

Abstract
Primary cell culture is a powerful tool commonly used by scientists to study cellular properties and mechanisms of isolated cells in a controlled environment. Despite vast differences in the physiology between mammals and fish, primary cell culture protocols from fish are often based on mammalian culture conditions, often with only minor modifications. The environmental differences affect not only body temperature, but also blood serum parameters such as osmolality, pH, and pH buffer capacity. As cell culture media and similar working solutions are meant to mimic characteristics of the extracellular fluid and/or blood serum to which a cell is adapted, it is crucial that these parameters are adjusted specifically to the animal in question.
The current protocol describes optimized primary culture conditions for medaka (Oryzias latipes). The protocol provides detailed steps on how to isolate and maintain healthy dissociated pituitary cells for more than one week and includes the following steps: 1. the adjustment of the osmolality to the values found in medaka blood plasma, 2. the adjustment of the incubation temperature to normal medaka temperature (here in the aquarium facility), and 3. the adjustment of the pH and bicarbonate buffer to values comparable to other fish species living at similar temperatures. The results presented using the described protocol promote physiologically meaningful results for medaka and can be used as a reference guide by scientists making primary cell cultures from other non-mammalian species.
Keywords: Neuroscience, Issue 138, Pituitary, endocrine cells, primary culture, fish, dissociation, temperature, osmolality, pH, pCO2, medaka
Introduction
Cell culture is one of the main tools used in molecular biological research, providing an excellent model system for answering different biological questions ranging from normal cellular physiology to drug screening and carcinogenesis1. Primary cells, isolated directly from the animal tissue using enzymatic and/or mechanical methods,are often considered more biologically relevant than cell lines as the biological response may be closer to the in vivo situation. Protocols for preparing primary cell cultures should be optimized for each species and cell type of interest in order to mimic the characteristics to which a cell is adapted and obtain physiologically meaningful results.
Numerous protocols describe culture conditions for mammalian cell systems, while similar protocols describing primary culture conditions for fish cells are rather scarce in comparison. Cells are vulnerable to rapid changes in temperature, pH, and osmolality, and are particularly fragile during the dissociation procedure. Commercial salt solutions and culture media used for mammalian cell cultures are not optimal for teleost fish, especially in terms of pH buffer system(s) and osmolality. It is, therefore, important to measure and adjust the solutions to physiologically relevant levels of these parameters in the species of interest.
Primary pituitary cultures have been made from several teleost fish species, including common carp (Cyprinus carpio)2,3, grass carp (Ctenopharyngodon idella)4, goldfish (Carassius auratus)5, rainbow trout (Oncorhynchus mykiss)6, European eel (Anguilla anguilla)7, tilapia (Oreochromis mossambicus)8, zebrafish (Danio rerio)9, and Atlantic cod (Gadus morhua)10. Apart from adjusting the incubation temperature to the species of interest, several of these protocols have incubated the cells at mammalian-like conditions that may be suboptimal for the species of interest, with a pH from 7.2 to 7.5 in a humidified atmosphere containing 3 - 5% CO2. In addition, it is unclear if the osmolality of solutions used for preparing several of these primary cell cultures were adjusted and stable between different solutions.
The current protocol is based on previous work with primary cultures from Atlantic cod10 and comprises adjustments of incubation temperature, osmolality, pH, and pH buffer systems, including the partial pressure of carbon dioxide (pCO2), to the physiology of medaka (O. latipes). Medaka is a small (3–4 cm) freshwater fish, native to East Asia. These days, it is used as a model species in many research laboratories around the world, as it is relatively easy to breed and highly resistant to many common fish diseases11. There are several advantages of using medaka as a model, including a temperature tolerance from 4–40 °C11, a short generation time, transparent embryos, a sex-determining gene12, and a sequenced genome13, as well as many other available genetic resources.
The primary culture conditions in this protocol are optimized to match the temperature of 26 °C that medakas are kept at in the fish facility. Further, the osmolality is reduced from 320 mOsm/kg from Atlantic cod living in salt water to 290 mOsm/kg for medaka living in fresh water and is in accordance with the normal osmolality of medaka plasma14. In comparison, the typical osmolality of mammalian plasma is in the range of 275–295 mOsm15. Fish lives in a variety of temperatures and have gills that are in direct contact with water, making the pH and buffer capacity of the blood and extracellular fluid in fish different from those in mammals. Mammalian culture media usually include buffer systems that result in a pH of around 7.4 when the media are equilibrated to a standard atmosphere of 5% CO2 in humidified air at 37 °C. The pH is temperature dependent and the value for neutral pH (in water) increases with a decreasing temperature16. Typical teleost fish plasma pH ranges from 7.7 to 7.917. The optimization of this protocol included a reduction from pH 7.85 for cod kept at 12 °C to pH 7.75 for medaka kept at 26 °C by increasing the CO2 from 0.5% to 1%.
