

Abstract
Studies in dynamic changes in protein translation require specialized methods. Here we examined changes in newly-synthesized proteins in response to ischemia and reperfusion using the isolated perfused mouse heart coupled with polysome profiling. To further understand the dynamic changes in protein translation, we characterized the mRNAs that were loaded with cytosolic ribosomes (polyribosomes or polysomes) and also recovered mitochondrial polysomes and compared mRNA and protein distribution in the high-efficiency fractions (numerous ribosomes attached to mRNA), low-efficiency (fewer ribosomes attached) which also included mitochondrial polysomes, and the non-translating fractions. miRNAs can also associate with mRNAs that are being translated, thereby reducing the efficiency of translation, we examined the distribution of miRNAs across the fractions. The distribution of mRNAs, miRNAs, and proteins was examined under basal perfused conditions, at the end of 30 min of global no-flow ischemia, and after 30 min of reperfusion. Here we present the methods used to accomplish this analysis—in particular, the approach to optimization of protein extraction from the sucrose gradient, as this has not been described before—and provide some representative results.
Keywords: Biochemistry, Issue 138, Ischemia and reperfusion, dynamic proteome, polysome profiling, miRNA, mRNA, newly-synthesized proteins, protein translation, sucrose density gradient sedimentation, isolated mouse heart perfusion, Langendorff perfusion, proteomics.
Introduction
The heart responds to the injury of ischemia (I) and reperfusion (R) in a dynamic fashion. However, there is little insight into acute changes in protein synthesis during the response. To address this, we took advantage of the well-established method of polysome profiling1 to identify changes in protein abundance that reflect redistribution of ribosomes and translational regulatory factors from cytosol to polysomes, and the increase in newly synthesized proteins (NSPs). In the setting of I/R, the increase in new protein synthesis occurs in a time frame that is inconsistent with transcription of new mRNAs2; moreover, discordance between mRNA expression levels and protein abundance has been reported3. For these reasons, we chose to analyze the changes in the dynamic proteome as reflected by protein translation. To do this, we quantify mRNA in the polysome fractions, and analyze the protein composition in the polysome fractions. Finally, because microRNAs (miRs) regulate availability of mRNAs for translation and can interfere with efficiency of protein translation4,5, we examined the distribution of miRs in the polysome fractions, focusing on the response to I/R.
We chose to use the isolated mouse Langendorff perfusion model and harvested tissue under basal conditions of continuous perfusion, after 30 min global no-flow ischemia, and after 30 min of ischemia followed by 30 min of reperfusion. We then solubilized the heart tissue and separated polysomes over a sucrose gradient, followed by proteomic analysis and selective detection of mRNAs and miRNAs by PCR and microarray, respectively. This combination of methods represents a powerful approach to understanding the dynamic proteome, enabling simultaneous detection of mRNA, miRNA, and NSPs, as well as the redistribution of regulatory proteins, miRNA, and mRNA between nontranslating fractions, low-efficiency polysomes, and high-efficiency polysomes (see Figure 1). Insights into the dynamic regulation of this process will be extended by further analysis of phosphorylation of key regulatory factors such as eIF2α or mTOR. These individual steps are now described in detail.
Protocol
All animal studies were performed in accordance with institutional guidelines and approved by the Institutional Animal Care and Use Committee of Cedars-Sinai Medical Center.
1. Langendorff Perfusion of Mouse Heart
- Langendorff perfusion of mouse heart with ischemia and reperfusion
- Administer intraperitoneal pentobarbital sodium 70 mg/kg to the adult mouse (8-week-old, male, C57BL6/j). Confirm deep anesthesia by lack of withdrawal to toe pinch.
- Anticoagulate with intraperitoneal heparin 500 U/kg.
- Open the chest via sternal incision. Open the diaphragm and cut along the costal border to the anterior axillary axis. Then, cut the anterior axillary axis up to the forelimb to open completely the thoracic cage. After, visualizing the heart, clamp the ascending aorta with forceps and rapidly excise the heart. The death is due to exsanguination. Place the heart in cold Krebs (NaCl 6.9 g/L, KCl 0.35 g/L, NaHCO3 2.1 g/L, KH2PO4 0.16 g/L, MgSO4 0.141 g/L, glucose 2 g/L and CaCl2 0.373 g/L).
- Cannulate the aorta with a blunt-tip needle and perfuse the heart retrogradely with Krebs solution in constant pressure mode.
- After a stabilization period of 15 min, subject the heart to a global no-flow ischemia for 30 min followed by reperfusion for 30 min.
- Harvest hearts after 15 min baseline perfusion, after 30 min ischemia, or after 30 min reperfusion. Trim off the atria, and snap-freeze the tissue in liquid nitrogen.
- Store at -80 °C until use
2. Tissue Homogenization, Solubilization, and Sucrose Density Gradient Sedimentation
- Gradient preparation
- Prepare 2 solutions (15% and 50%) of 100 mL sucrose, mixing 4 mL of NaCl 2.5 M; 0.8 mL MgCl2 1.25 M; 1 mL Tris-HCl 1 M, pH 7.5; 15 g or 50 g of sucrose (for 15% and 50%, respectively); ultrapure water to reach 100 mL; and xylene cyanol (0.02 mg/mL) to color the 15% solution
- Filter each solution (0.22 µm filters)
- The day the gradient is to be used, prepare 40 mL of each sucrose solution and add cycloheximide to each solution to achieve final concentration of 100 µg/mL
- Use a gradient maker to prepare a mix between the 15% and 50% sucrose solutions
- Fill the left compartment with 5 mL of the 15% sucrose gradient
- Fill the right compartment with the 50% sucrose gradient
- Open the tap and switch on the pump to stir the two small compartments with magnetic stirrer
- Use a tube linked to the second tap to allow the mixed sucrose solution to flow in an ultra-centrifugation tube with thin walls. Final volume will be around 10 mL.
