

Abstract
We herein report the case of a 76-year old man with aquaporin-4-Immunoglobulin-G(AQP4-IgG)-positive neuromyelitis optica spectrum disorder (NMOSD), in whom transient interstitial pulmonary lesions developed at the early stage of the disease. Chest X-ray showed multiple infiltrative shadows in both upper lung fields, and computed tomography revealed abnormal shadows distributed randomly in the lungs. Surgical lung biopsy showed features of unclassifiable interstitial pneumonia, characterized by various types of air-space organization, which resulted in obscure lung structure. This is the first report to describe the pathological findings of interstitial pneumonia, which may represent a rare extra-central nervous system complication of NMOSD.
Keywords: neuromyelitis optica spectrum disorder, hiccup, hyperCKemia, interstitial pneumonia
Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is a type of autoimmune disease that involves the central nervous system (CNS), particularly the spinal cord and optic nerve. Recently, immunoglobulin (Ig)G antibodies against the water channel protein aquaporin4 (AQP4) were discovered to play a key role in the pathogenesis of AQP4-IgG-positive NMOSD. The diagnostic criteria for NMOSD were renewed in 2015 (1), and various aspects of the disease have since been elucidated. A small number of reports have described NMOSD accompanied by interstitial pneumonia. In those cases, the pulmonary lesions were described as a part of other collagen vascular diseases accompanying NMOSD (2-5), or as occurring concomitantly with the transient elevation of creatine kinase (CK) in the early stage of the disease (6-8). None of those reports focused on the pulmonary lesions themselves.
We herein report a case of AQP4-IgG-positive NMOSD that was accompanied, in the early stage of the disease, by transient pulmonary interstitial lesions with elevated serum concentrations of CK and with brainstem symptoms such as intractable hiccups followed by vomiting. Lung biopsy was performed by video-associated thoracic surgery (VATS). Whether the pulmonary lesions represent a complication of AQP4-IgG-positive NMOSD itself has yet to be established. The present case may thus provide valuable insights into this autoimmune astrocytopathic disease.
Case Report
The patient was a 76-year-old man. Five days before admission, he experienced a sudden onset of intermittent hiccups followed by vomiting. He did not have any antecedent signs, such as flu-like symptoms. The day before his admission, he had symptoms of muscle weakness and numbness in the right leg, and was admitted to National Hospital Organization Saitama National Hospital. No respiratory symptoms or aspiration episodes were noted. His past medical history included hypertension, diabetes, dyslipidemia and angina. There had been no changes to any of the patient's medications prior to these episodes. He had no history of rheumatic disease, and denied any arthritis, uveitis or skin trouble indicative of collagen vascular disease. He had a 20 pack-year smoking history, but had quit smoking 20 years earlier.
On admission, his height was 164.1 cm and weight was 51.4 kg. His vital signs were as follows: body temperature, 37.1℃; blood pressure, 155/92 mmHg; heart rate, 83 beats/min; and oxygen saturation, 96%. His consciousness was alert, and despite dehydration due to intermittent, repetitive hiccups followed by vomiting, physical examinations yielded normal results, including respiratory sounds. A neurological examination revealed muscle weakness with enhanced tendon reflexes and hypesthesia in the right lower leg. Clinical tests of the cranial nerves and cerebellum showed no abnormalities. Spontaneous pain or grasp pain, which would have been indicative of myositis, were absent. In laboratory investigations, an increased urea nitrogen level (11.8 mmol/L) was seen as a result of dehydration, as well as elevated hemoglobin A1c (8.9%), indicative of diabetes. An analysis of the patient's cerebrospinal fluid revealed slightly elevated glucose (1.29 g/L) and protein (1.24 g/L) concentrations with a normal cell count (1/3 /μL). The patient was negative for anti-nuclear antibodies and other autoantibodies suggestive of collagen vascular disease. An elevated CK concentration of 719 IU/L (normal, 62-287 IU/L) and a Krebs von den Lungen (KL)-6 level of 1,854 U/mL (normal, <500 U/mL) were observed. No neuroelectrical examinations (i.e. electromyography) were performed.