In addition, the bicarbonate buffer capacity is quite different in fish and mammals. CO2 is easily exchanged over the gills in fish and the pCO2 in water is only a small fraction of the pCO2 in the lung18. Changing either the temperature or the pCO2 will change the pH and buffer of the medium. Consequently, neither the pH nor the pCO2 recommended for incubating mammalian cells is optimal for fish cells, and therefore, the culture media should be optimized with buffer systems containing physiologically relevant values for fish and the particular species of interest. This protocol describes how to prepare primary cell cultures from medaka pituitaries and include adjustments of the incubation temperature, osmolality, pH, and pH buffer system, in addition to other important parameters to consider when preparing primary cell cultures from non-mammalian species.
Protocol
The animal experiments performed in this study were approved by the Norwegian University of Life Sciences, following guidelines for the care and welfare of research animals.
1. Preparation of Solutions
Calibrate the osmometer and pH meter instruments according to the manufacturer’s instructions to ensure correct measurements.
- Prepare 500 mL of Ca2+- and Mg2+-free Dulbecco’s phosphate-buffered saline (dPBS), adjusting the pH to 7.75 (step 1.2.1) and the osmolality to 290 mOsm/kg (step 1.2.2) before a sterile filtration (step 1.2.3).
- Adjust the pH to 7.75 with a calibrated pH meter by carefully adding drops of a 1 M NaOH solution while carefully stirring the solution.
- Measure the osmolality of the solution using a calibrated osmometer and calculate the amount of mannitol needed to increase the osmolality to 290 mOsm/kg. Add the required amount of mannitol and then measure the osmolality of the solution again to ensure the correct adjustment. NOTE: The amount of mannitol needed depends on the osmolality measured, which usually varies between different batches. One molecule of mannitol equals 1 osmotic particle.
- Sterile-filter the dPBS solution using a 0.2 µm filter. NOTE: The solution may be stored at 4 °C for at least 3 months.
- Prepare 500 mL of Leibovitz (L-15) culture medium without L-glutamine and bicarbonate, and supplement it with 10 mM NaHCO3 (step 1.3.1), 4.5 mM glucose (step 1.3.2), and 2 mM commercially available glutamine (step 1.3.3). Adjust the solution to 290 mOsm/kg (step 1.3.4), sterile-filter it (step 1.3.5), and add 2.5 mL of a penicillin-streptomycin solution (step 1.3.6). NOTE: The solution may be stored at 4 °C for at least 4 weeks.
- Add 420 mg of NaHCO3 per 500 mL of culture medium to achieve a 10 mM solution.
- Add 405 mg of glucose per 500 mL of culture medium to achieve a 4.5 mM solution.
- Add 5 mL of 100x stock of a glutamine solution in 500 mL of culture medium to achieve a 2 mM solution. Stir until all solutes are dissolved.
- Adjust the solution to 290 mOsm/kg with mannitol using an osmometer (as performed in step 1.2.2).
- Perform a sterile filtration of the L-15 medium using a 0.2 µm filter.
- After the sterile filtration, add 2.5 mL of a penicillin-streptomycin solution, corresponding to 50 U/mL of penicillin and 50 µg/mL of streptomycin.
Prepare 50 mL of a trypsin solution using 1 mg/mL of (0.1%) trypsin type II-S dissolved in the modified dPBS (prepared in step 1.2). Make aliquots of 2 mL in sterile plastic tubes and store them at -20 °C until use. NOTE: Aliquots of 2 mL can be stored at -20 °C for at least 3 months.
Prepare 50 mL of a trypsin inhibitor solution using 1 mg/mL of (0.1%) trypsin inhibitor type I-S, and supplement it with 2 µg/mL of DNase I dissolved in the modified dPBS (prepared in step 1.2). Make aliquots of 2 mL in sterile plastic tubes and store them at -20 °C until use. Note: Aliquots of 2 mL can be stored at -20 °C for at least 3 months.
