 protein with gold crRNA and purple tracrRNA), the donor template plasmid containing the homology arms (HAs, shown in gold), and FP+linker sequence (shown in green), were electroporated. After 4 days, the FP-positive putatively edited cells were enriched by FACS and expanded as a population by seeding all of the sorted cells into a single well of a 96-well plate (~1,000 cells), and then expanded in culture until a working population of several million cells could be assayed as the "enriched population" (see Protocol step 3.8). The yield of FP-positive cells differs by experiment due to variable rates of HDR12; a successful enrichment may typically include ~300-5,000 cells after transfection of approximately 1.6 x 106 hiPS cells. Preliminary imaging studies confirmed the signal and localization of the fusion protein in the enriched population. Colonies were manually picked into a 96-well plate for expansion and cryopreservation. Further genomic quality control screening using droplet digital PCR (ddPCR) and other PCR-based assays was then used to identify properly edited clones, as previously described12. Please click here to view a larger version of this figure.
Figure 3. Enrichment of putatively edited cell populations. (A) Flow cytometry plots of the LMNB1 edited cells four days post-transfection. The y-axis displays GFP intensity and the x-axis displays forward scatter. Sorting gates were set based on the buffer-only control. Since hiPSCs are sensitive to perturbation, live/dead stain was omitted and a very conservative FSC/SSC gate was used instead. (B) After enrichment, the population of LMNB1 Cr1 edited cells showed ~90% of cells with GFP localizing to the nuclear envelope (expected LMNB1 localization). The population contained cells of varying GFP intensity as well as some GFP-negative cells. Scale bars are 10 microns. (C) After clonal line generation, cells showed a uniform GFP intensity, with some cell-cycle dependent differences. Scale bar is 20 microns.
Discussion
The method presented here for generating endogenously regulated fluorescent protein fusions in hiPSCs is a versatile and powerful approach for generating gene edited cell lines with applications ranging from live cell imaging to various functional studies and "disease in a dish" models using patient-derived hiPSC lines13,14,15. While this method has been used to introduce large FP tags to the N- or C-terminus of endogenous proteins, it could potentially be used to introduce other tags or small genetic changes to model or correct disease-causing mutations22,23. For smaller inserts, the size of the homology arm may be reduced, but the general approach to editing presented in this method may still apply24,25. While the use of hiPSCs is strongly encouraged for their vast utility, with careful optimization, this protocol may be adapted to edit the genomes of other mammalian cell lines.
When identifying a gene of interest for FP tagging, transcript abundance estimates (from microarray or RNA-Seq data) are a good starting point for assessing whether a gene or isoform of interest is expressed, although transcript levels do not always correlate with protein levels. The FACS-enrichment strategy described here will work best for genes that are at least moderately well expressed in the cell type of interest. This strategy has also been successful in selecting for fusions that show punctate and/or discrete localization patterns such as centrin, desmoplakin, and paxillin where the signal to background ratio is very low12,19. Genes that are not highly expressed or are only expressed in derivative cell types may require additional selection strategies.
The starting point for crRNA and donor template plasmid designs used in human cell lines should be the human reference genome (GRCh38). Because the genomes of different cell lines can vary within the same species, and because CRISPR/Cas9 is sequence-specific, it is extremely helpful to identify cell line-specific variants (single nucleotide polymorphisms or insertions/deletions (indels)) that differ from the reference genome and incorporate these into the design. This ensures that crRNAs will be compatible with the host genome and that the donor template plasmid homology arms will retain any cell-line specific variants. A suggested strategy is to incorporate homozygous variants into crRNAs and donor template plasmid homology arms during the design process. Incorporating heterozygous variants is optional. The specific reagents used for large knock-in experiments and other key considerations for this protocol are discussed below.
Cas9 Protein
The primary benefit of using Cas9 protein is that introducing the Cas9 and gRNA as an RNP complex has been shown to result in a limited duration of nuclease activity compared to plasmid-based approaches where expression of the Cas9 and gRNA may continue for days and lead to greater on- and off-target activity26,27. An additional benefit of using Cas9 protein is that it is readily available to cleave once inside the cells. This contrasts with more conventional methods of using Cas9 mRNA or Cas9/gRNA plasmid that require transcription, translation and protein processing26,28. Wildtype S. pyogenes Cas9 protein is now available from many commercial sources.