Chest X-ray (Fig. 1) showed multiple infiltrative shadows, mainly in the bilateral upper lobes of the lungs. However, the lungs appeared normal and these abnormal findings were not seen on a chest X-ray film taken approximately 10 months before the onset of these episodes. Computed tomography (CT) revealed multiple irregular, non-segmental, infiltrative shadows, predominantly in the bilateral upper lobes (Fig. 2). Multiple nodular and patchy shadows of various shades were also seen in the caudal parts of the lungs. At first glance, the abnormal shadows were mostly centered at the bronchovascular bundle, and lesions in some areas were spread out under the pleura and in areas unrelated to the blood vessels. These findings indicated that the shadows were distributed randomly. A bronchoscopic examination showed no remarkable findings, with normal bacterial flora. Neither acid-fast bacilli nor malignant cells were detected.
Figure 1.
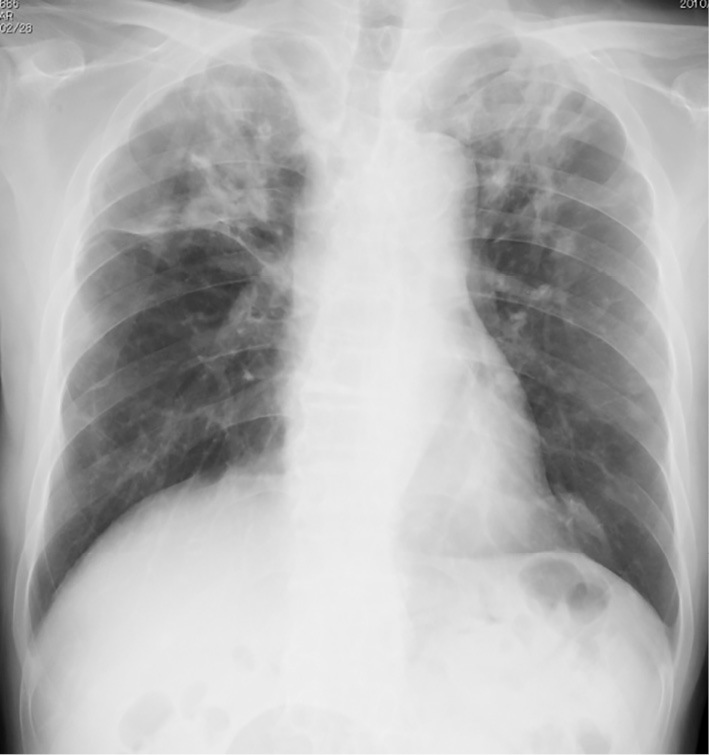
Chest X-ray on admission. Multiple infiltrative shadows were present in both upper lung fields. Small areas of nodular or patchy shadows were also seen in the mid-to-lower lung fields. These findings had not been present 10 months earlier.
Figure 2.

Chest CT. Two slices from CT performed at the time of admission. a) The upper lobes of both lungs showed numerous irregular and non-segmental infiltrative shadows. The distribution mainly appeared to be centered on the bronchovascular bundle. b) In the caudal parts, several nodular or patchy shadows of various shades were present. All of these shadows were randomly distributed in the subpleural area or in areas unrelated to vessels.
To confirm the diagnosis, a lung biopsy was performed under VATS on hospital day 23. Under thoracoscopic observation, the surface of the visceral pleura showed patchy areas of yellow-gray discoloration. Biopsies were performed based on the lesions in segments 4 and 5 of the right lung. A pathological examination showed various findings. Histologically, there was inflammatory thickening of the alveolar wall together with various types of intraluminal organization with indistinct and rather distinct margins. Due to the various types of intraluminal organization, the lung structure became obscure (Fig. 3). These lesions were not characteristic of organizing pneumonia or non-specific interstitial pneumonia, and were diagnosed as unclassifiable interstitial pneumonia.
Figure 3.