- Keep the gradients at 4 °C (overnight if needed, but not longer)
- Homogenization of heart tissue
- Homogenize fresh or frozen heart with a polytron in filtered lysis buffer (modified for sucrose gradient fractionation) containing 100 mM KCl, 20 mM Tris pH 7.5, 5 mM MgCl2, 0.4% NP-40. Include cycloheximide 100 µg/mL to maintain polysome structure, 0.1 U RNase inhibitor and protease inhibitor cocktail (1 tablet for 50 mL).
- Incubate the lysate on ice 15 min.
- Centrifuge at 18,000 x g for 15 min at 4 °C to remove insoluble material and collect the supernatant.
- Reserve 50 µL for protein determination, RNA isolation and RNA integrity analysis (see Figure 2).
- Layer the supernatant (500–100 µL) on top of the gradients and balance the tubes with lysis buffer.
- Sedimentation and collection of fractions
- Perform ultracentrifugation for 120 min at 37,000 rpm at 4 °C in a swinging bucket rotor. According to the manufacturer, this corresponds to 228,000 x g at maximal radius.
- Collect each gradient as 17 fractions (around 600 µL each fraction) in microcentrifuge tubes with continuous monitoring of absorbance at 254 nm (OD254).
- Program the fraction collector as follows:
- Discard the first 3 min of collection (corresponding to the liquid contained in the tubes before the gradient)
- From 4 to 19 min, collect the fractions at the speed of 1 mL/min in 17 fractions.
- Place the fractions on ice as soon as each is collected. Freeze samples at -80 °C, if they are not being processed immediately. A typical UV densitometric tracing of the gradient as it is collected is shown in Figure 1.
3. Isolation and Analysis of mRNAs from Polysome and Nontranslating Fractions
- Extraction of mRNA
- Thaw the fractions on ice, if they were flash-frozen after collection
- Transfer 200 µL of each fraction to a fresh tube NOTE: At this step, two or more consecutive fractions can be pooled together. It should be done carefully after analyzing the OD254 graph so that polysome and non-polysome fractions don't get mixed.
- Add 10 ng of luciferase RNA as internal control to each sample for normalization purpose. NOTE: Luciferase primers should be used as an internal control to normalize the results.
- Add 700 µL of RNA extraction reagent (monophasic solution of phenol and guanidine isothiocyanate; see Table of Materials) to each tube. Mix well and incubate the tubes for 5 min at RT (room temperature).
- Add 140 µL chloroform and vortex for 15 s. Incubate for 2-3 min at RT.
- Centrifuge the samples for 15 min at 12,000 x g at 4 °C.
- Carefully transfer the upper aqueous phase to a new tube.
- Since the RNA content of the fractions is not high, add 10 µg of glycogen as carrier to each tube.
- Add 350 µL isopropanol to each tube. Mix well and incubate at -20 °C for 1 h to increase the yield.
- Centrifuge for 15 min at 12,000 x g at 4 °C.
- Discard the supernatant and add 700 µL ethanol 75% to wash the RNA pellet.
- Centrifuge for 5 min at 7,500 x g at 4 °C.
- Remove ethanol and let the pellet air dry for 10–15 min. NOTE: Over-drying the RNA pellet will prevent its complete solubilization in water.
- Resuspend the RNA pellet in 50 μL of RNase-free ultrapure H2O.
- Incubate the samples for 10 min at 55 °C.
- Use 2 μL of each sample to measure the quality and quantity of the extracted RNA and store the remainder at -80 °C.
- Reverse transcription and qPCR
- Use 4 μL of reverse transcription cDNA synthesis kit (see Table of Materials) for a 20 μL reaction. Prepare the supermix according to the number of samples. Transfer the required volume to each tube and add 5 μL of the input RNA to each tube. This volume can be corrected to be relevant for the range of the Taq polymerase working conditions.
- Incubate the tubes in a thermal cycler with the following program recommended by the manufacturer: 5 min at 25 °C, 20 min reverse transcription for 20 min at 46 °C, and reverse transcriptase inactivation for 1 min at 95 °C.
- Run qPCR with the optimized program for each pair of primers. NOTE: To detect the presence of certain mRNAs in fractions, equal volume from each fraction (of the isolated RNA) should be used for reverse transcription.
- Analysis of the results
- Use the Ct value of the interested gene and the luciferase to calculate the delta Ct values
- Express each fraction delta Ct value as the percentage of the total mRNA value contained in all the fractions to visualize the distribution of the mRNA across the different fractions (Table 1)
4. Isolation and Analysis of miRNAs from Polysome Fractions and Nontranslating Fractions
- Extraction of mRNA and miRNA NOTE: This protocol follows manufacturer’s instructions, with a few changes.
- Thaw the glycogen and the spike-in control. Keep on ice.
- Add 700 µL of miRNA extraction reagent to 200 µL of pooled sample.
- Add 1 µg/µL of glycogen and homogenize it using the pipette. Incubate it for 5 min at room temperature.
- Add 3.5 µL of 1.6 x 108 spike-in control and mix it.
- Add 200 µL of chloroform and vortex it for at least 15 sec.
- Incubate for 3 min at room temperature.
- Centrifuge at 12,000 x g for 15 min at 4 °C.
- Using a pipette, transfer the supernatant to a new 1.5 mL tube. Discard the middle and bottom phases.
- Add 1.5x volume of 100% ethanol to the supernatant and mix it.
- Transfer 700 µL of this mixture to the miRNA extraction column and centrifuge it at full speed for 15 s at RT and discard the flow-through; repeat this step with any remaining sample.
- Add 700 µL of buffer 1 to the column and centrifuge it at full speed for 15 s at RT; discard the flow-through.
- Add 500 µL of buffer 2 to the column and centrifuge it at full speed for 15 s at RT; discard the flow-through.
- Add 500 µL of 80% ethanol to the column and centrifuge it at full speed for 2 min at RT; discard the flow-through.