2. Preparation of Equipment
Prepare glass pipettes by fire polishing the tip for the cell dissociation. Make tips with 2 different size categories (a medium opening of 0.6–0.8 µm and a small opening of 0.4–0.6 µm) by playing with the time and the distance from the flame while simultaneously turning the pipette. Place the glass pipettes in a clean glass container and autoclave them (using the solid mode at 105 °C for 45 min).
Prepare a polystyrene box with ice.
Prepare a plastic tube with 1.5 mL of modified dPBS (prepared in step 1.2) to transfer the pituitaries to during the dissection and keep it on ice.
Prepare a 15 mL tube with 10 mL of modified dPBS (prepared in step 1.2) and keep it on ice until the cell dissociation.
Prepare fresh or thaw aliquots of the trypsin and trypsin inhibitor solutions (prepared in steps 1.4 and 1.5) and keep them on ice until use.
Set the water bath to 26 °C before starting the dissection to allow the water to reach the desired temperature before the chemical dissociation of the pituitaries.
Clean all dissection tools with 70% ethanol before, during, and after use, including fine forceps to use for the dissection and fine needles and a wax plate to pin the fish. Use clean gloves (change and clean frequently) during the entire procedure to avoid a potential contamination of the cells.
3. Pituitary Dissection and Cell Dissociation
Euthanize the fish by immersing it in ice slush (0 °C). Leave the fish in the ice slush for at least 1 min to ensure an irreversible hypothermic shock. NOTE: Immobilization and equilibrium loss will occur after a few seconds. Make sure the fish is completely submerged in the ice water and does not lie on top of the ice, as the latter will lead to suffocation and potential skin burns instead of a rapid and humane hypothermic shock.
Quickly transfer the fish in a net to a wax plate under the dissection microscope and destroy the spine by a needle punch through the neck.
Pin the head of the fish to the wax plate using fine needles, by inserting one needle in the front (over the mouth) and one on each side of the head (behind the brain) to stabilize the head before the dissection.
View the fish under the dissection microscope and gently scrape off the scales at the top of the head using fine forceps with an angled tip.
Carefully insert the angled tip of the fine forceps under the skin from the side of the head and remove the skull roof by slowly moving the forceps in the direction of the mouth, while holding a firm grip to the forceps enclosing the skull roof.
Cut the spinal cord completely off using forceps with an angled tip. NOTE: Time is an important factor, so work efficiently and accurately to limit the time spent from the dissection of the pituitaries until the cells are plated in the dish. After some practice, pituitary dissection should take around 2–3 min per fish, of which only around 10 s are needed to pick the pituitary when the brain is already exposed.
Carefully flip the brain over towards the mouth to expose the pituitary and dissect it with clean forceps with a straight tip. Transfer the pituitary to a plastic tube containing 1.5 mL modified dPBS (for details, see step 2.3) placed on ice. Check the tip of the forceps under the microscope to ensure that the pituitary is successfully added to the tube and nothing is left on the forceps. NOTE: Dissect as many pituitaries as needed, depending on the number of culture dishes that are going to be prepared simultaneously. As a rule of thumb, calculate at least 10 pituitaries from adult fish per dish to have an appropriate density of cells (which depends on the downstream application).
Spin down the pituitaries in a tabletop centrifuge for 1–2 s at room temperature. Carefully remove the dPBS using a glass pipette and take care to avoid removing any pituitaries.
Add 1 mL of the trypsin solution (prepared in step 1.4) to wash the pituitaries, spin them down in a tabletop centrifuge for 1–2 s, and remove most of the liquid using a glass pipette before adding an additional 1 mL of trypsin solution.
Incubate the tube in a water bath at 26 °C for 30 min, preferably with gentle shaking, or alternatively, flick the tube a couple of times during the incubation period.
Spin down the pituitaries in a tabletop centrifuge for 1–2 s at room temperature and make sure all pituitaries are located at the bottom of the tube. Remove most of the trypsin solution with a glass pipette and add 1 mL of the trypsin inhibitor solution (prepared in step 1.5) to inactivate the trypsin and wash the pituitaries.
Collect the pituitaries at the bottom of the tube by spinning them down in a tabletop centrifuge for 1–2 s at room temperature. Remove most of the liquid with a glass pipette and add 1 mL of the trypsin inhibitor solution (prepared in step 1.5). Place the tube on a water bath at 26 °C for 20 min, preferably with gentle shaking, or alternatively, flick the tube a couple of times during the incubation period to inactivate the trypsin.