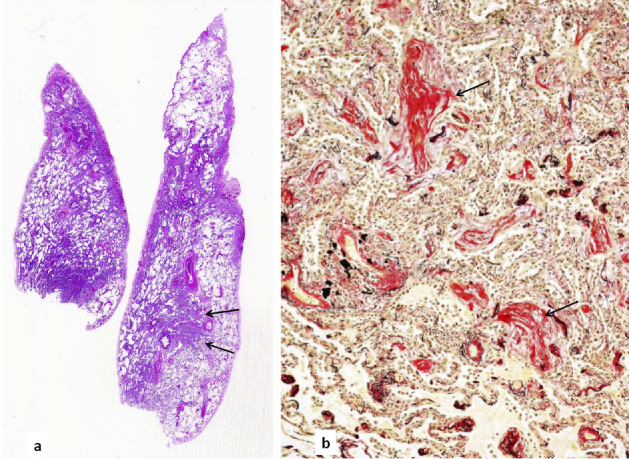
The histology of the biopsied lung. a) Panoramic images of Hematoxylin and Eosin staining lung specimens. On the left side (biopsied from segment 4a of the right lung), diffusely spreading interstitial pneumonia was obvious. On the right side (from the diaphragmatic part of segment 5), areas of relatively distinctly partitioned healthy lung and nodule-shaped intraluminal organization were clear (arrows). b) The nodular area was stained for elastic fibers. The lung structure was indistinct because of interstitial thickening and intraluminal organization (arrows). Magnification: ×100
With regard to the clinical course, a diagnosis of NMOSD could not be made for more than 4 weeks after admission. Detailed examinations were performed while using metoclopramide and anticonvulsants to treat the severe hiccups. Brain magnetic resonance imaging (MRI) performed on day 2 after admission revealed no abnormalities. Thereafter, the patient's hiccups gradually improved with the improvement of the pulmonary lesion, over the course of approximately 5 weeks. Despite the effective treatment of dehydration due to vomiting at the time of admission, the patient's CK levels remained high (500-700 IU/L), with gradual normalization paralleling the improvement of his hiccups. However, from around this time, the muscle weakness of the upper and lower extremities worsened, and painful lower leg spasms occurred. The left lower extremity weakness then progressed, causing paraplegia and gait disturbance. Sensory disturbance was evident from the level of the sixth cervical nerve to the caudal area. Bladder and bowel disturbances also developed from 7 weeks after admission. Cervical MRI performed after the worsening of the patient's neurological symptoms showed high-intensity signals at the C1-2 and C4-6 levels on T2-weighted imaging, which confirmed longitudinal extensive transverse myelitis (LETM) (Fig. 4). At the same time, an in-house cell-based assay performed at Tohoku University revealed that the patient was positive for AQP4-IgG. The combination of severe hiccups and vomiting, neurological symptoms with LETM on cervical MRI, and AQP4-IgG positivity indicated a diagnosis of AQP4-IgG-positive NMOSD. The patient's neurological symptoms gradually improved after the administration of high-dose steroids and immunoadsorption therapy. Following several courses of immunoadsorption therapy and rehabilitation with the tapering of the doses of steroid, the patient's bladder and bowel disturbances improved and he regained the ability to walk, leading to his discharge at 4 months after the induction of therapy (Fig. 5). At the time of discharge, a chest X-ray revealed apparently normal findings. After the confirmation of AQP4-IgG-positive NMOSD, we attempted to stain lung tissue specimens for AQP4 using immunohistochemical methods, but could not obtain sufficient staining of the lung tissue.
Figure 4.