- Transfer the column to a new 2 mL uncapped tube, open the lid and centrifuge it at RT for 5 min at full speed; discard the flow-through.
- Add 16 µL of RNAse-free water to the column and centrifuge it at full speed for 1 min at RT. Keep the eluate on ice or store at -80 °C for future analysis. NOTE: Two microliters of each sample can be used for OD analysis.
- Reverse Transcription
- Prepare on ice. For each 20 µL RT-PCR reaction, add 4 µL of the miRNA RT buffer, 2 µL of nucleic acids, 9 µL of total RNA, 2 µL of enzyme and 3 µL of RNAse-free water; mix it and spin it down briefly.
- Set the Thermal Cycler as follows: 37 °C for 60 min; 95 °C for 5 min; 4 °C hold.
- Remove the tubes from the thermal cycler and add 200 µL of RNAse-free water. Store at -20 °C or proceed to the real-time PCR.
- Real-time PCR NOTE: This protocol follows 96 well plate format. Prepare the reaction mix on ice.
- Prepare PCR reaction mix and add cDNA (from miRNAs above) according to the range accepted by the protocol. Multiple reactions can be prepared in batch.
- Add a total of 25 µL of PCR mix + cDNA in each well.
- Seal the plate using an optical plate seal.
- Centrifuge the plate for 1 min at 1,000 x g at room temperature.
- Program the real-time cycler as follows: Initial activation step at 95 °C for 15 min; 3-step cycling: denaturation at 94 °C for 15 s; annealing at 55 °C for 30 s; extension at 70 °C for 30 s (perform fluorescence data collection); add 40 cycles; Melting-curve according to cycler default.
- Comparative analysis
- Example of normalization (see Table 2):
- Find the SCM minimum value.
- Normalize all the SCM values to this minimum.
- Subtract this value from the Ct values of miRNAs of interest (MOI) and reference gene (REF).
- Analyze data using the 2-ΔCt formula.
5. Proteomic Analysis
- Extraction of proteins from polysome fractions
- Combine collected sucrose fractions into three final fractions. Mark the fractions as heavy (high efficiency polysomes in pooled fractions 4, 5, 6 and 7 with the highest concentration of sucrose; bottom of the gradient tube), light (low efficiency polysomes in pooled fraction 8, 9, 10 and 11; middle of the gradient tube) and non-translating (pooled fractions 12, 13, 14 and 15 with the lowest concentration of sucrose; top of the gradient tube). Perform protein assay on pooled fractions.
- Prepare 100% stock of trichloroacetic acid (TCA) and store it at 4 °C in dark. Mix 0.5 mL of sample with 60 µL of 100% TCA and incubate at -20 °C overnight in the dark. NOTE: Use 1.5 mL low protein retention tubes (see Table of Materials).
- After overnight incubation, thaw the sample on ice and centrifuge at 15,000 x g, 4 °C for 30 min. Discard supernatant. NOTE: 100 µg of total protein in sample gives visible protein pellet at the tube bottom.
- Pre-chill acetone in -20 °C freezer. Add 0.5 mL of cold acetone to the protein pellet and centrifuge at 15,000 x g, 4 °C for 15 min. Discard supernatant. Repeat this step twice.
- Air dry the pellet. Resuspend the pellet in 360 µL of 50 mM Tris-HCl buffer (pH 8). NOTE: If needed, use a pulse sonicator to resuspend the pellet.
- Add 40 µL of 0.1 M dithiothreitol (final concentration 10 mM) and incubate at 55 °C on shaker for 45 min. Add 50 µL of 0.15 M iodoacetamide (final concentration 17 mM) and incubate at room temperature on shaker for 30 min in dark.
- Prepare trypsin solution: resuspend 20 µg of trypsin in 100 µL of 50 mM ammonium bicarbonate. Add corresponding volume of trypsin solution to sample at 1:50 (w/w) trypsin:protein ratio, ensure that pH is 8 and incubate on shaker at 37 °C overnight. NOTE: Add 1 M ammonium bicarbonate to bring pH to 8.
- After trypsin digestion, cool the sample, centrifuge briefly in bench centrifuge, add 10% formic acid to pH 2–3 to quench the trypsin activity, and proceed with sample desalting. NOTE: Use µ-elution plate for sample desalting.
- Condition the sorbent in the wells of 96 well plate with 200 µL of methanol using vacuum three times followed by conditioning by 200 µL of 0.1% formic acid three times. Load sample and perform a sample wash using 200 µL of 0.1% formic acid three times. Elute the sample with 100 µL of 50% acetonitrile/0.1% formic acid twice into low protein retention 0.5 mL tube. NOTE: Elute the sample slowly at first using gravitation followed by low vacuum on.
- Evaporate sample eluate to dryness using concentrator and either directly carry out mass spectrometry analysis or store at -80 °C until use.
- Mass spectrometry analysis
- Reconstitute each pellet (consisting of the tryptic peptides) in 50–100 µL and inject 1 µL into mass spectrometer. Note: Optimize reconstitution and injection volumes to obtain maximum intensity LC/MS/MS signal. In our case, the maximum signal (total ion current; TIC) in the heart of the scan was in range of 1E8 to 2E9.
- Use a trap column C18 (300 µm i.d. x 5 mm, 5 µm, 100Å) followed by peptide separation using C18 column (75 µm i.d. x 250 mm, 2 µm, 100Å) with a flow rate setting at 300 nL/min. Set the nano-source capillary temperature to 275 °C and the spray voltage to 2 kV.
- Apply a linear gradient of 5–35% B for 90 min, 35–95% B for 3 min, holding at 95% B for 7 min and equilibration at 5% B for 25 min (A: 0.1% formic acid/water and B: 0.1% formic acid in acetonitrile). Acquire MS2 spectra for the 15 highest intensity ions from each MS1 scan using CID mode. Use 3 mass spectrometry replicates for each sample analyzed. NOTE: Optimize and use the experimental conditions and parameters that are suitable for your samples on the particular mass spectrometry instrument. The conditions/parameters mentioned in section 5.2. were used and optimized in our laboratory.