Prepare a 35 mm poly-d-lysine-coated cell culture dish with a central glass bottom by adding 2 mL of L-15 medium and letting that incubate for at least 10 min at 26 °C and with 1% CO2 so that it can reach the right temperature and pH before plating the cells. NOTE: A plastic cell culture dish can also be used. The main advantage of using a dish with a centralized glass bottom is that the area where the cells are plated is smaller, and therefore, fewer pituitary cells are required per dish. Some downstream applications might also require a glass bottom (i.e., some imaging techniques).
Set the cooling centrifuge for the 15 mL tubes to 4 °C to allow it to reach the desired temperature before starting the mechanical dissociation.
Collect the pituitaries at the bottom of the tube by spinning down the tube with the trypsin inhibitor solution (from step 3.11) in a tabletop centrifuge for 1–2 s at room temperature and make sure all pituitaries are located at the bottom of the tube before carefully removing most of the trypsin inhibitor solution using a glass pipette. Note: Perform all the following steps of the protocol in a laminar flow bench (LAF) certified for cell work to avoid a contamination of the cells.
Add 1 mL of ice-cold modified dPBS (prepared in step 2.4) to the pituitary tissue pieces and gently suck the tissue pieces up and down (6–7x) in a fire-polished glass pipette (prepared in step 2.1). NOTE: Temperature is an essential aspect, so keep all solutions at 4 °C. From this point onward, perform all steps on ice when possible.
Spin down the tissue pieces in a tabletop centrifuge for 1–2 s at room temperature and carefully transfer the upper part of the dPBS containing the dissociated cells (almost 1 mL) to a new 15 mL tube placed on ice. Avoid moving the pipette towards the bottom of the tube where the tissue pieces are left.
Repeat step 3.16–3.17, dissociating the cells in 1 mL of ice-cold dPBS at a time, until all 10 mL of dPBS is used. Start using a glass pipette with a medium-sized opening and switch to a glass pipette with a smaller opening towards the end (for the last 3–4 mL of dPBS added) of the dissociation. If there are still visible tissue pieces towards the end of the dissociation procedure, increase the pipetting to approximately 10x towards the 2 last steps of dPBS for dissociation, and finally leave the residual tissue pieces behind. Resuspend the cells gently and avoid creating bubbles as it may cause cell damage when the cells attach to the surface of the bubbles. NOTE: This is a critical step of this protocol and requires some training to achieve a good result.
Balance the tube in the centrifuge (pre-cooled to 4 °C) and spin down the cells at 200 x g for 10 min at 4 °C. Make sure to mark the side of the tube where the cell pellet will be.
Carefully remove the tube from the centrifuge and gently transfer the supernatant with a glass pipette to an empty 15 mL plastic tube directly after the centrifugation. Be sure to remove most of the supernatant but take care to avoid losing the small (often invisible) cell pellet.
Carefully resuspend the cell pellet in 50–100 µL of L-15 culture medium. NOTE: At this step, it is possible to count the cells and/or perform a viability test (such as a cell counting in the presence of trypan blue using a hemocytometer), but beware that using part of the cell suspension for this purpose will lower the yield. Avoid using a large suspension volume as it will increase the risk of cells diffusing outside of the center of the dish.
Carefully drip the dissociated cells in the middle of the cell culture dish containing 2 mL of the L-15 medium (prepared in step 3.13). Allow the cells to sink to the bottom of the dish for approximately 5 min before moving them to the incubator, to avoid having the cells spread outside of the sunken part of the glass bottom.
Incubate the dish at 26 °C and 1% CO2 to allow all the cells to sink thoroughly and attach to the bottom of the dish.
After 30 min, look at the cells in the microscope to make sure they have attached to the culture dish.
Continue to incubate the cells at 26 °C and 1% CO2.
Carefully change most of (but not all) the medium every 3–4 days. Note: Avoid changing the medium more frequently, as it may disturb the cells and make them detach from the glass bottom. Use the cells for experiments within a week after seeding them.
Representative Results
This protocol describes the preparation of a primary cell culture from medaka pituitaries and provides healthy cells that can be maintained in a culture for at least one week. The protocol is based on physiological relevant values for medaka14 and is additionally optimized to the pituitary tissue in adult fish, using a pH of 7.75 and an osmolality of 290 mOsm/kg during the entire procedure from the tissue harvesting to the plated cells in culture (Figure 1 and Figure 2).