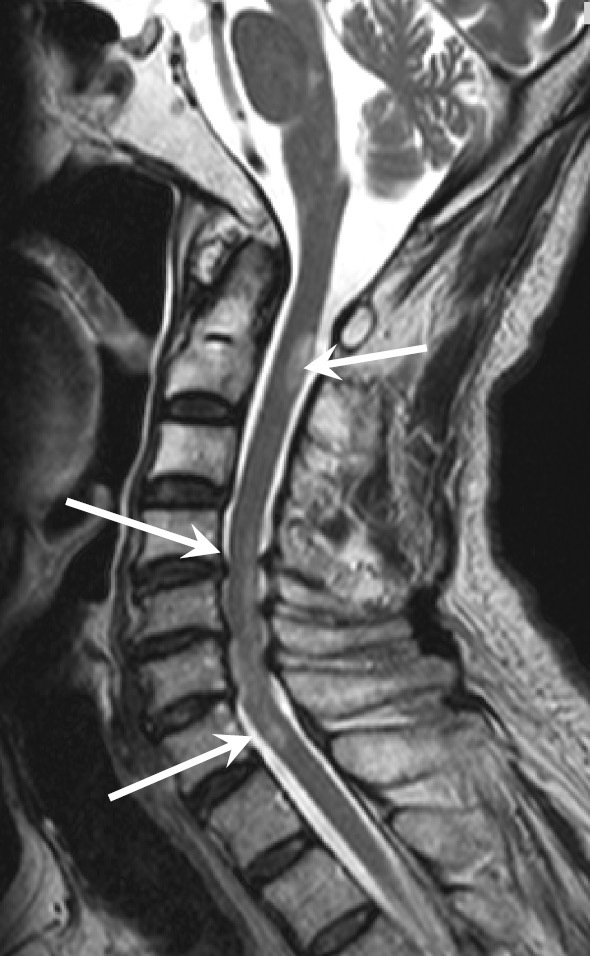
Cervical MRI. Cervical MRI was performed after the worsening of the patient's neurological symptoms. Arrows show high-intensity signals on T2-weighted imaging.
Figure 5.

The clinical course. Pulmonary lesion, intractable hiccups and vomiting, and hyperCKemia improved before the worsening of myelopathy which was a trigger for the diagnosis of NMOSD.
Discussion
NMO has previously been called Devic's disease, and has been considered a subtype of multiple sclerosis (MS). However, after autoantibody targeting AQP4 was discovered to play an important role in the diagnosis and pathogenesis, NMO was recognized as a different disease from MS. In 2015, the diagnostic criteria were revised and unified under the term NMOSD (1). In adults, two subtypes were classified according to the presence or absence of AQP4-IgG: NMOSD with AQP4-IgG and NMOSD without AQP4-IgG, respectively. According to these new criteria, the LETM and intractable hiccups with vomiting (area postrema syndrome) in our patient matched some of the “six core clinical characteristics”. The detection of AQP4-IgG antibodies confirmed the diagnosis of NMOSD with AQP4-IgG in this case.
In the human lung, AQP4 has been recognized as being broadly present at the basolateral membrane of the ciliated epithelium from the upper to lower respiratory tract (9). The function of AQP4 in the respiratory epithelium remains unclear, although the regulation of humidity at the mucosal membrane surface has been suggested as one possible role. However, no marked changes were observed in AQP4-knockout mice (10).
In some cases of lung cancer, AQP4 is known to be strongly expressed in cancer cells (11). AQP1 is known to play an important role in pedicle formation when cancer cells protrude or invade into the adjacent tissue (12). Whether AQP4 plays a role in making the foot processes and regulating invasion, similar to AQP1, has not been confirmed. On the other hand, NMOSD can occur as a paraneoplastic syndrome of non-small cell lung cancer (13). Among cases of NMOSD in elderly patients, the risk of malignancy may be increased (14). These findings may suggest a relationship between AQP4 in respiratory epithelium and NMOSD. The accumulation of further insights is necessary to clarify the function of AQP4 in the respiratory tract.
Some reports have described NMOSD complicated with other autoimmune disorders, such as Sjögren's syndrome (2), systemic lupus erythematosus (3), scleroderma (4), dermatomyositis (5), and myasthenia gravis (15). In some of those reports, interstitial pneumonia was described as a complication of collagen vascular disease. However, because those reports did not focus on pulmonary lesions, we cannot compare the pulmonary findings from our case due to the lack of imaging and pathological observations.