- Protein identification/quantitation
- After mass spectrometry analysis, convert the raw spectra files into compatible mzXML files using MSConvert software (http://proteowizard.sourceforge.net/tools/msconvert.html).
- Submit the converted files into SEQUEST search engine with the search criteria, e.g., UniProtKB/Swiss-Prot mouse database (most up to date canonical reviewed); semi-enzyme digest using trypsin (after KR/-); with up to 2 missed cleavages; precursor mass range: 400 to 4,500 amu; static modification: carbamidomethyl (C) 57.021465 amu; peptide mass tolerance: 50 ppm; fragment mass type: monoisotopic. NOTE: Other protein database search engines and data pipelines can be used. If using database for e.g. rat, which is less complete than databases for mouse and human, you may need to search against mouse (and/or human) databases to increase proteome coverage. In this case, redundancy in protein names will need to be removed.
- Perform a post-search analysis. Set the filters for viewing the data. For example, set a minimum protein probability (protein threshold) as 95% or 99%, i.e., only the proteins for which a statistical analysis implies 95% or 99% probability of being present in the sample will be displayed. Set the filter for peptide threshold (e.g., 95%), i.e., a minimum probability that will be used in determining whether a spectrum identifies a peptide. Select the minimum number of peptides that will determine if a protein is present (2 peptides as a minimum for protein identification is recommended).
- Evaluate identified proteins for quality/quantity. Ensure that proteotypic peptides are used for quantification. If isoform is identified ensure it is bases on observation of a tryptic peptide comprised of an amino acid sequence unique to the specific isoform. Any “similar to” or hypothetical proteins should undergo comparison to see if the observed peptides correspond to a known protein.
Representative Results
mRNA analysis mRNA results can be expressed as a distribution of a particular mRNA in each fraction (Figure 3A); for quantification, combine polyribosomal translating fractions and compare to the non-translating fraction (Figure 3B), presenting a ratio of mRNA abundance in translating to nontranslating fractions. Additional information is gained by examining the high efficiency polysome fractions separately from low-efficiency polysome fractions (and separate from nontranslating fractions). This is particularly important when analyzing miRNAs, which are enriched in the low-efficiency fractions (where they interfere with protein translation of their target mRNAs).
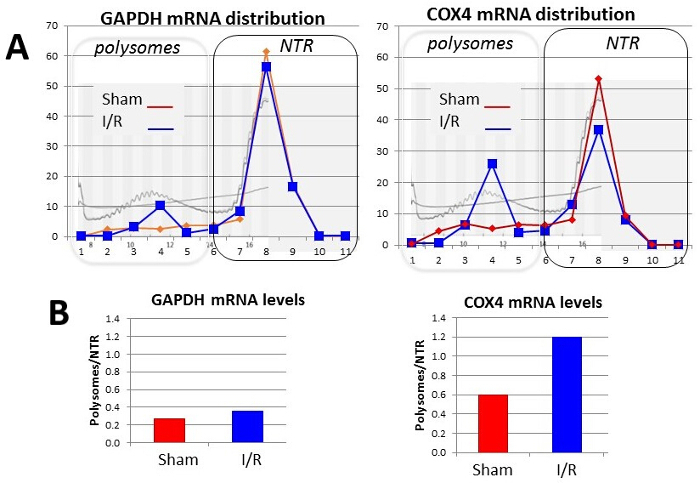
Representative results for a particular mRNA that changes its distribution across the translating and nontranslating fractions after the mouse hearts were subjected to ischemia and ischemia/reperfusion compared to sham are shown in Figure 4.
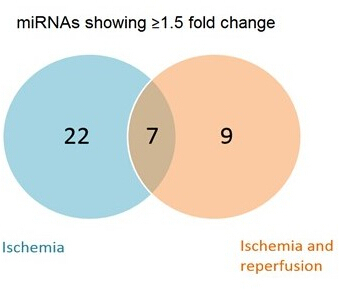
miRNA array analysis Representative results from a miRNA array analysis are shown in Figure 5. Here, a subset of miRNAs (custom-designed array plate) were analyzed in the pooled heavy fractions indicating that 22 miRNAs were associated with polysomes during ischemia, 9 miRNas during ischemia and reperfusion, with 7 that were common to both conditions. This shows that the association of miRNAs is dynamic in response to the stresses of ischemia and reperfusion.
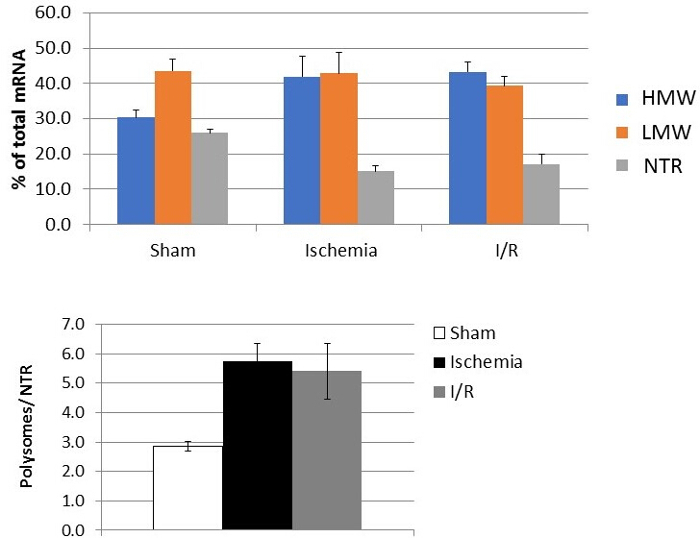
Proteomic analysis Analysis of heavy, light and non-translating fractions of sham (control) sample: 881 of total proteins were identified by mass spectrometry; 3, 46 and 208 proteins of total were unique to heavy, light and non-trans fractions, respectively (Figure 6A). The majority (88%) of mitochondrial ribosomal proteins (28S and 39S) were identified in light fraction (36 out of 41) and a majority (88%) of cytosolic ribosomal proteins (40S and 60S) were identified commonly in both heavy and light fractions (53 out of 60) confirming efficient separation and proteomic ability to identified heavier cytosolic vs lighter mitochondrial ribosomal proteins. The subset of identified mitochondrial and cytosolic ribosomal proteins is shown in Figure 6B.