Guide RNA
There are many publicly available tools for finding crRNA targets near the desired FP insertion site that have zero or few predicted off-targets in the host genome29,30,31,32. Efficiencies in HDR and the precision of the HDR outcome vary widely between crRNA targets used at a given locus12. For this reason, testing several crRNAs (2-4 and preferably within 50 bp of the desired insertion site) per locus is recommended as this may increase the probability of a successful editing experiment. Current possibilities for delivering gRNA include synthetic 2-part crRNA and tracrRNA, synthetic single gRNAs (sgRNAs), in vitro transcribed sgRNAs, or delivering a plasmid to cells expressing the sgRNA from a U6 promoter. This protocol was not optimized for high cleavage activity. Unmodified 2-part crRNA and tracrRNA (see Table of Materials) were used with the goal of generating mono-allelic FP-tagged cell lines while causing the least potential perturbation to the cells.
Donor Template Plasmid
Because some of the homology arm sequence provided in the donor template plasmid will be incorporated into the host genome during the HDR event, point mutations to the crRNA recognition sites should be introduced to prevent further cleavage by Cas9 following HDR. Often the simplest disruptive change is to mutate the PAM sequence. Because some non-canonical PAM sequences can still be recognized by wild type S. pyogenes Cas9, it is best to avoid using NGG, NAG or NGA33. When mutating the homology arm, avoid non-synonymous mutations and the introduction of rare codons. If a synonymous change to the PAM sequence is not possible, consider making three synonymous point mutations in the seed region (10 bp proximal to PAM) of the crRNA binding site. Extreme care should be taken when making these changes in the 5ʹ untranslated region (UTR) since these regions may contain important regulatory sequences. Consulting a genetic conservation database such as the UCSC Genome Browser's Comparative Genomics tracks can provide guidance in these cases, as changes to non-conserved bases may be better tolerated than changes to highly conserved bases17. Sometimes the mere insertion of the FP sequence is enough to disrupt the crRNA binding site (as in Figure 1); however, the newly appended sequence should be checked for the persistence of crRNA binding and PAM sequences.
Amino acid linkers between the FP and the native protein are recommended to conserve the function of the fusion protein34. Often an amino acid linker may be chosen for its particular charge or size. If a cDNA fusion with a design similar to the targeted endogenous fusion protein has been well studied, that same linker sequence can be used for the CRISPR/Cas9 knock-in experiment12,19. If such information is unavailable, a short linker such as GTSGGS has also been used successfully12. Other studies have demonstrated success with a generic small 3-amino acid linker sequence for a variety of targets35.
Transfection and FACS Enrichment
Many commercially available transfection reagents are formulated for delivery of certain types of molecules to cells, whereas an electroporation system can be used to deliver reagents with a wide range of size, charge, and composition. In addition to being a common transfection method for hard-to-transfect cells like hiPSCs, electroporation also carries the benefit of delivering all three components for CRISPR/Cas9-mediated FP knock-in as described in this method. Electroporation was found to produce the best results when compared to other commercially available reagents when developing this method (data not shown), and has also been used by others for RNP delivery26,28,36.
When using this protocol for editing hiPSCs, special care should be taken to ensure gentle handling of the cells before and after the gene editing process for optimal cell survival and minimal spontaneous differentiation. In particular, the FACS enrichment methods should be adapted for sorting stem cells by using the largest nozzle possible (130 µm), a low flow-rate (≤24 µL/min), preservative-free sheath fluid (such as saline, see Table of Materials), and low sample pressure (10 psi). Instead of single cell sorting, which results in suboptimal viability in stem cells, the FACS-enriched hiPSCs are sorted in bulk and expanded as a population to optimize cell viability and stem cell integrity. However, single cell sorting may be appropriate for less sensitive cell types. To promote cell survival, cells are returned to culture no longer than one hour after harvesting for the FACS enrichment and kept at room temperature throughout the sorting process. For some cell types, cell survival may also be enhanced by incubating cells on ice (4°C) throughout the sorting process.
The bulk expansion of FP-positive cells provides an opportunity to evaluate the population by imaging analysis for fusion protein localization prior to generating clonal lines. While the resulting enriched population of cells may be sufficient for some studies, these populations frequently display FP signal of varying intensity. The isolated clonal lines have uniform signal (Figure 3), making them more appropriate for functional experiments12.
Clonal Cell Line Generation
Throughout the editing and clonal line generation process, it is important to monitor cell morphology. hiPSC colonies grown in feeder-free conditions should exhibit smooth edges and an even, well-packed center12,18,19. Differentiated cells should be observed in less than 5% of the culture. When picking individual colonies, choose those that exhibit good morphology. During the 96-well plate passaging events, check clones for morphology and discontinue those that have overgrown as this may lead to differentiation or be an indication of genetic instability.