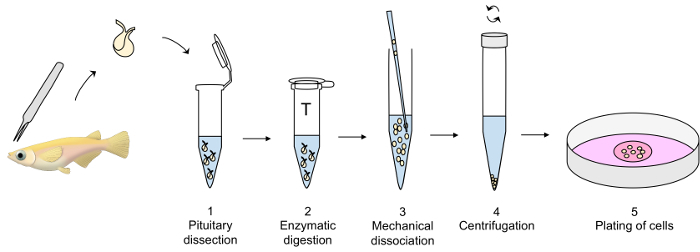
Representative results from experiments on primary cell cultures produced by this protocol are displayed in Figure 3, Figure 4, and Figure 5. For the results presented here, a transgenic medaka line, where luteinizing hormone (Lh)-gonadotrope cells express the green fluorescent protein (GFP), was utilized19. Figure 3 shows a representative field in the dish of a primary cell culture from day 0 to day 7 after seeding, including both bright-field and fluorescent microscopy images, showing the GFP-expressing Lh-gonadotrope cells. Using around 10 adult medaka pituitaries per dish usually results in a density of around 5 x 104 – 1 x 105 cells per dish after seeding. The viability of the pituitary cells in culture is calculated based on the total number of GFP-expressing cells in the culture at day 1, 3, and 7 after plating, as compared to day 0 (Figure 4). The graph shown in Figure 4 indicates that the number of dying cells is nearly constant over time, with over 95% of the cells alive after 1 day, while over 90% is still alive after 3 days. Although there is a small decline in cell viability with time, most cells (around 75%) are still alive after a week in culture. Electrophysiological recordings of GFP-expressing Lh-gonadotrope cells (performed as described in Strandabø, et al.)20 show that the cells are able to fire action potentials spontaneously after 4 days in culture (Figure 5A). Further, upon stimulation of the gonadotropin-releasing hormone, an initial transient hyperpolarization of the cell membrane is followed by a dramatic increase in firing frequency that is concomitant with a depolarization and increased duration of the action potentials (Figure 5B).

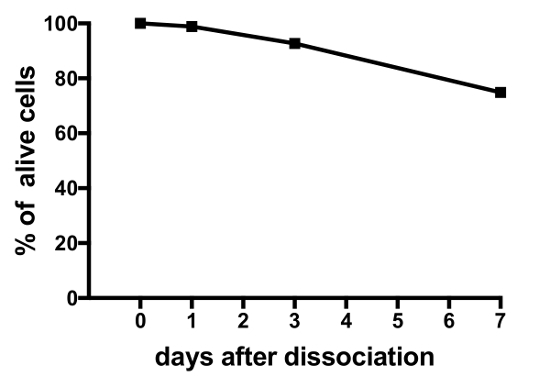
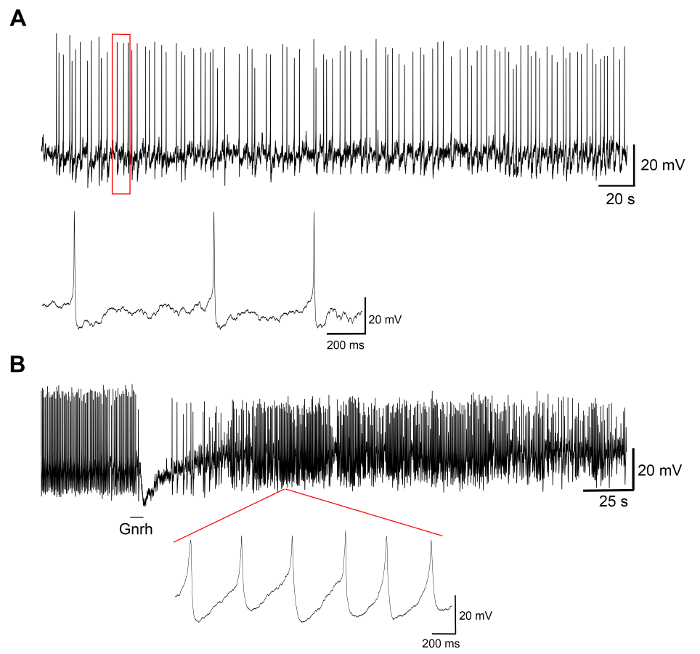
Figure 1: Overview of the pituitary primary cell culture technique. Pituitaries are dissected with fine forceps and transferred to a plastic tube containing modified dPBS on ice (step 1 of the protocol) until the enzymatic digestion of the pituitaries with trypsin at 26 °C (step 2), followed by an inactivation with a trypsin inhibitor at 26 °C (not shown). The mechanical dissociation of the enzymatically digested pituitaries with a glass pipette is carefully performed in dPBS on ice (step 3), followed by a centrifugation at 4 °C (step 4). Finally, the cell pellet is dissolved in culture medium and the cells plated out in a cell culture dish (step 5). Please click here to view a larger version of this figure.