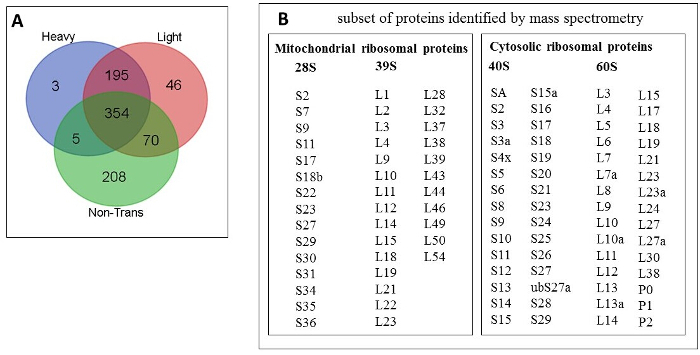
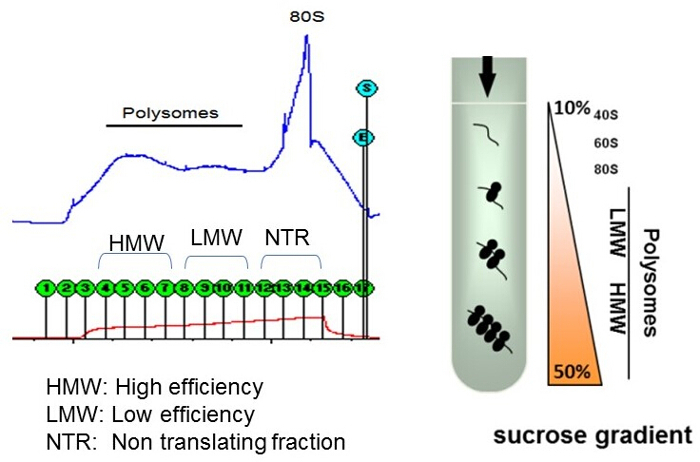
Figure 1: Typical UV absorbance profile of polysome fractions on sucrose gradient. The first three fractions represent void volume in the tubing. The high efficiency (high molecular weight, HMW) fractions are collected in fractions 4-7, the low efficiency (low molecular weight, LMW) fractions in 8-11, and the non-translating (and remaining cytosolic material) is collected in fractions 12-15 (nontranslating, NTR). The red line indicates conductivity, and the blue trace indicates UV absorbance at 254 nm. On the right is a cartoon of a test tube with sucrose gradient, showing distribution of polysomes after sedimentation. Please click here to view a larger version of this figure.
Figure 2: RNA integrity control analysis on Bioanalyzer. RNA integrity number (RIN) of total RNA isolated from heart whole lysate. RIN is calculated according to 18S and 28S peaks. RIN range: 1-10, and RIN = 10 represents a very good integrity. Please click here to view a larger version of this figure.
Figure 3: Representative profile of mRNA distribution in sucrose gradient. Hearts were subjected to basal Langendorff perfusion or ischemia and reperfusion (I/R). (A) Heart lysates were resolved on sucrose gradient and mRNA content for GAPDH (left) and COX4 (right) were determined in each fraction. Plot shows relative mRNA abundance in each fraction (fraction number on x-axis) for basal perfusion (sham, red line and diamonds) and ischemia/reperfusion (I/R, blue line and squares). Superimposed on the plot are UV absorbance indicating total mRNA content (light gray curve) and conductance (straight gray sloping line). Boxes indicate how fractions were pooled. (B) Bar graph plot shows ratio of mRNA abundance in translating (polysomes) vs nontranslating (NT) fractions for GAPDH (left) and COX4 (right) mRNAs. Please click here to view a larger version of this figure.
Figure 4: Representative mRNA results. Quantitation of one mRNA in the pooled fractions (HMW, LMW, NTR) showing changes according to the experimental group. Polysome fractionation of mouse hearts was performed after Langendorff perfusion. Results are plotted as % of total mRNA (upper panel) and as ratio of polysomes (HMW+LMW) to nontranslating fraction (NTR). Error bars represent results from 5 hearts per group (sham, ischemia, or I/R). Please click here to view a larger version of this figure.
Figure 5: Representative miRNA results. Venn Diagram of a pathway focused miRNA analysis showing up and downregulated miRNAs in heavy fraction pools of mice samples after heart ischemia or ischemia and reperfusion. Cut-off value: 1.5 fold. Please click here to view a larger version of this figure.
Figure 6: Proteins identified by mass spectrometry. (A) Venn diagram of the proteins common and unique to heavy, light and non-trans fractions. (B) Subset of mitochondrial and cytosolic ribosomal proteins identified. Please click here to view a larger version of this figure.