Generation of clonal cell lines allows for genetic confirmation of precise editing, which is important because Cas9-induced double strand breaks in the genome are often repaired imprecisely despite incorporation of the tag at the desired locus. Previously described PCR-based assays showed that cumulatively across ten unique genomic loci many (45%) of the FP-expressing clones suffered from donor plasmid backbone integration at the targeted locus or (rarely) randomly in the genome12. Additionally, 23% of GFP-positive clones (n=177) across ten unique loci were found to harbor mutations at or near the anticipated crRNA cutting site in the untagged allele, most likely due to NHEJ12. This genetic analysis of many clonal lines (~100 clones/edit) underscored the importance of genetic validation that is not possible in a cell population since FP-expression and expected fusion protein localization alone do not guarantee precise editing12. Additionally, these PCR-based assays cannot be performed on an enriched population of cells with any certainty, warranting the need for clonal line generation before meaningful analysis can be completed. Genetic confirmation of the inserted FP tag and verification of the genetic integrity of the unedited allele (in a mono-allelic edited clone) are both necessary to ensure precise editing at the targeted locus beyond tag expression.
A low rate of bi-allelic edits and lack of off-target mutations (as assayed by Sanger and exome sequencing) have been observed to date using this method (unpublished data)12. This is consistent with previous studies describing the use of short-lasting RNP for CRISPR/Cas9 experiments26,27. The lack of clonal cell lines with bi-allelic edits may also be locus specific or due to the inability of the cell to tolerate two tagged copies of an essential protein as suggested from previously published experiments where putative bi-allelic edited cells were observed for one locus (LMNB1), but not another (TUBA1B)12. Bi-allelic fully validated clonal cell lines have been generated using this method to tag ST6 beta-galactoside alpha-2, 6-sialyltransferase 1 (ST6GAL1), and RAB5A member RAS oncogene family (RAB5A) with mEGFP19.
Beyond confirming precision of the edit in the genome, there are a variety of quality control assays that can be used to further characterize the clonal line and identify clones that fulfill all stem cell, genomic, and cell biological criteria for use in future studies. Cell biological and functional assays can be used to confirm appropriate expression, localization, and function of the fusion protein12. The comparison to unedited parental controls will help evaluate the influence of the editing process on localization, dynamics, and function. Other assays such as growth analysis and tests for genomic stability can also help determine if the tagged protein is perturbing to the cell. When using hiPSC in this protocol, evaluation of pluripotency markers and differentiation potential can be critical in determining a clone that is valuable for downstream studies12. Because extended culture of hiPSC has been shown to lead to genetic instability, monitoring the growth rate and karyotype of clonal cell lines is also important12,37. However, the final intended use of the edited cells will ultimately determine the level and breadth of quality control analysis and will vary based on the application.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Daphne Dambournet for many insightful discussions and advice on gene editing, Thao Do for illustration, Angelique Nelson for critical reading of the manuscript, and Andrew Tucker for generating the mEGFP-tagged Lamin B1 cell line. We wish to acknowledge the Stem Cells and Gene Editing and Assay Development teams at the Allen Institute for Cell Science for their contributions to the gene editing and quality control process. The WTC line that we used to create our gene-edited cell line was provided by the Bruce R. Conklin Laboratory at the Gladstone Institutes and UCSF. We thank the Allen Institute for Cell Science founder, Paul G. Allen, for his vision, encouragement, and support.