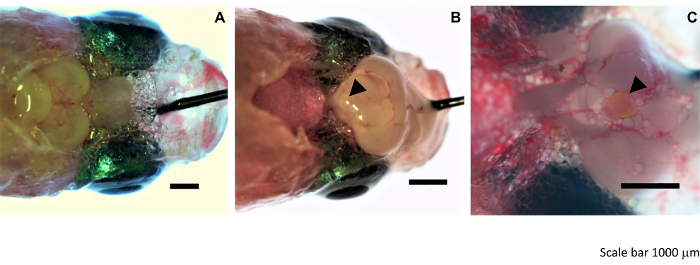
Figure 2: Details of the medaka pituitary dissection. (A) The head of an immobilized medaka (dorsal view) is pinned to a wax plate, and the top of the skull roof is removed with fine forceps to expose the top of the brain. (B) The spinal cord is severed, and the brain flipped over toward the front of the head such that the pituitary is exposed. (C) The pituitary is collected with clean, fine forceps. Arrowhead points to the pituitary in panel B and C. Scale bar = 1,000 µm. Please click here to view a larger version of this figure.
Figure 3: Medaka pituitary primary cell culture. These panels show high-resolution confocal images of a representative medaka pituitary primary cell culture at day 0, day 3, and day 7 after seeding. The upper panel shows bright-field microscopy images of a representative field in the dish, the middle panel shows fluorescent microscopy images of the same field of view displaying GFP-expressing Lh-gonadotrope cells, and the lower panel is an overlay of the two. Scale bar = 20 µm. Please click here to view a larger version of this figure.
Figure 4: Viability of medaka pituitary cells in culture. This figure shows the viability of the pituitary primary cell cultures measured at day 0, 1, 3, and 7 after seeding. The numbers are calculated using the open-source software CellProfiler V2 and are based on the total number of GFP-expressing Lh-gonadotrope cells per dish at the different time points, compared to time 0. Please click here to view a larger version of this figure.
Figure 5: Electrophysiological recordings of pituitary cells. These panels show electrophysiological recordings from a GFP-expressing Lh-gonadotrope cell in a primary cell culture performed at day 4 after seeding, showing (A) a representative trace of spontaneous action potentials, followed by (B) a stimulation with the gonadotropin-releasing hormone Gnrh1 for 10 s. The Gnrh1 stimulation induces a biphasic response with a hyperpolarization of the cell membrane, followed by a depolarization and increased duration of the action potentials (measured at 50% peak) from 8.9 ± 3.0 ms before the stimulation to 29.2 ± 9.9 ms after the stimulation. Please click here to view a larger version of this figure.
Discussion
In vitro cell culture systems provide powerful tools for researchers to answer a plethora of different biological questions if used in the right way1. It is important to remember that dissociated cells that have lost their connections to the neighboring cells may have obtained different functional properties than they originally had in vivo. To avoid being at the risk of misinterpreting the results obtained from in vitro experiments, it is important to consider adjusting the primary cell culture procedure to the species and cell type of interest. Even moderate adjustments of standard culture procedures and solutions used are often sufficient to profoundly alter the culture conditions of a specific cell type.
This protocol describes optimized conditions for preparing and maintaining primary cell cultures from adult medaka pituitaries, which can be achieved by adjusting parameters such as the temperature, osmolality, pH, and pCO2 of the solutions used, to match the physiological conditions of this species. The pH is temperature dependent and should, therefore, also be adjusted between different species of teleost fish living at different temperatures17. The osmolality of all solutions used during cell dissociation and culturing should also be adjusted in order to improve the condition of the cells. Especially, if there is a discrepancy in osmolality between different solutions used during the isolation procedure, it can have devastating effects on the culture quality, leading to a decreased viability (the authors' own unpublished results). Hyperosmotic solutions will cause the cells to shrink (and vice versa), thereby interfering with the membrane integrity and membrane protein function21. An important point in this context is that parameters such as osmolality and pH are not usually measured nor stable between different batches of commercial buffers and culture media used in mammalian culture conditions. Differences in osmolality and pH between different solutions used during the isolation procedure should be avoided at all times.