Figure 7: Schematic workflows for protein extraction methods tested. The protein extraction methods were tested by using sham (control) sample and heavy (high efficiency polysomes) fraction with highest sucrose content. Numbers in red indicate the number of proteins that were identified by mass spectrometry; where two numbers are shown, they are from two separate experiments. The detailed description is given in the text. Please click here to view a larger version of this figure.
| FRACTIONS | GENE Ct | LUCIFERASE Ct | ΔCt | 2ΔCt | SUM OF ALL FRACTIONS | % OF EACH FRACTION | Polysome/NTR Ratio |
| High efficiency (HMW) | 28.75 | 22.79 | 5.96 | 0.016 | 0.046 | 34.64 | 2.77 |
| Low efficiency (LMW) | 28.43 | 22.63 | 5.80 | 0.018 | 38.81 | ||
| Non Translating (NTR) | 29.12 | 22.77 | 6.35 | 0.012 | 26.55 | ||
| Polysome/NTR Ratio = (HMW + LMW)/NTR |
Table 1:Sample analysis of mRNA. Method of analysis of one mRNA is shown for the different pooled fractions (HMW, LMW, NTR) after quantitative PCR. ΔCt = gene Ct - luciferase Ct, and the 2ΔCt is 2ΔCt.
| Find the SCM minimum value: 22.77 | |||
| Sample 1 | Sample 2 | Sample 3 | |
| Ct MOI | 22.92 | 22.36 | 22.58 |
| Ct SCM | 22.81 | 22.77 | 22.9 |
| Ct REF | 23.27 | 23.55 | 23.19 |
| Normalize all the SCM values to this minimum | |||
| Sample 1 | Sample 2 | Sample 3 | |
| Ct SCM | 0.05 | 0 | 0.13 |
| Subtract these values from the Ct values of MOI and REF | |||
| Sample 1 | Sample 2 | Sample 3 | |
| Ct MOI | 22.87 | 22.36 | 22.45 |
| Ct REF | 23.22 | 23.55 | 23.06 |
Table 2: Sample real-time PCR calculation for miRNA expression comparison in polysome and nontranslating fractions. miRNAs of interest (MOI) and reference gene (REF) were analyzed in the pooled heavy fractions of mice after ischemia or ischemia/reperfusion. The Ct values were first normalized to the spike-in control (SCM) Ct value, then the 2-ΔCt formula was used to compare miRNA expression.
| Method | Sample volume [mL] | # of proteins identified | Comments |
| FASP | 1 | 227 | bulk of sucrose precipitated on filter |
| acetone precipitation | 0.6 | 372 | viscous sludge of sucrose at the tube bottom; 4:1 (reagent:sample) volumes; no visible protein pellet |
| chloroform/methanol precipitation | 0.32 | 389 | viscous sludge of sucrose at the tube bottom; 8:1 (reagent:sample) volumes; no visible protein pellet |
| TCA precipitation (at 4 °C) | 0.5 | 405, 415 | no sucrose precipitation; 0.12:1 (reagent:sample) volumes; visible pellet at the tube bottom |
| re-precipitation | 85, 60 | additional TCA precipitation of supernatant after first pellet was collected | |
| TCA precipitation (at -20 °C) | 0.5 | 440, 430 | no sucrose precipitation; 0.12:1 (reagent:sample) volumes; visible pellet at the tube bottom |
| re-precipitation | 81, 55 | additional TCA precipitation of supernatant after first pellet was collected |
Table 3:Comparison of protein extraction methods. FASP, acetone precipitation, chloroform/methanol precipitation, and TCA precipitation at 4 °C and -20 °C were evaluated. The goal was to maximize the number of proteins identified. TCA precipitations were evaluated on a second batch of material to confirm reproducibility.
Discussion
Polysome profile analysis allows for the study of protein translation by analyzing the translational state of a specific mRNA or the whole transcriptome6,7. It is also of great help when local translation needs to be studied such as synaptosomes8. Traditionally, this method involves the separation of mono- and polyribosomes and the associated mRNAs on a sucrose gradient which could be coupled with genomic or proteomic techniques to obtain the intended results6,9. For instance, a recent study performed by our group has revealed a post-transcriptional regulatory mechanism executed in the heart of patients undergoing CPB, which mimics ischemia/reperfusion injury. Taking advantage of polysome profile analysis, we have shown an increase in translation of nuclear-encoded mitochondrial proteins in heart after CPB procedure2.
The dynamic proteome polyribosomal profiling approach presented here can reveal changes in the population of mRNAs associated with polyribosomes and the proteins that are being actively translated. As translation of mRNAs is governed in part by miRNAs, analysis of miRNAs in these fractions can reveal additional insights. In fact, several studies have modified this method and proved it to be a suitable and reliable approach to investigate the miRNA mode of regulation10,11.
Many studies have been conducted in the last decade delving into the role of miRNAs, considering their importance in various biological functions12,13. Since miRNAs act through base-pairing with counterpart mRNAs so as to mark them for degradation, polysome profile analysis has been performed and shown that miRNAs are in fact found in the polysome fractions, targeting translating mRNAs14,15,16. miR-21 is a good example where it is associated with a low repression and weak polysomes binding in normal cells, while in cancer cells, the association with polysomes is increased and the repressive effect is much stronger17.
In this study, Real-time PCR analysis was done using the Ct values of the miRNAs of interest (MOI), the Ct values of the spike-in control miRNA (SCM) to reduce technical variations and the Ct value of a reference gene or miRNA (REF) which is stable among samples. The first step was to normalize the Ct values of the MOI and the REF to the Ct values of the SCM. Then there was a second step normalization of the MOI to the REF and the resulting value was used to calculate the 2-ΔCt formula. These values or fold-change values can be used to demonstrate differences between groups.
Since the extraction of proteins is difficult from polysome fractions, most of the studies performed until now, have used the fractions directly for western blot analysis. They simply sediment the protein content of each fraction and mix it with SDS-loading buffer to use for SDS-PAGE analysis8,18. Here, we have developed a method which allows us to not only extract mRNAs and miRNAs after fractionation, but also to obtain peptides and proteins with a high quality to proceed with Mass Spectrometry analysis.
This approach could be extended further by actively measuring the newly synthesized proteins using metabolic labeling such as biorthogonal amino acid homolog, such as azidohomoalanine (AHA) for methionine19,20. AHA incorporation followed by click chemistry allows for incorporation of a biotin tag followed by streptavidin purification of all proteins that incorporated AHA. This allows definitive identification of newly synthesized proteins — the other part of the dynamic proteome. Detection of miRNAs that redistribute among the translating and nontranslating fractions in response to a stimulus (here with I/R) allows interrogation of the dynamic regulation of protein translation. However, the factors that recruit miRNAs to the vicinity of ribosome-bound mRNA remain to be identified and systematically investigated.