In our case, intractable hiccups and vomiting, as area postrema syndrome, were prominent in the early stage of NMOSD. At the same time, CK elevation and interstitial pulmonary lesions were observed. It is notable that despite the presence of these infiltrates on a chest X-ray, the patient had no respiratory symptoms. CK elevation is reported as one of the extra-CNS signs of NMOSD (6). Several case reports have described CK elevation and pulmonary infiltration accompanying early-stage NMOSD (6-8). Suzuki et al. reported three cases of NMOSD with hyperCKemia. In this report, two cases had pulmonary lesions, and in one case, pulmonary infiltration spontaneously disappeared before treatment, as occurred in our case (6). Malik et al. reported two cases of NMO with CK elevation. In one case, pulmonary infiltrates were observed, but were considered to represent pneumonia (i.e., infectious causes), and the report did not include a description of the clinical course of the pulmonary lesions (7). Langille et al. also reported a case with pulmonary left lower lobe consolidation as an extra-CNS manifestation of NMOSD (8). In these reports, as in our case, hyperCKemia or pulmonary lesions only appeared in the initial phase of the disease, but no definitive pathophysiological explanation for this clinical feature was found. Ratelade et al. offered a good suggestion for understanding this initial feature of NMOSD using an animal model (16). They intravenously injected recombinant monoclonal human NMO-IgG into mice and stained the peripheral organs and CNS for AQP4 and NMO-IgG after the injection. They described NMO-IgG binding to the AQP4 present on cell membrane in the skeletal muscle, kidney (collecting duct), trachea (epithelial cells), and stomach (parietal cells). In their experiments, NMO-IgG was only stained on astrocytes in the area postrema of the brain, and not elsewhere in other areas of the CNS. AQP4 is present in many organs at birth, and immunological tolerance to AQP4 should thus be established. Once AQP4-IgG is produced by the disruption of such immunological tolerance, hyperCKemia and area postrema syndrome or pulmonary lesions could occur in the initial stage of the disease as hypothesized based on the experiments of Ratelade et al. (16). However, many questions remain, such as how AQP4-IgG targets other sites of the CNS in the late stage, why hyperCKemia and area postrema syndrome regress within a limited period, and what differences exist between cases with or without hyperCKemia. The further accumulation of cases and experimental results will be necessary to answer these questions.
On pathological investigation, this pulmonary lesion was classified as unclassifiable interstitial pneumonia. Organizing pneumonia can be denied because of the unpreserved lung structure and non-specific interstitial pneumonia can be denied because of the high degree of intraluminal organization. We attempted to investigate AQP4 in the lung tissue immunohistochemically; however, the specimens obtained by VATS were not sufficiently stained to allow evaluation. The reason for the poor stainability was the small number of bronchial epithelial cells with AQP4 on the surface. In the CNS, AQP4 is strongly expressed on the foot processes of astrocytes, and AQP4 stainability is lost in lesions from NMOSD. The poor stainability of our lung biopsy specimen made it impossible to determine whether the lesion had lost AQP4 stainability. Thus, we therefore could not confirm immunohistochemically whether anti-AQP4 IgG played a definitive role in the pathogenesis of this pulmonary interstitial lesion. However, the pulmonary lesion developed as NMOSD began, accompanying the CK elevation and brainstem symptoms in the early stage of disease. Because other autoimmune disorders or drug-induced pneumonitis can be ruled out based on the patient's medical history, and for the reasons described above, we concluded that this pulmonary lesion may represent an extra-CNS sign of NMOSD. Further investigations are necessary to confirm this phenomenon.
Finally, we must consider whether severe hiccups may produce pulmonary edema via strong negative intrathoracic pressure. In the medical literature, one report described a case of pulmonary edema due to severe hiccups (17). However, the pulmonary edema in that report demonstrated a central pattern, and resembled the pulmonary edema induced by cardiac failure. Another possibility is that tiny aspirations due to strong hiccups may induce pulmonary lesions. As our case showed neither respiratory symptoms nor C-reactive protein or white blood cell elevation, and no inflammatory processes were evident on bronchoscopic examination, aspiration was not considered a cause of the patient's pulmonary lesions. Thus, we do not consider that the pulmonary lesions occurred due to strong hiccups.