The success rate of this protocol is improved by training, so it is expected researchers need some time to get acquainted with the different steps of the technique. For instance, the time from dissection of the pituitaries until the cells are plated in the dish is inversely related to cell health. The mechanical dissociation of the pituitaries using a glass pipette is a particularly critical step of the protocol and it requires some training to achieve a good result. The dissociation procedure can introduce osmotic and mechanical stress that is harmful to the cells. The addition of small volumes of modified dPBS in repeated steps for the mechanical dissociation ensures a gentler dissociation procedure where already dissociated cells are transferred to a new tube and left untouched, while the researcher continues to dissociate additional cells from the tissue in a new volume of dPBS.
This protocol is optimized for adult medaka pituitary cells. Different species, tissues, or even different life stages of the animal within the same tissue require optimizations of the culture conditions. For instance, cells obtained from immature animals may be more fragile and, therefore, more susceptible towards apoptosis and cell death and, thus, may necessitate the need to adjust the protocol, especially toward a gentler dissociation procedure. The mechanical dissociation using a glass pipette is a very critical step of the procedure, as a too careful mechanical dissociation will lead to a lower gain, while a too rough one may induce apoptosis and/or cell death. If different enzymes are considered for dissociation, it might be necessary to use a different digestion time and enzyme concentration. Medaka pituitaries are very small and relatively easy to dissociate using the described protocol. However, optimizations for other species might require the use of a mesh filter after the dissociation to remove debris. The success of this protocol also depends largely on performing the steps under sterile conditions to avoid contaminating the cells.
The number of cells to seed per dish depends on the number of pituitaries at the start, as well as the success of the dissociation procedure. There are roughly around 15,000 cells in an intact adult medaka pituitary. However, cells are lost during the dissociation process and this should be accounted for when calculating the number of pituitaries needed for an experiment. Using at least 10 pituitaries per dish will usually result in a density of around 50,000 – 100,000 cells per dish. At this density, aggregation does not form in any significant amount, even after 7 days in culture. Noticeably, aggregation seems to be density dependent, as the denser the cells are seeded, the more they seem to aggregate. Certain procedures may require a denser culture and, therefore, the optimal seeding density should be considered depending on the downstream application. It is an important point to take into consideration when planning the number of pituitaries to use per dish.
Most common downstream applications use primary cell cultures within the first few days after seeding. The results presented here, from the cell viability to the electrophysiological recordings, show that it is possible to obtain good results from primary cell cultures prepared using the described protocol for up to 1 week after seeding. Nevertheless, cultures may be kept longer, and healthy cells are still present after over 2 weeks in culture (the authors' own unpublished results).The optimized conditions presented in this protocol facilitates physiologically meaningful results and may be beneficial for other scientists preparing primary cell cultures from non-mammalian cells.
Disclosures
The authors have nothing to declare.
Acknowledgments
This project was funded by the Norwegian University of Life Sciences and the Research Council of Norway, grant number 243811 and 244461 (Aquaculture program). We are grateful to Lourdes Carreon Tan at the Norwegian University of Life Sciences for maintaining the fish facility.
References
- Freshney RI. Culture of Animal Cells: A Manual of Basic Technique and Specialized Applications. Hoboken, NJ: John Wiley & Sons; 2010. [Google Scholar]
- Ribeiro LP, Ahne W. Fish cell culture: initiation of a line of pituitary cells from carp (Cyprinus carpio) to study the release of gonadotropin in vitro. In Vitro. 1982;18(5):419–420. doi: 10.1007/BF02796467. [DOI] [PubMed] [Google Scholar]
- Ribeiro L, Ahne W, Lichtenberg V. Primary culture of normal pituitary cells of carp (Cyprinus carpio) for the study of gonadotropin release. In Vitro. 1983;19(1):41–45. doi: 10.1007/BF02617992. [DOI] [PubMed] [Google Scholar]