In our study, in addition to TCA precipitation, we tested three other methods for protein extraction from polysome fractions: i) FASP (filter-aided sample preparation)21, ii) acetone precipitation, and iii) chloroform/methanol precipitation. The suitability of the methods was evaluated based on both the ability to process fractions with high concentration of sucrose (up to 50%) and number of proteins identified by mass spectrometry (Figure 7 and Table 3).

For FASP method, we followed the protocol of sample preparation given by a FASP Protein Digestion Kit that employed an ultrafiltration unit with a relative molecular mass cut-off of 30 kDa. This filter unit served as a tool for detergent removal, buffer exchange, protein digestion and peptide elution with the ability to retain high-molecular-weight substances (proteins and DNA) that would otherwise interfere with subsequent peptide separation21. Briefly, the sample was mixed with urea-containing buffer and loaded on unit spin filter with spinning steps as iodoacetamide solution, trypsin solution and sodium chloride solution (elution) were subsequently added. Final filtrate that contained digested peptides was acidified by TFA, desalted and stored for mass spectrometry analysis. Using this method, however, the bulk of precipitated sucrose steadily accumulated on the top of the filter, rendering the centrifugation and washing steps difficult.
For acetone precipitation and chloroform/methanol precipitation, multiple volumes of solvents were needed to precipitate proteins from a single volume of sample. This limited the size of sample volume applied when the precipitation was performed in 1.5 mL tubes (protein low-retention tubes). As well, the viscous sludge of sucrose accumulated at the bottom of tubes after precipitation for both methods and there were no visible pellets at the tube bottom and at the liquid interface after acetone precipitation and chloroform/methanol precipitation, respectively.
Table 3 and Figure 7 show the comparison of four protein extraction methods we used to optimize our experimental workflow. The red numbers given at each method (Figure 7) represent the number of proteins that were identified by mass spectrometry (see Section 5.2 and 5.3). Although sample volumes used for each method were different, chloroform/methanol and TCA precipitations resulted in highest number of proteins identified. However, due to disadvantages mentioned above (sample volume limitation and sucrose precipitation), we opted for TCA precipitation for subsequent experiments.
To find the optimum conditions for the sample processing, TCA precipitation was carried out at various temperatures and TCA concentrations22,23,24. Thus, we performed a precipitation with 10.7% TCA at 4 °C and -20 °C and further precipitated the supernatants with additional 60 µL of TCA (final 19.4% TCA) after pellets were collected. This resulted in four pellets that were analyzed separated by mass spectrometry. In addition, we repeated the whole protocol twice to ensure that obtained results were reproducible. The workflow and the number of proteins identified in the pellets at both 4 °C and -20 °C conditions with two repeated experiments are shown in Figure 7 (numbers in red). Although the analysis of the pellets derived from the supernatants yielded an additional number of proteins (85 and 60 at 4 °C; 81 and 55 at -20 °C); a majority of the proteins were already identified in first pellets and thus, we opted to use 10.7% TCA for all subsequent experiments. Although chloroform/methanol precipitation resulted in a similar number of proteins identified compared to TCA precipitation, we preferred TCA precipitation due to several advantages: i) small volume of solvent needed to precipitate the proteins (60 µL of TCA vs 1.28 mL of chloroform/methanol/water) enabling the use of larger sample volumes if needed while conveniently using 1.5 mL tubes for precipitation, ii) visibility of the pellet at the tube bottom, and iii) no accumulation of sucrose during precipitation steps.
Despite offering in-depth functional information regarding the translational state, a major drawback of this method is the need for fresh and large starting material. Many studies have tried to optimize the conditions using small volume of cells or tissues or add steps to increase the efficiency of extracting RNA and proteins25. Still, tissues obtained from different animals under the same experimental conditions could be pulled together, if need be.
In conclusion, this protocol allows for simultaneous analysis of mRNA, miRNA, and protein from fractions obtained from polysome profiling to study the regulatory mechanisms involved in ischemia/reperfusion. However, further studies are required to see whether the translating mRNAs are induced after ischemia or reperfusion and if they will be turned off upon the removal of stress.
Disclosures
The authors have nothing to disclose.
Acknowledgments
NIH P01 HL112730 (RAG, JVE), NIH R01 HL132075 (RAG, JVE), Barbra Streisand Women's Heart Center (RAG, JVE), Dorothy and E. Phillip Lyon Chair in Molecular Cardiology (RAG), Erika Glazer Endowed Chair in Woman's Heart Health (JVE) and Czech Academy of Sciences Institutional Support RVO: 68081715 (MS).