Conclusion
We encountered a case of AQP4-IgG-positive NMOSD with pulmonary interstitial lesions in the early stage of the disease. Whether AQP4-IgG contributed to the pulmonary lesions was unclear. Our case and others suggest that NMOSD may occasionally be accompanied by pulmonary lesions. We expect that further insights into the relationship between AQP4 and NMOSD in the human airway will continue to be reported.
The authors state that they have no Conflict of Interest (COI).
References
- 1.Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85: 177-189, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qiao L, Wang Q, Fei Y, et al. The clinical characteristics of primary Sjögren's syndrome with neuromyelitis optica spectrum disorder in China: A STROBE-compliant article. Medicine (Baltimore) 94: e1145, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polgar A, Rozsa C, Muller V, Matolesi J, Poor G, Kiss EV. Devic's syndrome and SLE: challenges in diagnosis and therapeutic possibilities based on two overlapping cases. Autoimmun Rev 10: 171-174, 2011. [DOI] [PubMed] [Google Scholar]
- 4.Franciotta D, Zardini E, Caporali R, et al. Systemic sclerosis in aquaporin-4 antibody-positive longitudinally extensive transverse myelitis. J Neurol Sci 303: 139-141, 2011. [DOI] [PubMed] [Google Scholar]
- 5.Delman D, Peng X, Zedek DC, Jewells V, Chahin N, Markovic-Plese S. Dermatomyositis as a presentation of neuromyelitis optica spectrum disorder. J Neuroimmunol 278: 108-111, 2015. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki N, Takahashi T, Aoki M, et al. Neuromyelitis optica preceded by hyperCKemin episode. Neurology 74: 1543-1545, 2010. [DOI] [PubMed] [Google Scholar]
- 7.Malik R, Lewis A, Cree BAC, et al. Transient hyperCKemia in the setting of neuromyelitis optica (NMO). Muscle Nerve 50: 859-862, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langille MM, Desai J. Multisystem involvement in neuromyelitis optica. Ann Indian Acad Neurol 18(Suppl 1): S56-S58, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mobasheri A, Marples D, Young IS, Floyd RV, Moskaluk CA, Frigeri A. Distribution of the AQP4 water channel in normal human tissues: protein and tissue microarrays reveal expression in several new anatomical locations, including the prostate gland and seminal vesicles. Channels (Austin) 1: 29-38, 2007. [PubMed] [Google Scholar]
- 10.Verkman AS. Role of aquaporins in lung liquid physiology. Respir Physiol Neurobiol 159: 324-330, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iorio R, Rindi G, Erra C, Damato V, Ferilli M, Sabatelli M. Neuromyelitis optica spectrum disorder as a paraneoplastic manifestation of lung adenocarcinoma expressing aquaporin-4. Multi Scler 21: 791-794, 2015. [DOI] [PubMed] [Google Scholar]
- 12.Hu J, Verkman AS. Increased migration and metastatic potential of tumor cells expressing aquaporin water channels. FASEB J 20: 1892-1894, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Vershuur CVM, van der Kooi AJ, Troost D. Anti-aquaporin 4 related paraneoplastic neuromyelitis optica in the presence of adenocarcinoma of the lung. Clin Neuropathol 34: 232-235, 2015. [DOI] [PubMed] [Google Scholar]
- 14.Ontaneda D, Fox RJ. Is neuromyelitis optica with advanced age of onset a paraneoplastic disorder? Int J Neurosci 124: 509-511, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uzawa A, Mori M, Iwai Y, et al. Association of anti-aquaporin-4 antibody-positive neuromyelitis optica with myasthenia gravis. J Neurol Sci 287: 105-107, 2009. [DOI] [PubMed] [Google Scholar]
- 16.Ratelade J, Bennett JL, Verkman AS. Intravenous neuromyelitis optica autoantibody in mice targets aquaporin-4 in peripheral organs and area postrema. PLoS One 6: e27412, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stuth EAE, Stucke AG, Berens RJ. Negative-pressure pulmonary edema in a child with hiccups during induction. Anesthesiology 93: 282-284, 2000. [DOI] [PubMed] [Google Scholar]