- Wong AO, Ng S, Lee EK, Leung RC, Ho WK. Somatostatin inhibits (d-Arg6, Pro9-NEt) salmon gonadotropin-releasing hormone- and dopamine D1-stimulated growth hormone release from perifused pituitary cells of chinese grass carp, Ctenopharyngodon idellus. General and Comparative Endocrinology. 1998;110(1):29–45. doi: 10.1006/gcen.1997.7045. [DOI] [PubMed] [Google Scholar]
- Chang JP, et al. Use of a pituitary cell dispersion method and primary culture system for the studies of gonadotropin-releasing hormone action in the goldfish, Carassius auratus. I. Initial morphological, static, and cell column perifusion studies. General and Comparative Endocrinology. 1990;77(2):256–273. doi: 10.1016/0016-6480(90)90310-i. [DOI] [PubMed] [Google Scholar]
- Weil C, Hansen P, Hyam D. Use of pituitary cells in primary culture to study the secretion in rainbow-trout - Setting up and validating the system as assessed by its responsiveness to mammalian and salmon gonadotropin-releasing hormone. General and Comparative Endocrinology. 1986;62(2):202–209. doi: 10.1016/0016-6480(86)90110-3. [DOI] [PubMed] [Google Scholar]
- Montero M, LeBelle N, Vidal B, Dufour S. Primary cultures of dispersed pituitary cells from estradiol-pretreated female silver eels (Anguilla anguilla L): Immunocytochemical characterization of gonadotropic cells and stimulation of gonadotropin release. General and Comparative Endocrinology. 1996;104(1):103–115. doi: 10.1006/gcen.1996.0146. [DOI] [PubMed] [Google Scholar]
- Xu SH, Cooke IM. Voltage-gated currents of tilapia prolactin cells. General and Comparative Endocrinology. 2007;150(2):219–232. doi: 10.1016/j.ygcen.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Lin SW, Ge W. Differential regulation of gonadotropins (FSH and LH) and growth hormone (GH) by neuroendocrine, endocrine, and paracrine factors in the zebrafish-An in vitro approach. General and Comparative Endocrinology. 2009;160(2):183–193. doi: 10.1016/j.ygcen.2008.11.020. [DOI] [PubMed] [Google Scholar]
- Hodne K, von Krogh K, Weltzien FA, Sand O, Haug TM. Optimized conditions for primary culture of pituitary cells from the Atlantic cod (Gadus morhua). The importance of osmolality, pCO2, and pH. General and Comparative Endocrinology. 2012;178(2):206–215. doi: 10.1016/j.ygcen.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Wittbrodt J, Shima A, Schartl M. Medaka - a model organism from the far East. Nature Reviews Genetics. 2002;3:53–64. doi: 10.1038/nrg704. [DOI] [PubMed] [Google Scholar]
- Matsuda M, et al. DMY is a Y-specific DM-domain gene required for male development in the medaka fish. Nature. 2002;417(6888):559–563. doi: 10.1038/nature751. [DOI] [PubMed] [Google Scholar]
- Kasahara M, et al. The medaka draft genome and insights into vertebrate genome evolution. Nature. 2007;447(7145):714–719. doi: 10.1038/nature05846. [DOI] [PubMed] [Google Scholar]
- Miyanishi H, Inokuchi M, Nobata S, Kaneko T. Past seawater experience enhances seawater adaptability in medaka, Oryzias latipes. Zoological Letters. 2016;2(12) doi: 10.1186/s40851-016-0047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waymouth C. Osmolality of mammalian blood and of media for culture of mammalian cells. In Vitro. 1970;6(2):109–127. doi: 10.1007/BF02616113. [DOI] [PubMed] [Google Scholar]
- Cameron JN. Acid-base homeostasis - past and present perspectives. Physiological Zoology. 1989;62(4):845–865. [Google Scholar]
- Claiborne JB, Edwards SL, Morrison-Shetlar AI. Acid-base regulation in fishes: cellular and molecular mechanisms. Journal of Experimental Zoology. 2002;293(3):302–319. doi: 10.1002/jez.10125. [DOI] [PubMed] [Google Scholar]
- Perry SF, Tufts BL. Fish respiration. San Diego, CA: Academic Press; 1998. [Google Scholar]
- Hildahl J, et al. Developmental tracing of luteinizing hormone β-subunit gene expression using green fluorescent protein transgenic medaka (Oryzias latipes) reveals a putative novel developmental function. Developmental Dynamics. 2012;241(11):1665–1677. doi: 10.1002/dvdy.23860. [DOI] [PubMed] [Google Scholar]
- Strandabø RAU, et al. Signal transduction involved in GnRH2-stimulation of identified LH-producing gonadotropes from lhb-GFP transgenic medaka (Oryzias latipes) Molecular and Cellular Endocrinology. 2013;372(1-2):128–139. doi: 10.1016/j.mce.2013.03.022. [DOI] [PubMed] [Google Scholar]
- Verbalis JG. Brain volume regulation in response to changes in osmolality. Neuroscience. 2010;168(4):862–870. doi: 10.1016/j.neuroscience.2010.03.042. [DOI] [PubMed] [Google Scholar]