References
- Pourpirali S, Valacca C, Merlo P, Rizza S, D’Amico S, Cecconi F. Prolonged pseudohypoxia targets ambra1 mrna to p-bodies for translational repression. PLoS ONE. 2015;10:e0129750. doi: 10.1371/journal.pone.0129750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres AM, Tucker KC, Thomas A, Taylor DJ, Sengstock D, Jahania SM, Dabir R, Pourpirali S, Brown JA, Westbrook DG, Ballinger SW, Mentzer RM, Jr, Gottlieb RA. Mitophagy and mitochondrial biogenesis in atrial tissue of patients undergoing heart surgery with cardiopulmonary bypass. JCI Insight. 2017;2:e89303. doi: 10.1172/jci.insight.89303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislinger T, Cox B, Kannan A, Chung C, Hu P, Ignatchenko A, Scott MS, Gramolini AO, Morris Q, Hallett MT, Rossant J, Hughes TR, Frey B, Emili A. Global survey of organ and organelle protein expression in mouse: Combined proteomic and transcriptomic profiling. Cell. 2006;125:173–186. doi: 10.1016/j.cell.2006.01.044. [DOI] [PubMed] [Google Scholar]
- Kren BT, Wong PY, Shiota A, Zhang X, Zeng Y, Steer CJ. Polysome trafficking of transcripts and micrornas in regenerating liver after partial hepatectomy. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2009;297:G1181–G1192. doi: 10.1152/ajpgi.90636.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb RA, Pourpirali S. Lost in translation: Mirnas and mrnas in ischemic preconditioning and ischemia/reperfusion injury. Journal of Molecular and Cellular Cardiology. 2016;95:70–77. doi: 10.1016/j.yjmcc.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudert L, Adjibade P, Mazroui R. Analysis of translation initiation during stress conditions by polysome profiling. Journal of Visualized Experiments. 2014. [DOI] [PMC free article] [PubMed]
- Lorsch J. Methods in enzymology. Laboratory methods in enzymology: Rna. Preface. Methods in Enzymology. 2013;530:xxi. doi: 10.1016/B978-0-12-420037-1.09986-3. [DOI] [PubMed] [Google Scholar]
- Kuzniewska B, Chojnacka M, Milek J, Dziembowska M. Preparation of polysomal fractions from mouse brain synaptoneurosomes and analysis of polysomal-bound mrnas. Journal of Neuroscience Methods. 2018;293:226–233. doi: 10.1016/j.jneumeth.2017.10.006. [DOI] [PubMed] [Google Scholar]
- He SL, Green R. Polysome analysis of mammalian cells. Methods in Enzymology. 2013;530:183–192. doi: 10.1016/B978-0-12-420037-1.00010-5. [DOI] [PubMed] [Google Scholar]
- Molotski N, Soen Y. Differential association of micrornas with polysomes reflects distinct strengths of interactions with their mrna targets. RNA. 2012;18:1612–1623. doi: 10.1261/rna.033142.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroney PA, Yu Y, Fisher J, Nilsen TW. Evidence that micrornas are associated with translating messenger rnas in human cells. Nature Structural and Molecular Biology. 2006;13:1102–1107. doi: 10.1038/nsmb1174. [DOI] [PubMed] [Google Scholar]
- Paul P, Chakraborty A, Sarkar D, Langthasa M, Rahman M, Bari M, Singha RS, Malakar AK, Chakraborty S. Interplay between mirnas and human diseases. Journal of Cellular Physiology. 2018;233:2007–2018. doi: 10.1002/jcp.25854. [DOI] [PubMed] [Google Scholar]
- Rupaimoole R, Slack FJ. Microrna therapeutics: Towards a new era for the management of cancer and other diseases. Nature Reviews Drug Discovery. 2017;16:203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Hatzigeorgiou AG, Mourelatos Z. Mirnp:Mrna association in polyribosomes in a human neuronal cell line. RNA. 2004;10:387–394. doi: 10.1261/rna.5181104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottrott S, Simard MJ, Richter JD. Human let-7a mirna blocks protein production on actively translating polyribosomes. Nature Structural and Molecular Biology. 2006;13:1108–1114. doi: 10.1038/nsmb1173. [DOI] [PubMed] [Google Scholar]
- Kim J, Krichevsky A, Grad Y, Hayes GD, Kosik KS, Church GM, Ruvkun G. Identification of many micrornas that copurify with polyribosomes in mammalian neurons. Proceedings of the National Academy of Sciences of the United States of America. 2004. pp. 360–365. [DOI] [PMC free article] [PubMed]
- Androsavich JR, Chau BN, Bhat B, Linsley PS, Walter NG. Disease-linked microrna-21 exhibits drastically reduced mrna binding and silencing activity in healthy mouse liver. RNA. 2012;18:1510–1526. doi: 10.1261/rna.033308.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraushar ML, Thompson K, Wijeratne HR, Viljetic B, Sakers K, Marson JW, Kontoyiannis DL, Buyske S, Hart RP, Rasin MR. Temporally defined neocortical translation and polysome assembly are determined by the rna-binding protein hu antigen r. Proceedings of the National Academy of Sciences of the United States of America. 2014. pp. E3815–E3824. [DOI] [PMC free article] [PubMed]
- McClatchy DB, Ma Y, Liu C, Stein BD, Martinez-Bartolome S, Vasquez D, Hellberg K, Shaw RJ, Yates JR., 3rd Pulsed azidohomoalanine labeling in mammals (palm) detects changes in liver-specific lkb1 knockout mice. Journal of Proteome Research. 2015;14:4815–4822. doi: 10.1021/acs.jproteome.5b00653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiapparelli LM, McClatchy DB, Liu HH, Sharma P, Yates JR, 3rd, Cline HT. Direct detection of biotinylated proteins by mass spectrometry. Journal of Proteome Research. 2014;13:3966–3978. doi: 10.1021/pr5002862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nature Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Rajalingam D, Loftis C, Xu JJ, Kumar TK. Trichloroacetic acid-induced protein precipitation involves the reversible association of a stable partially structured intermediate. Protein Science. 2009;18:980–993. doi: 10.1002/pro.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fic E, Kedracka-Krok S, Jankowska U, Pirog A, Dziedzicka-Wasylewska M. Comparison of protein precipitation methods for various rat brain structures prior to proteomic analysis. Electrophoresis. 2010;31:3573–3579. doi: 10.1002/elps.201000197. [DOI] [PubMed] [Google Scholar]
- Sashital DG, Greeman CA, Lyumkis D, Potter CS, Carragher B, Williamson JR. A combined quantitative mass spectrometry and electron microscopy analysis of ribosomal 30s subunit assembly in e. Coli. Elife. 2014;3 doi: 10.7554/eLife.04491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Bellato HM, Lorent J, Lupinacci FCS, Oertlin C, van Hoef V, Andrade VP, Roffé M, Masvidal L, Hajj GNM, Larsson O. Polysome-profiling in small tissue samples. Nucleic Acids Research. 2018;46:e3. doi: 10.1093/nar/gkx940. [DOI] [PMC free article] [PubMed] [Google Scholar]