References
- Wood AJ, et al. Targeted genome editing across species using ZFNs and TALENs. Science. 2011;333(6040):307. doi: 10.1126/science.1207773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dambournet D, Hong SH, Grassart A, Drubin DG. Tagging endogenous loci for live-cell fluorescence imaging and molecule counting using ZFNs, TALENs, and Cas9. Methods in Enzymology. 2014. pp. 139–160. [DOI] [PubMed]
- Ratz M, Testa I, Hell SW, Jakobs S. CRISPR/Cas9-mediated endogenous protein tagging for RESOLFT super-resolution microscopy of living human cells. Sci Rep. 2015;5:9592. doi: 10.1038/srep09592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks WT, Warren CR, Cowan CA. Genome Editing in Human Pluripotent Stem Cells: Approaches, Pitfalls, and Solutions. Cell Stem Cell. 2016;18(1):53–65. doi: 10.1016/j.stem.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Jaenisch R. Induced Pluripotent Stem Cells Meet Genome Editing. Cell Stem Cell. 2016;18(5):573–586. doi: 10.1016/j.stem.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon JB, et al. Rapid and efficient clathrin-mediated endocytosis revealed in genome-edited mammalian cells. Nature Cell Biology. 2011;13(3):331–337. doi: 10.1038/ncb2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho WK, et al. Super-resolution imaging of fluorescently labeled, endogenous RNA Polymerase II in living cells with CRISPR/Cas9-mediated gene editing. Sci Rep. 2016;6:35949. doi: 10.1038/srep35949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CW, Vanyai HK, See HB, Johnstone EKM, Pfleger KDG. Using nanoBRET and CRISPR/Cas9 to monitor proximity to a genome-edited protein in real-time. Sci Rep. 2017;7(1):3187. doi: 10.1038/s41598-017-03486-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts B, et al. Systematic gene tagging using CRISPR/Cas9 in human stem cells to illuminate cell organization. Mol Biol Cell. 2017;28(21):2854–2874. doi: 10.1091/mbc.E17-03-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146(2):318–331. doi: 10.1016/j.cell.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JE, Goldstein LS. Alzheimer's disease in a dish: promises and challenges of human stem cell models. Human Molecular Genetics. 2012;21(R1):R82–R89. doi: 10.1093/hmg/dds319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares FA, Sheldon M, Rao M, Mummery C, Vallier L. International coordination of large-scale human induced pluripotent stem cell initiatives: Wellcome Trust and ISSCR workshops white paper. Stem Cell Reports. 2014;3(6):931–939. doi: 10.1016/j.stemcr.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene, NCBI- National Center for Biotechnology Information. U.S. National Library of Medicine; 2017. https://www.ncbi.nlm.nih.gov/gene. [Google Scholar]
- Gene, NCBI- National Center for Biotechnology Information. UCSC Genome Browser; 2017. https://genome.ucsc.edu/ [Google Scholar]
- Allen Cell Methods: Single cell passaging human iPS cells. 2018. https://www.youtube.com/watch?v=wao2UcMFPMc.
- Allen Cell Explorer. 2017. http://www.allencell.org/
- Lingeman E, Jeans C, Corn JE. Production of Purified CasRNPs for Efficacious Genome Editing. Curr Protoc Mol Biol. 2017;120:31–31. doi: 10.1002/cpmb.43. [DOI] [PubMed] [Google Scholar]
- Michael Davidson Fluorescent Protein Collection. 2017. https://www.addgene.org/fluorescent-proteins/davidson/
- Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nature Medicine. 2015;21(2):121–131. doi: 10.1038/nm.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas SA, Dettmer V, Cathomen T. Therapeutic genome editing with engineered nucleases. Hamostaseologie. 2017;37(1):45–52. doi: 10.5482/HAMO-16-09-0035. [DOI] [PubMed] [Google Scholar]
- Beumer KJ, Trautman JK, Mukherjee K, Carroll D. Donor DNA Utilization During Gene Targeting with Zinc-Finger Nucleases. G3 (Bethesda) 2013;3(4):657–664. doi: 10.1534/g3.112.005439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando SJ, et al. Zinc-finger nuclease-driven targeted integration into mammalian genomes using donors with limited chromosomal homology. Nucleic Acids Research. 2010;38(15):e152. doi: 10.1093/nar/gkq512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt MA, Corn JE, Carroll D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. 2017;121-122:9–15. doi: 10.1016/j.ymeth.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 2014;24(6):1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 2014;3:e04766. doi: 10.7554/eLife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labun K, Montague TG, Gagnon JA, Thyme SB, Valen E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Research. 2016;44(W1):W272–W276. doi: 10.1093/nar/gkw398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montague TG, Cruz JM, Gagnon JA, Church GM, Valen E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Research. 2014;42(Web Server issue):W401–W407. doi: 10.1093/nar/gku410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Park J, Kim JS. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30(10):1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRISPOR V4.3. 2017. http://crispor.tefor.net/
- Zhang Y, et al. Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci Rep. 2014;4:5405. doi: 10.1038/srep05405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zaro JL, Shen WC. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65(10):1357–1369. doi: 10.1016/j.addr.2012.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonetti MD, Sekine S, Kamiyama D, Weissman JS, Huang B. A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc Natl Acad Sci U S A. 2016;113(25):E3501–E3508. doi: 10.1073/pnas.1606731113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. Journal of Biotechnology. 2015;208:44–53. doi: 10.1016/j.jbiotec.2015.04.024. [DOI] [PubMed] [Google Scholar]
- Baker D, et al. Detecting Genetic Mosaicism in Cultures of Human Pluripotent Stem Cells. Stem Cell Reports. 2016;7(5):998–1012. doi: 10.1016/j.stemcr.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]